Graphene Oxide Oxygen Content Affects Physical and Biological Properties of Scaffolds Based on Chitosan/Graphene Oxide Conjugates


 , ,
, ,  , , and
, , and
Abstract
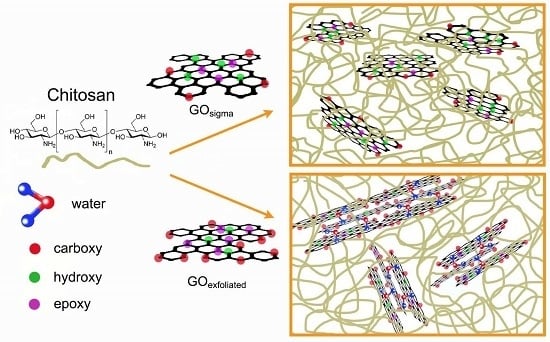
1. Introduction
2. Results and Discussion
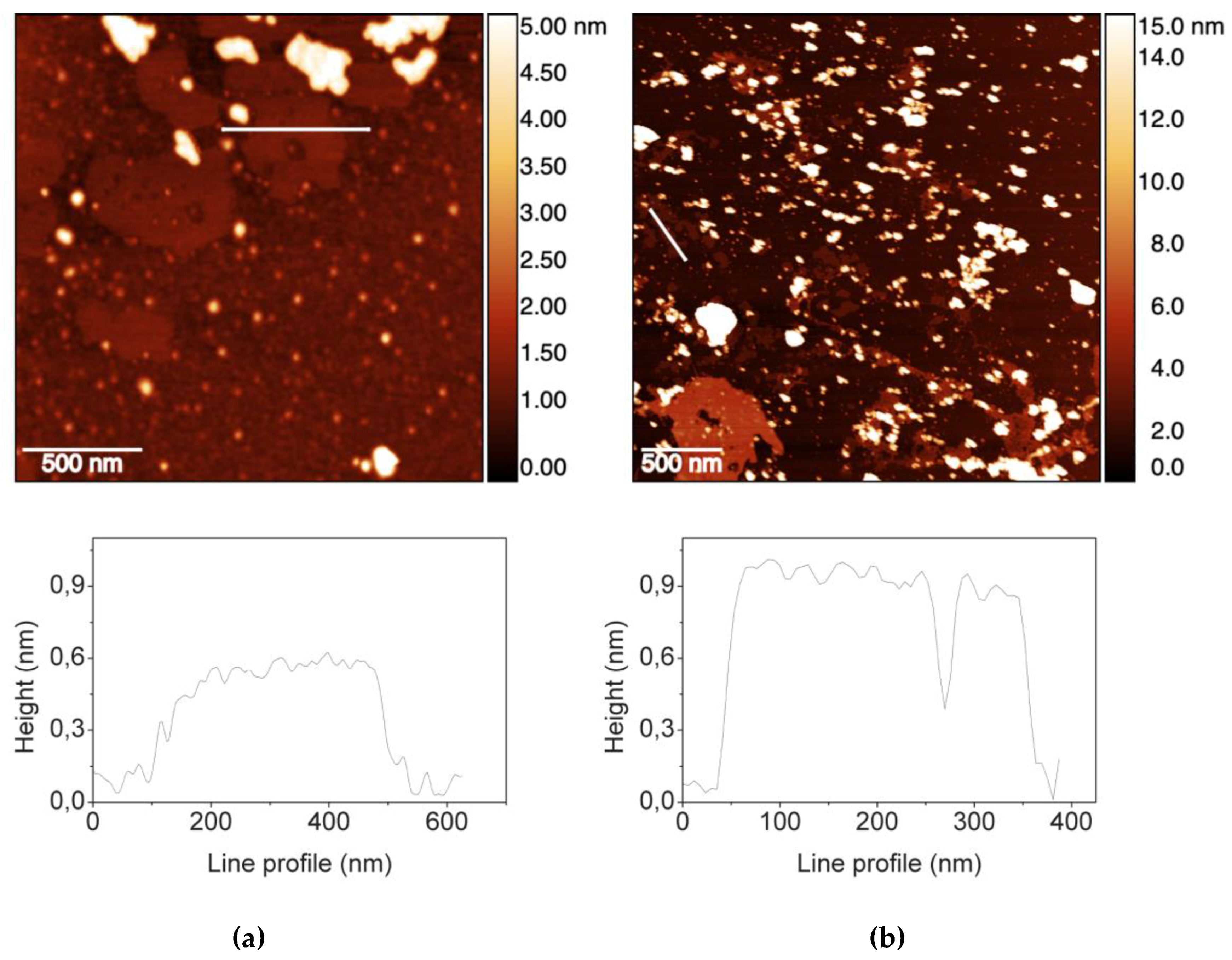
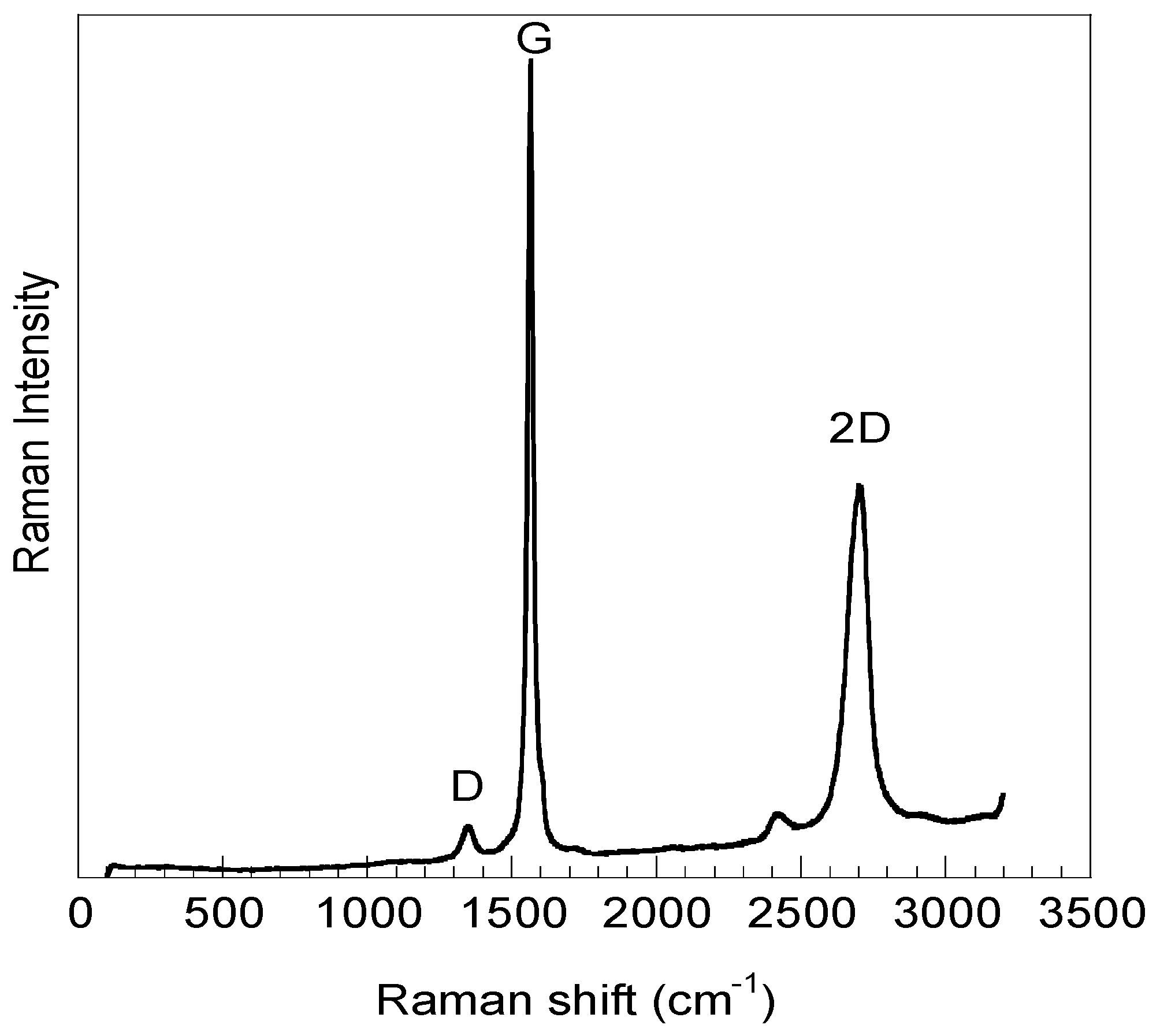
2.1. GO Characterization
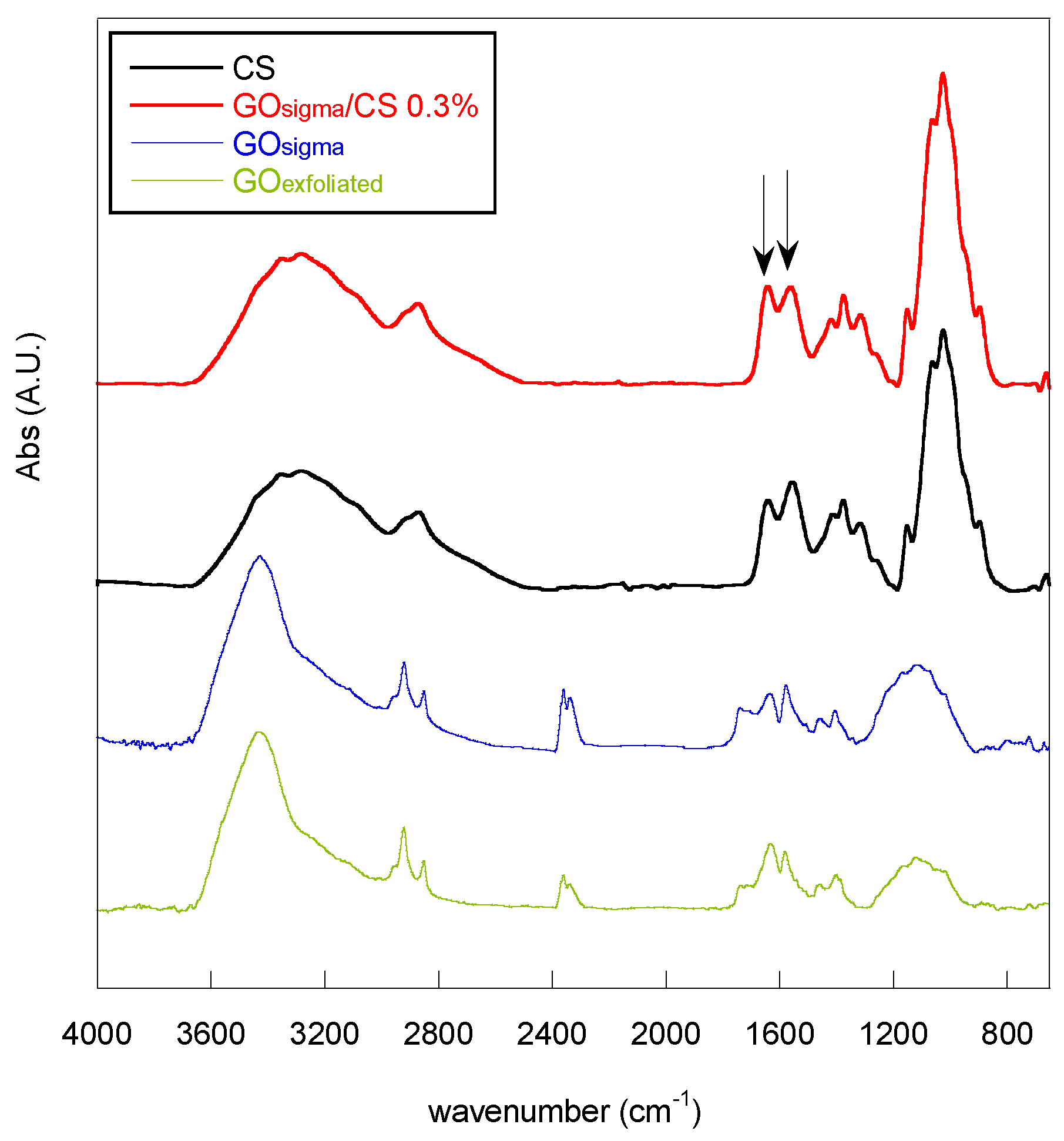
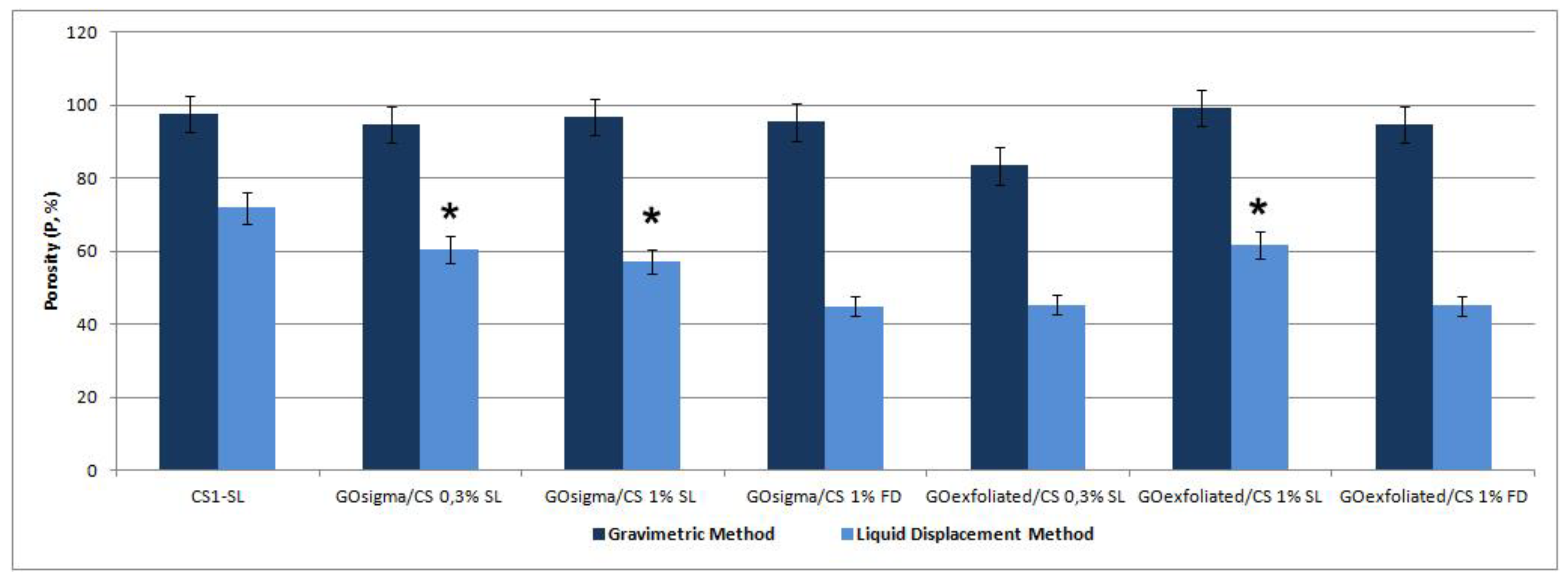
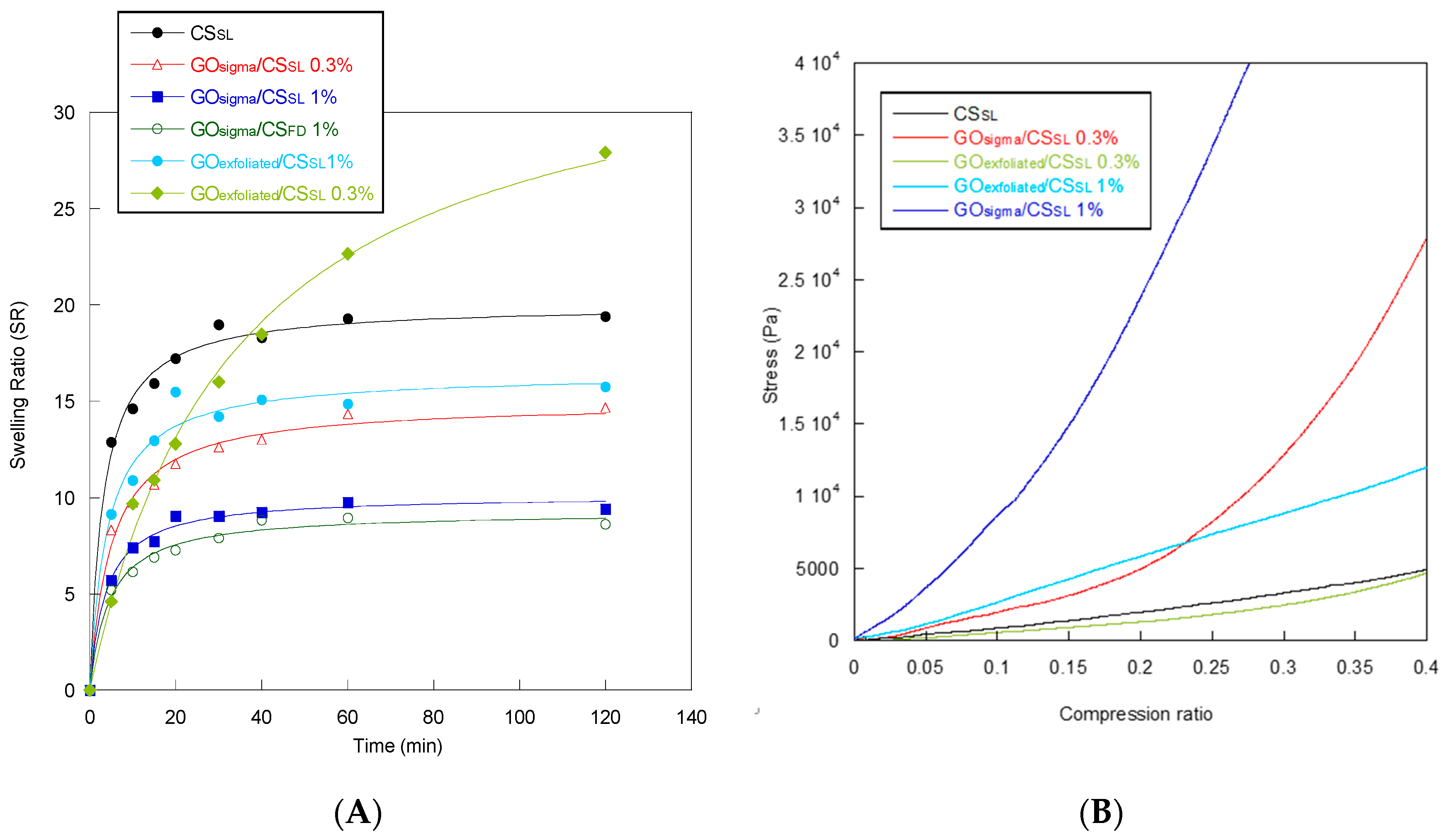
