Thermal Expansion and Other Thermodynamic Properties of α2-Ti3Al and γ-TiAl Intermetallic Phases from First Principles Methods




Abstract
:1. Introduction
2. Methods
3. Results
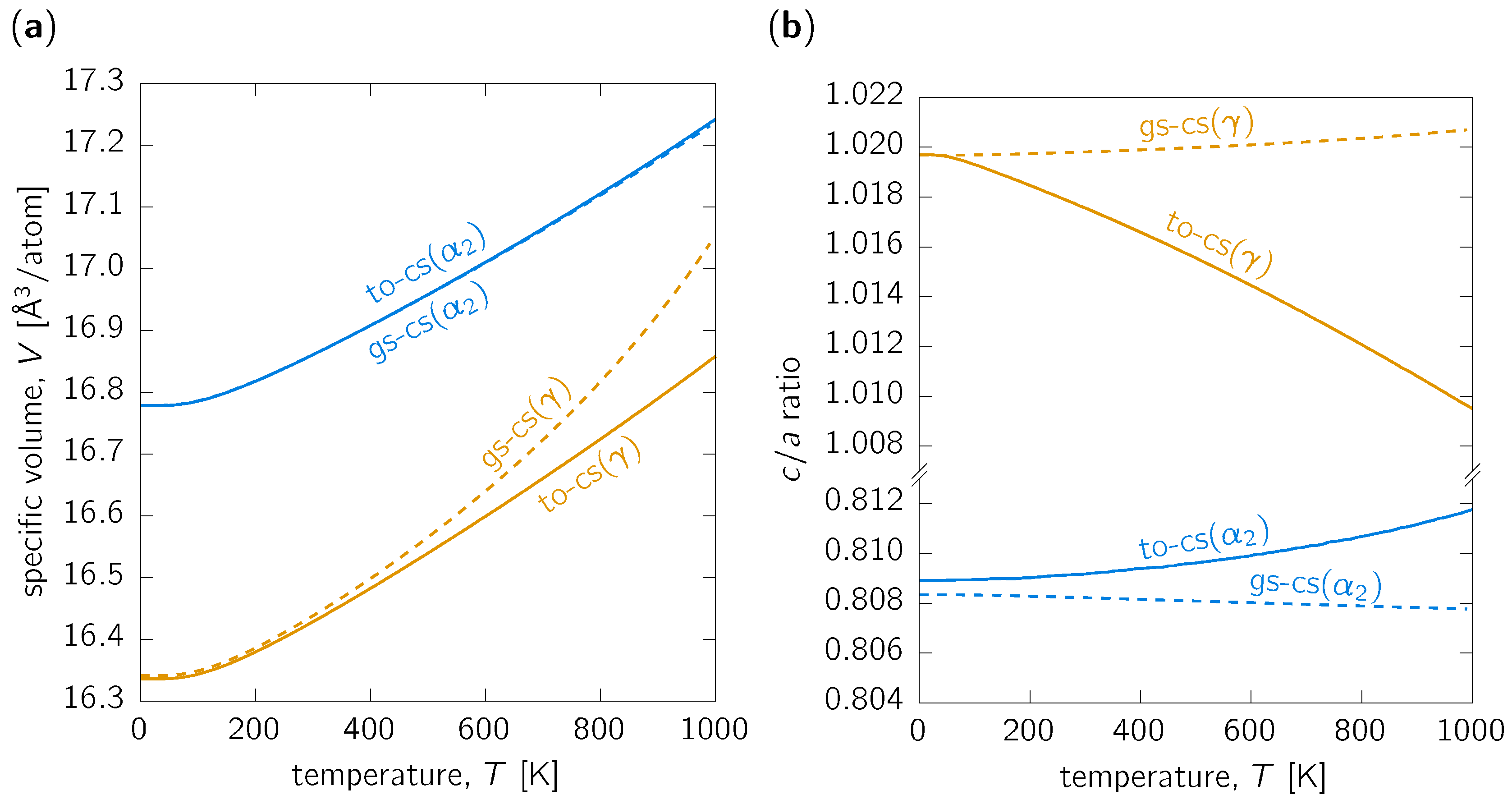
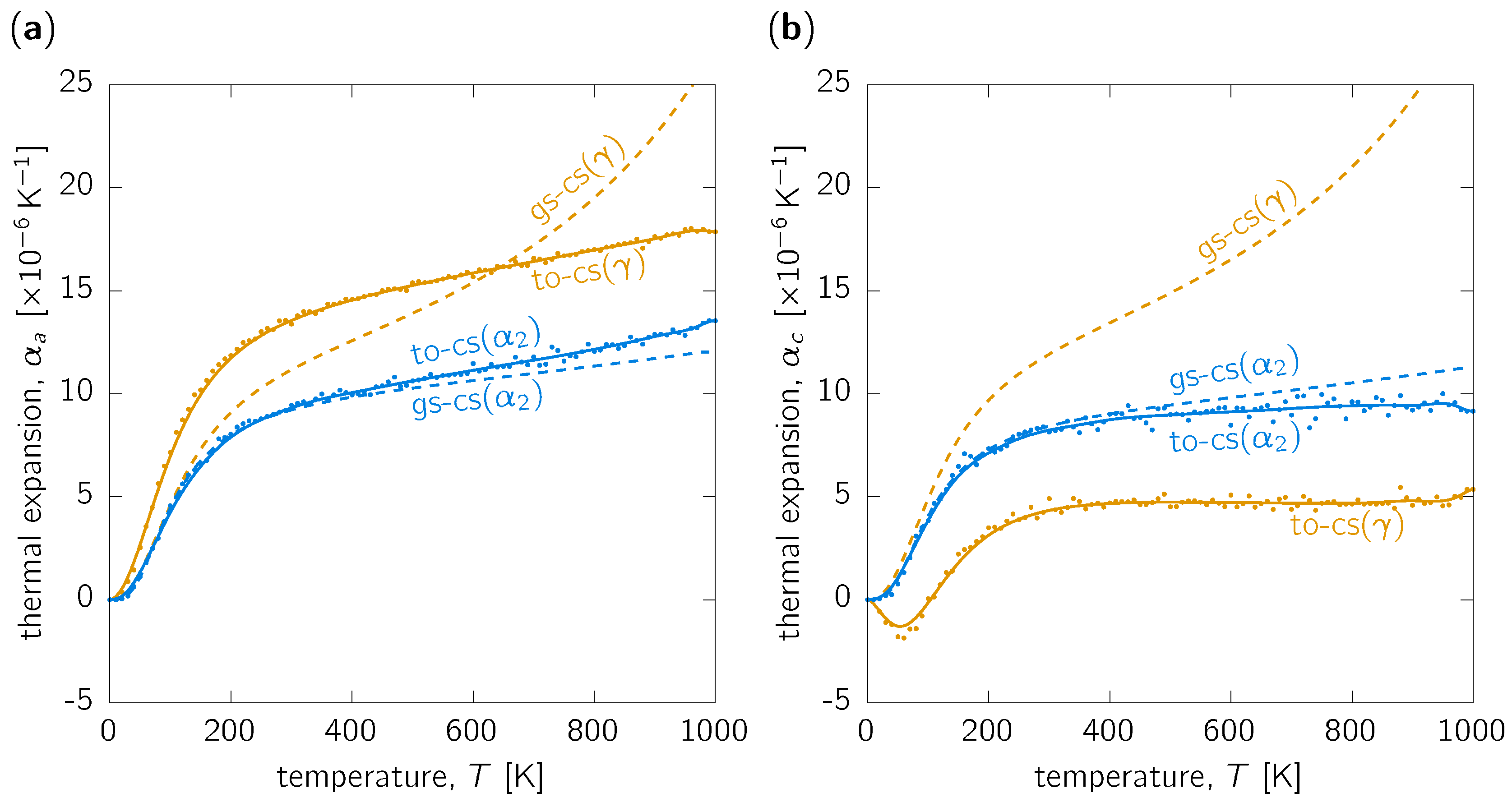
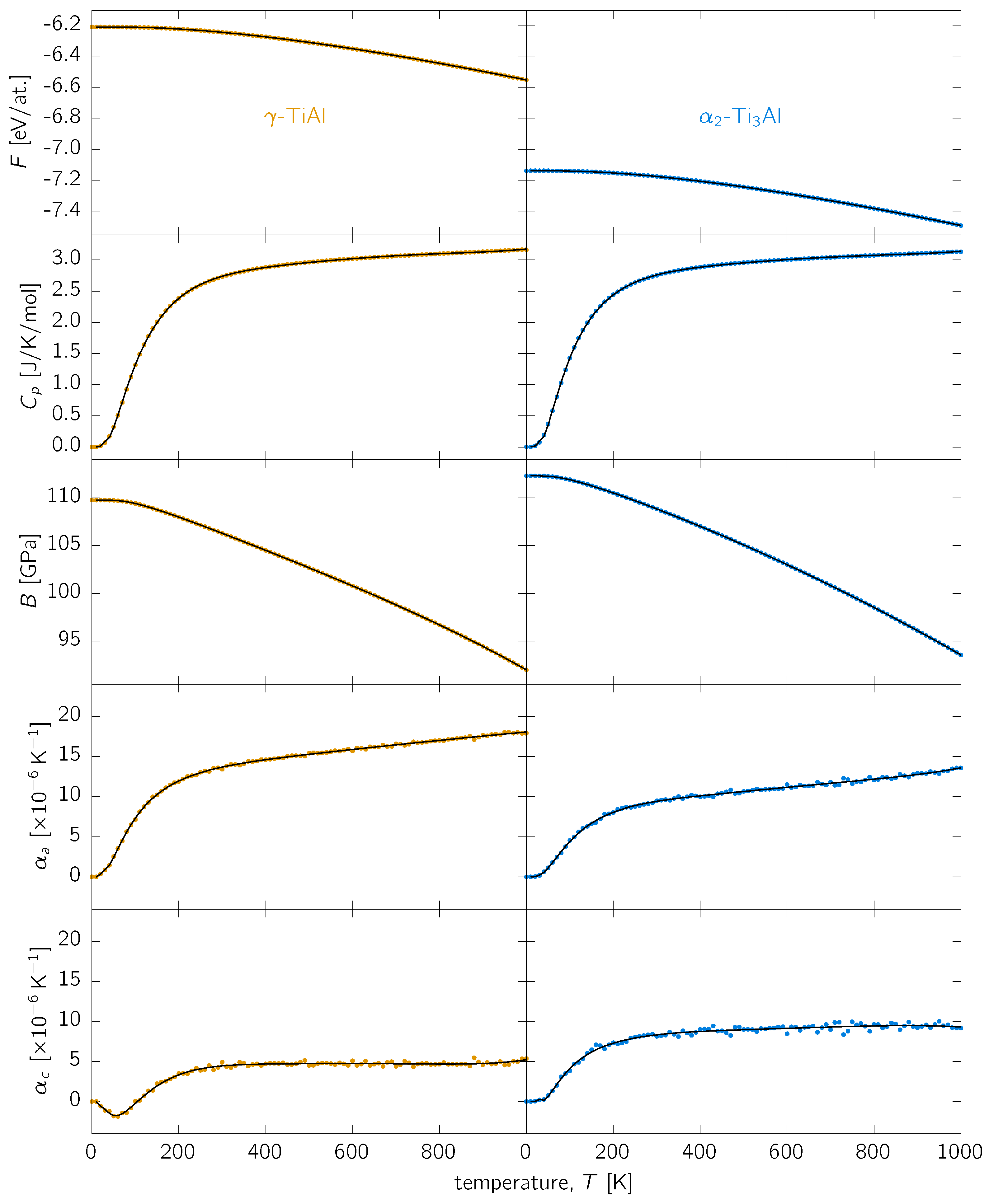
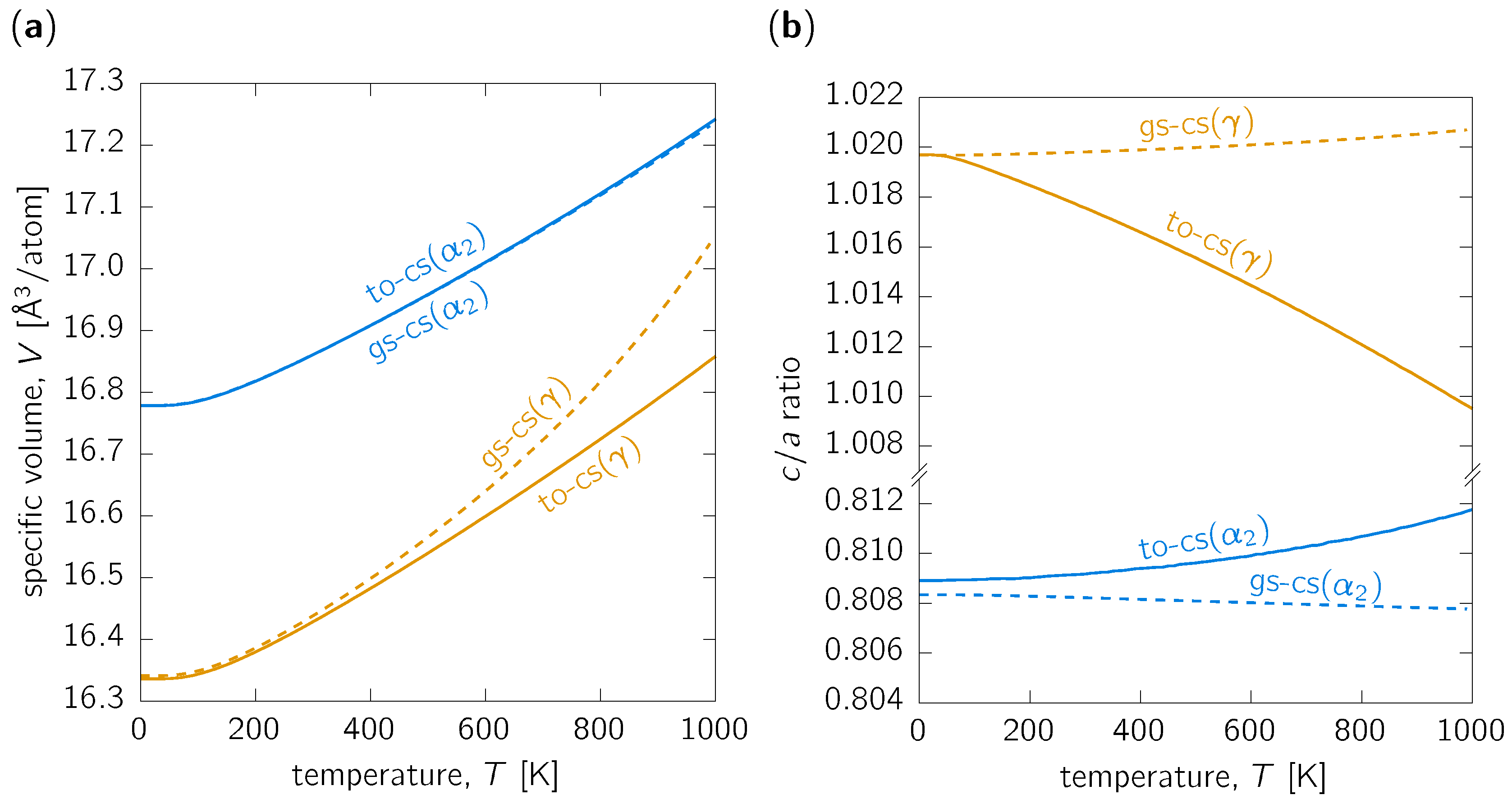
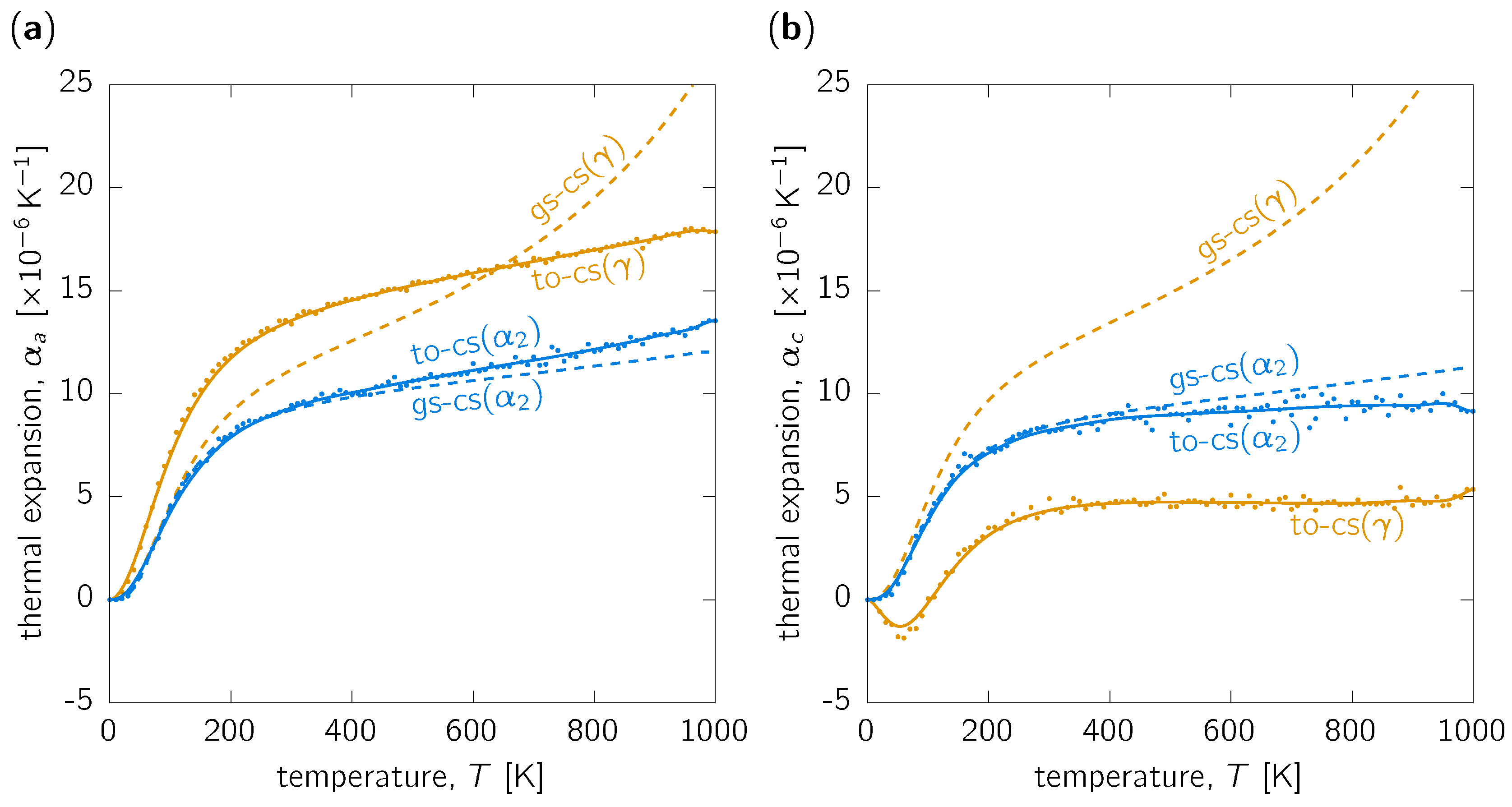
3.1. Thermal Expansion
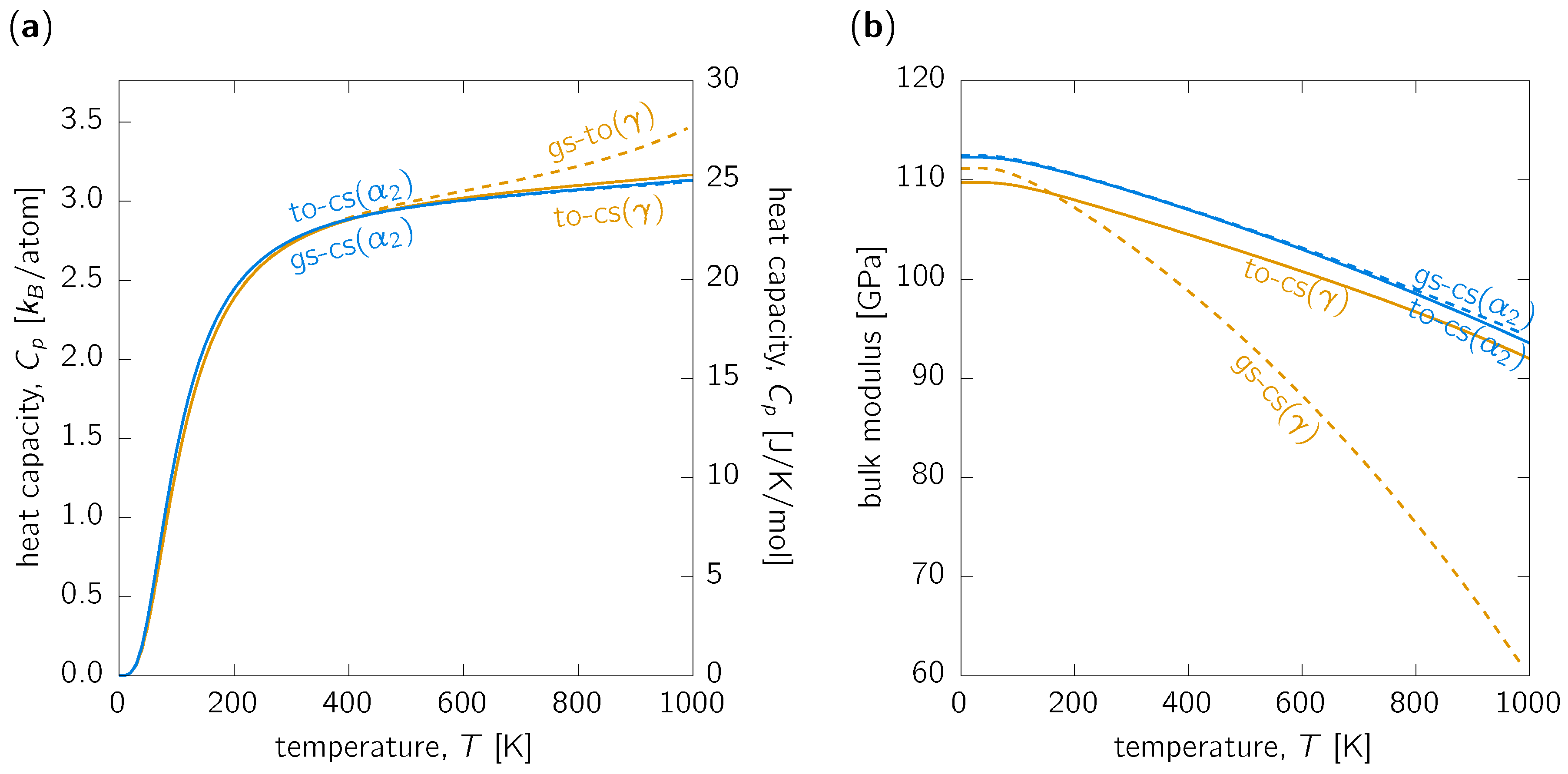
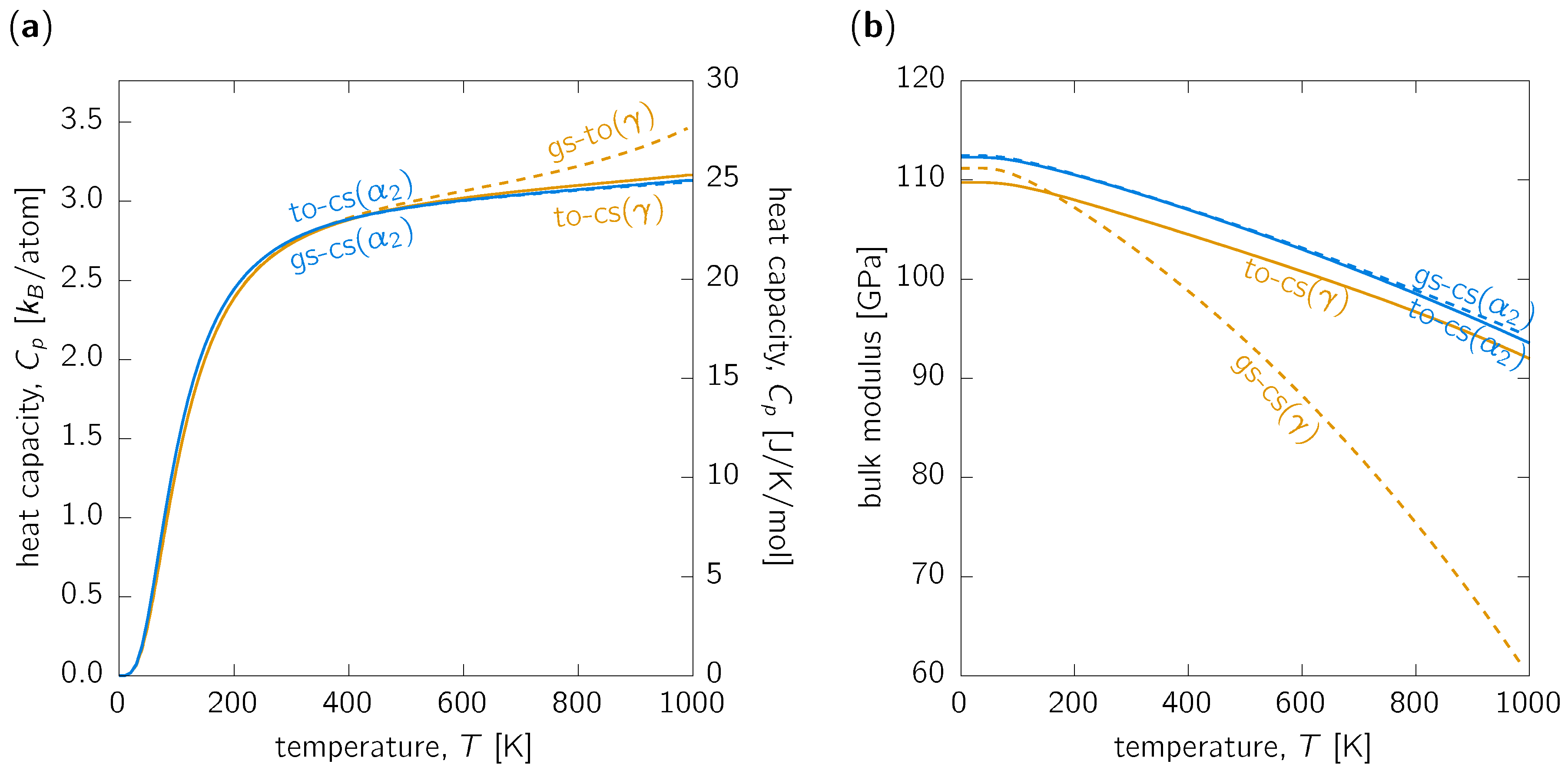
3.2. Other Thermodynamic Properties
3.3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| GGA | Generalised Gradient Approximation |
| gs-cs | ground state optimised cell shape |
| HA | harmonic approximation |
| QHA | quasi-harmonic approximation |
| RT | room temperature |
| TEC | coefficient of thermal expansion |
| to-cs | temperature optimised cell shape |
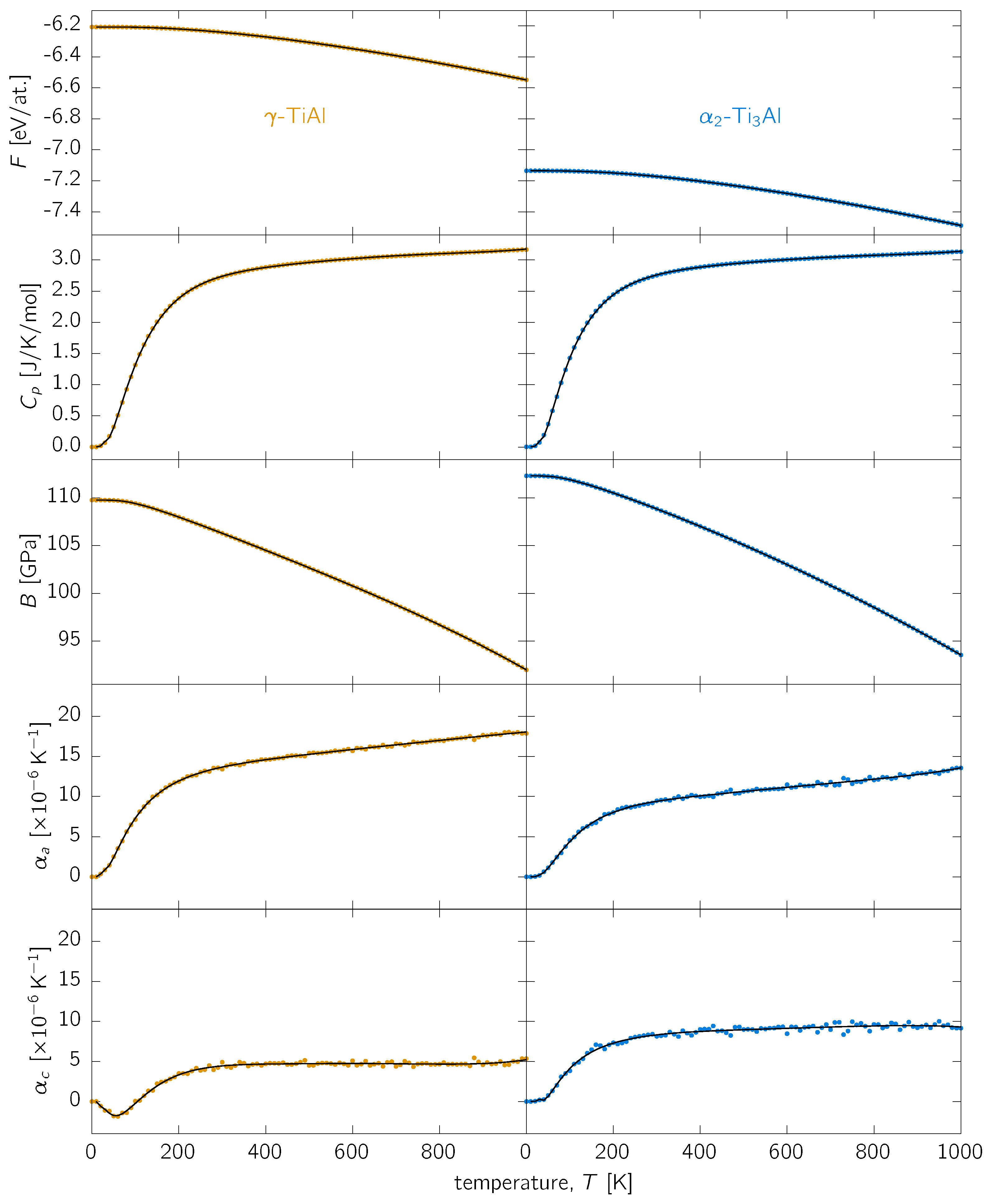
Appendix A. Analytical Fits

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F | B | ||||
|---|---|---|---|---|---|
| [eV/at.] | [J/K/mol] | [GPa] | [] | [] | |
| c |
| F | B | ||||
|---|---|---|---|---|---|
| [eV/at.] | [J/K/mol] | [GPa] | [] | [] | |
| c |
References
- Holec, D.; Zhou, L.; Riedl, H.; Koller, C.M.; Mayrhofer, P.H.; Friák, M.; Šob, M.; Körmann, F.; Neugebauer, J.; Music, D.; et al. Atomistic modeling-based design of novel materials. Adv. Eng. Mater. 2017, 19, 1600688. [Google Scholar] [CrossRef]