2.2. Preparation and Characterization of GO/CS Composite Scaffolds
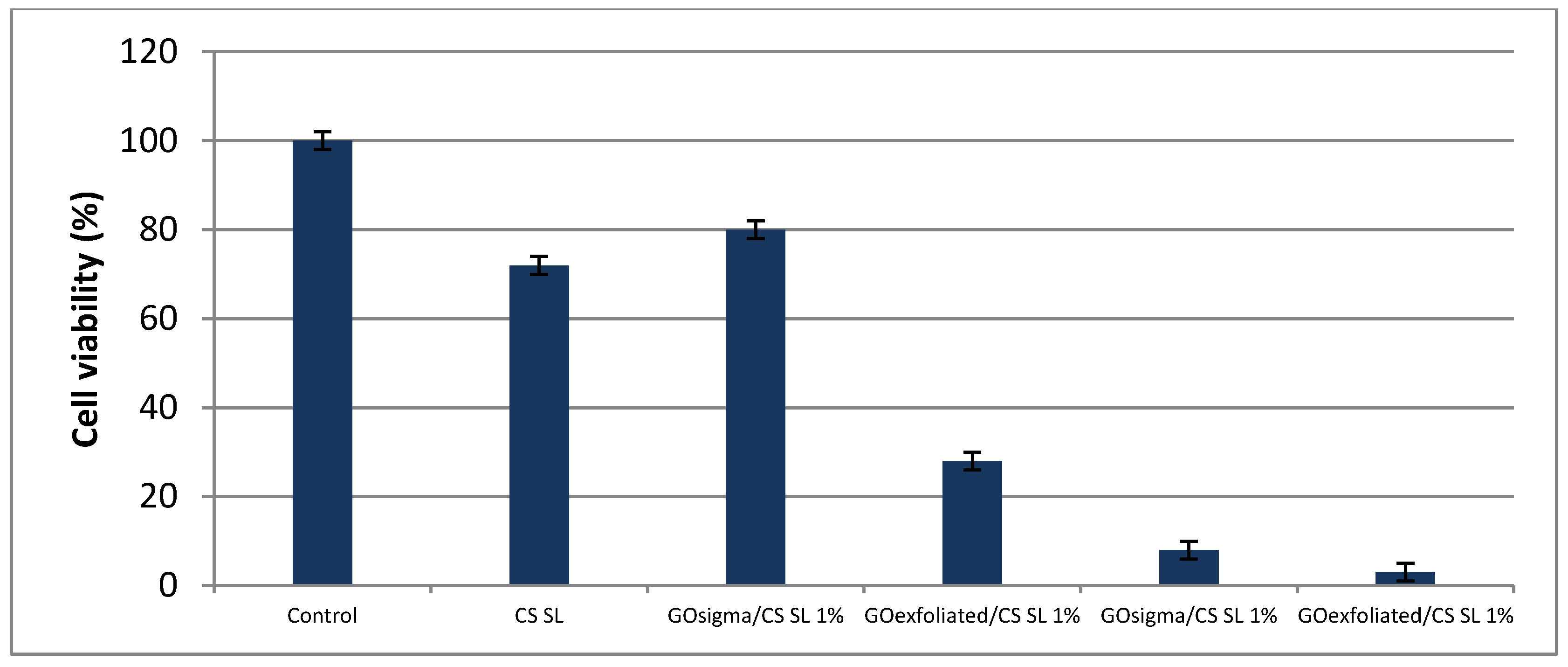
2.3. Assessment of Cell Viability in the Scaffolds
3. Materials and Methods
3.1. Graphene Oxide
3.2. Characterization of Graphene Oxide Samples
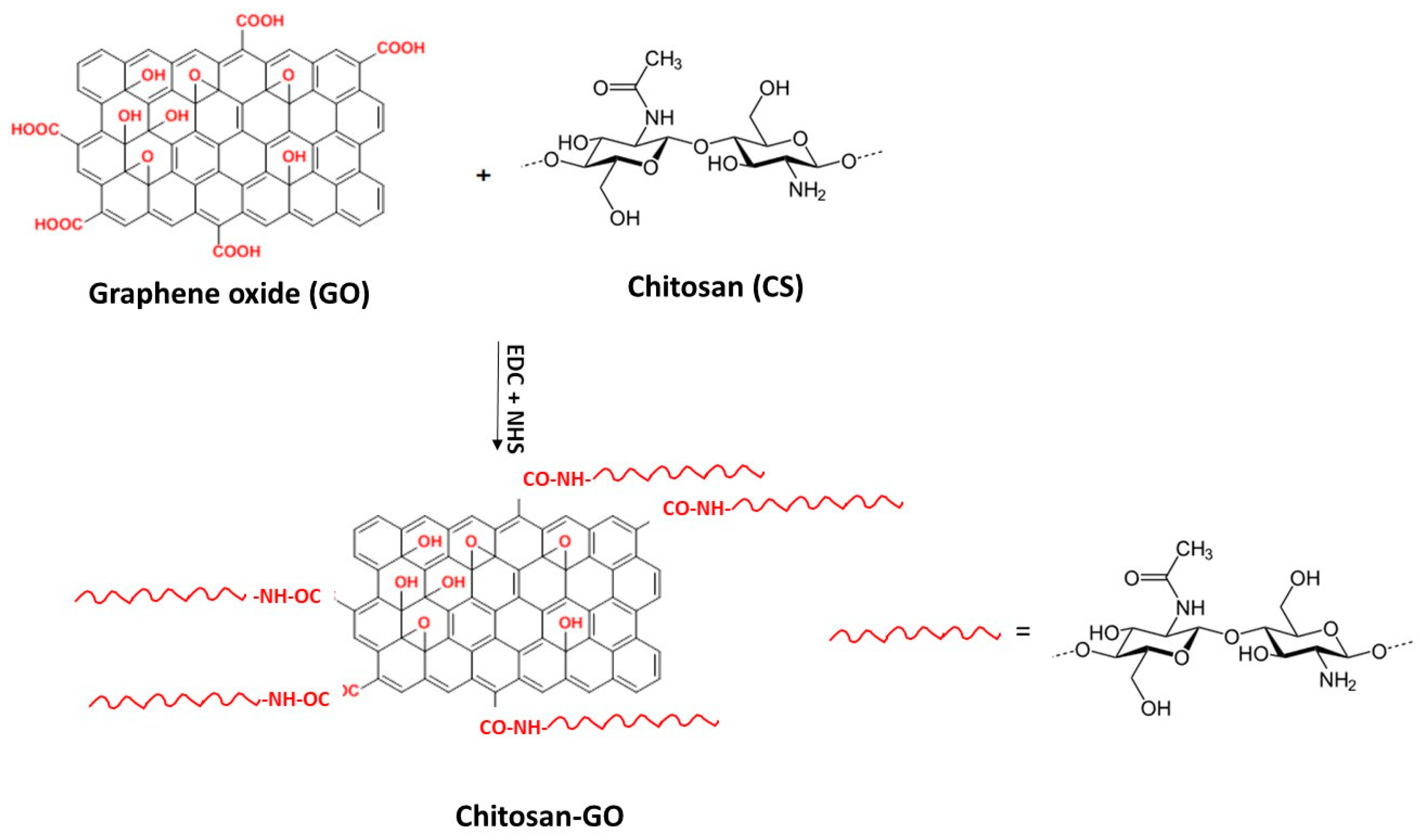
3.3. Functionalization of Chitosan with GO
3.4. Preparation of GO/CS Composite Scaffolds
3.5. Characterization of GO/CS Composite Scaffolds
3.6. Assessment of Cell Viability in Scaffolds by MTS Assay