- Curtarolo, S.; Hart, G.L.W.; Nardelli, M.B.; Mingo, N.; Sanvito, S.; Levy, O. The high-throughput highway to computational materials design. Nat. Mater. 2013, 12, 191–201. [Google Scholar] [CrossRef]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef]
- Draxl, C.; Scheffler, M. NOMAD: The FAIR concept for big data-driven materials science. MRS Bull. 2018, 43, 676–682. [Google Scholar] [CrossRef]
- Koutná, N.; Erdely, P.; Zöhrer, S.; Franz, R.; Du, Y.; Liu, S.; Mayrhofer, P.H.; Holec, D. Experimental chemistry and structural stability of AlNb enabled by antisite defects formation. Materials 2019, 12, 1104. [Google Scholar] [CrossRef]
- Grabowski, B.; Hickel, T.; Neugebauer, J. Ab initio study of the thermodynamic properties of nonmagnetic elementary fcc metals: Exchange-correlation-related error bars and chemical trends. Phys. Rev. B Condens. Matter 2007, 76, 024309. [Google Scholar] [CrossRef]
- Körmann, F.; Dick, A.; Grabowski, B.; Hallstedt, B.; Hickel, T.; Neugebauer, J. Free energy of bcc iron: Integrated ab initio derivation of vibrational, electronic, and magnetic contributions. Phys. Rev. B Condens. Matter 2008, 78, 033102. [Google Scholar] [CrossRef]
- Grabowski, B.; Ismer, L.; Hickel, T.; Neugebauer, J. Ab initio up to the melting point: Anharmonicity and vacancies in aluminum. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 79, 134106. [Google Scholar] [CrossRef]
- Hickel, T.; Grabowski, B.; Körmann, F.; Neugebauer, J. Advancing density functional theory to finite temperatures: Methods and applications in steel design. J. Phys. Condens. Matter 2012, 24, 053202. [Google Scholar] [CrossRef]
- Palumbo, M.; Fries, S.G.; Corso, A.D.; Kürmann, F.; Hickel, T.; Neugebauer, J. Reliability evaluation of thermophysical properties from first-principles calculations. J. Phys. Condens. Matter 2014, 26, 335401. [Google Scholar] [CrossRef]
- Glensk, A.; Grabowski, B.; Hickel, T.; Neugebauer, J. Understanding anharmonicity in fcc materials: From its origin to ab initio strategies beyond the quasiharmonic approximation. Phys. Rev. Lett. 2015, 114, 195901. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Cui, W.F.; Liu, C.M.; Bauer, V.; Christ, H.J. Thermomechanical fatigue behaviours of a third generation γ-TiAl based alloy—Advanced intermetallic alloys and bulk metallic glasses 6th international workshop on advanced intermetallic and metallic materials. Intermetallics 2007, 15, 675–678. [Google Scholar] [CrossRef]
- Clemens, H.; Mayer, S. Design, processing, microstructure, properties, and applications of advanced intermetallic TiAl alloys. Adv. Eng. Mater. 2013, 15, 191–215. [Google Scholar] [CrossRef]
- Lasalmonie, A. Intermetallics: Why is it so difficult to introduce them in gas turbine engines? Intermetallics 2006, 14, 1123–1129. [Google Scholar] [CrossRef]
- Appel, F.; Paul, J.; Oehring, M. Gamma Titanium Aluminide Alloys: Science and Technology; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Clemens, H.; Mayer, S. Intermetallic titanium aluminides in aerospace applications—Processing, microstructure and properties. Mater. High Temp. 2016, 33, 560–570. [Google Scholar] [CrossRef]
- Kim, Y.W.; Clemens, H.; Rosenberger, A.H. Gamma Titanium Aluminides 2003; Minerals, Metals, & Materials Society: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Yamaguchi, M.; Inui, H.; Ito, K. High-temperature structural intermetallics. Acta Mater. 2000, 48, 307–322. [Google Scholar] [CrossRef]
- Appel, F.; Wagner, R. Microstructure and deformation of two-phase γ-titanium aluminides. Mater. Sci. Eng. R Rep. 1998, 22, 187–268. [Google Scholar] [CrossRef]
- Kestler, H.; Clemens, H. Production, processing and application of gamma (TiAl)-based alloys. In Titanium and Titanium Alloys: Fundamentals and Applications; Leyens, C., Peters, M., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003; pp. 351–392. [Google Scholar]
- Wu, X. Review of alloy and process development of TiAl alloys. Intermetallics 2006, 14, 1114–1122. [Google Scholar] [CrossRef]
- Kainuma, R.; Fujita, Y.; Mitsui, H.; Ohnuma, I.; Ishida, K. Phase equilibria among α (hcp), β (bcc) and γ (L10) phases in Ti–Al base ternary alloys. Intermetallics 2000, 8, 855–867. [Google Scholar] [CrossRef]
- Tetsui, T.; Shindo, K.; Kobayashi, S.; Takeyama, M. A newly developed hot worked TiAl alloy for blades and structural components. Scr. Mater. 2002, 47, 399–403. [Google Scholar] [CrossRef]
- Shi, J.D.; Pu, Z.; Zhong, Z.; Zou, D. Improving the ductility of γ(TiAl) based alloy by introducing disordered β phase. Scr. Metall. Mater. 1992, 27, 1331–1336. [Google Scholar] [CrossRef]
- Mayer, S.; Erdely, P.; Fischer, F.D.; Holec, D.; Kastenhuber, M.; Klein, T.; Clemens, H. Intermetallic β-solidifying γ-TiAl based alloys—From fundamental research to application. Adv. Eng. Mater. 2017, 19, 1600735. [Google Scholar] [CrossRef]
- Clemens, H.; Wallgram, W.; Kremmer, S.; Güther, V.; Otto, A.; Bartels, A. Design of novel β-solidifying TiAl alloys with adjustable β/B2-phase fraction and excellent hot-workability. Adv. Eng. Mater. 2008, 10, 707–713. [Google Scholar] [CrossRef]
- Loretto, M.H.; Godfrey, A.B.; Hu, D.; Blenkinsop, P.A.; Jones, I.P.; Cheng, T.T. The influence of composition and processing on the structure and properties of TiAl-based alloys. Intermetallics 1998, 6, 663–666. [Google Scholar] [CrossRef]
- Holec, D.; Legut, D.; Isaeva, L.; Souvatzis, P.; Clemens, H.; Mayer, S. Interplay between effect of Mo and chemical disorder on the stability of β/βo-TiAl phase. Intermetallics 2015, 61, 85–90. [Google Scholar] [CrossRef]
- Witusiewicz, V.T.; Bondar, A.A.; Hecht, U.; Stryzhyboroda, O.M.; Tsyganenko, N.I.; Voblikov, V.M.; Petyukh, V.M.; Velikanova, T.Y. Thermodynamic re-modelling of the ternary Al–Mo–Ti system based on novel experimental data. J. Alloys Compd. 2018, 749, 1071–1091. [Google Scholar] [CrossRef]
- Nabarro, F. Two-phase materials for high-temperature service. Intermetallics 2000, 8, 979–985. [Google Scholar] [CrossRef]
- Schuh, C.; Dunand, D.C.; Wanner, A.; Clemens, H. Thermal-cycling creep of γ-TiAl-based alloys. Intermetallics 2000, 8, 339–343. [Google Scholar] [CrossRef]
- Zupan, M.; Hemker, K.J. High temperature microsample tensile testing of γ-TiAl. Mater. Sci. Eng. A 2001, 319–321, 810–814. [Google Scholar] [CrossRef]
- He, Y.; Schwarz, R.B.; Darling, T.; Hundley, M.; Whang, S.H.; Wang, Z.M. Elastic constants and thermal expansion of single crystal γ-TiAl from 300 to 750 K—4th conference on high-temperature intermetallics. Mater. Sci. Eng. A 1997, 239–240, 157–163. [Google Scholar] [CrossRef]
- Bittorf, C.; Matthies, S.; Priesmeyer, H.G.; Wagner, R. Diffractive determination of thermo-elastic single crystal constants using polycrystalline samples. I. Thermal expansion of γ-TiAl from 300 to 900 K. Intermetallics 1999, 7, 251–258. [Google Scholar] [CrossRef]
- Stone, W.; Kurfess, T. Titanium Aluminide-Thermal Diffusivity, Heat Capacitance, and Coefficient of Thermal Expansion as a Function of Temperature; Technical Paper; Society of Manufacturing Engineers: Dearborn, MI, USA, 2002; number MR02-143; pp. 1–5. [Google Scholar]
- Zhang, W.J.; Reddy, B.V.; Deevi, S.C. Physical properties of TiAl-base alloys. Scr. Mater. 2001, 45, 645–651. [Google Scholar] [CrossRef]
- Novoselova, T.; Malinov, S.; Sha, W.; Zhecheva, A. High-temperature synchrotron X-ray diffraction study of phases in a gamma TiAl alloy. Mater. Sci. Eng. A 2004, 371, 103–112. [Google Scholar] [CrossRef]
- Li, X.; Dippenaar, R.; Shiro, A.; Shobu, T.; Higo, Y.; Reid, M.; Suzuki, H.; Akita, K.; Funakoshi, K.I.; Liss, K.D. Lattice parameter evolution during heating of Ti-45Al-7.5Nb-0.25/0.5C alloys under atmospheric and high pressures. Intermetallics 2018, 102, 120–131. [Google Scholar] [CrossRef]
- Fu, H.; Zhao, Z.; Liu, W.; Peng, F.; Gao, T.; Cheng, X. Ab initio calculations of elastic constants and thermodynamic properties of γTiAl under high pressures. Intermetallics 2010, 18, 761–766. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Togo, A.; Chaput, L.; Tanaka, I.; Hug, G. First-principles phonon calculations of thermal expansion in Ti3SiC2, Ti3AlC2, and Ti3GeC2. Phys. Rev. B Condens. Matter 2010, 81, 174301. [Google Scholar] [CrossRef]
- Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. 1947, 71, 809–824. [Google Scholar] [CrossRef]
- Nakano, T.; Negishi, A.; Hayashi, K.; Umakoshi, Y. Ordering process of Al5Ti3, h-Al2Ti and r-Al2Ti with f.c.c.-based long-period superstructures in rapidly solidified Al-rich TiAl alloys. Acta Mater. 1999, 47, 1091–1104. [Google Scholar] [CrossRef]
- Daniel, R.; Holec, D.; Bartosik, M.; Keckes, J.; Mitterer, C. Size effect of thermal expansion and thermal/intrinsic stresses in nanostructured thin films: Experiment and model. Acta Mater. 2011, 59, 6631–6645. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holec, D.; Abdoshahi, N.; Mayer, S.; Clemens, H. Thermal Expansion and Other Thermodynamic Properties of α2-Ti3Al and γ-TiAl Intermetallic Phases from First Principles Methods. Materials 2019, 12, 1292. https://doi.org/10.3390/ma12081292
Holec D, Abdoshahi N, Mayer S, Clemens H. Thermal Expansion and Other Thermodynamic Properties of α2-Ti3Al and γ-TiAl Intermetallic Phases from First Principles Methods. Materials. 2019; 12(8):1292. https://doi.org/10.3390/ma12081292
Chicago/Turabian StyleHolec, David, Neda Abdoshahi, Svea Mayer, and Helmut Clemens. 2019. "Thermal Expansion and Other Thermodynamic Properties of α2-Ti3Al and γ-TiAl Intermetallic Phases from First Principles Methods" Materials 12, no. 8: 1292. https://doi.org/10.3390/ma12081292
APA StyleHolec, D., Abdoshahi, N., Mayer, S., & Clemens, H. (2019). Thermal Expansion and Other Thermodynamic Properties of α2-Ti3Al and γ-TiAl Intermetallic Phases from First Principles Methods. Materials, 12(8), 1292. https://doi.org/10.3390/ma12081292