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ramya, R.; Venkatesan, J.; Kim, S.K.; Sudha, P.N. Biomedical applications of chitosan: An overview. J. Biomim. Biomater. Tissue Eng. 2012, 2, 100–111. [Google Scholar] [CrossRef]
- Al-Jbour, N.D.; Beg, M.D.; Gimbun, J.; Alam, A.K.M.M. An overview of chitosan nanofibers and their applications in drug delivery process. Curr. Drug Deliv. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cuzzucoli Crucitti, V.; Migneco, L.M.; Piozzi, A.; Taresco, V.; Garnett, M.; Argent, R.H.; Francolini, I. Intermolecular interaction and solid state characterization of abietic acid/chitosan solid dispersions possessing antimicrobial and antioxidant properties. Eur. J. Pharm. Biopharm. 2018, 125, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.; Migneco, L.M.; Martinelli, A.; Pietrelli, L.; Piozzi, A.; Francolini, I. Antimicrobial activity of catechol functionalized-chitosan versus Staphylococcus epidermidis. Carbohydr. Polym. 2018, 179, 273–281. [Google Scholar] [CrossRef]
- Singh, R.; Shitiz, K.; Singh, A. Chitin and chitosan: Biopolymers for wound management. Int. Wound J. 2017, 14, 1276–1289. [Google Scholar] [CrossRef]
- Moura, M.J.; Brochado, J.; Gil, M.H.; Figueiredo, M.M. In situ forming chitosan hydrogels: Preliminary evaluation of the in vivo inflammatory response. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 75, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, D.; Bosi, S.; Melchionna, M.; Da Ros, T.; Marchesan, S. The Glitter of carbon nanostructures in hybrid/composite hydrogels for medicinal use. Curr. Top. Med. Chem. 2016, 16, 1976–1989. [Google Scholar] [CrossRef]
- Zhou, G.; Niepel, M.S.; Saretia, S.; Groth, T. Reducing the inflammatory responses of biomaterials by surface modification with glycosaminoglycan multilayers. J. Biomed. Mater. Res. A 2016, 104, 493–502. [Google Scholar] [CrossRef]
- Moraes, P.C.; Marques, I.C.S.; Basso, F.G.; Rossetto, H.L.; Pires-de-Souza, F.C.P.; Costa, C.A.S.; Garcia, L.D.F.R. Repair of bone defects with chitosan-collagen biomembrane and scaffold containing calcium aluminate cement. Braz. Dent. J. 2017, 28, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Haifei, S.; Xingang, W.; Shoucheng, W.; Zhengwei, M.; Chuangang, Y.; Chunmao, H. The effect of collagen-chitosan porous scaffold thickness on dermal regeneration in a one-stage grafting procedure. J. Mech. Behav. Biomed. Mater. 2014, 29, 114–125. [Google Scholar] [CrossRef]
- Chen, C.K.; Chang, N.J.; Wu, Y.T.; Fu, E.; Shen, E.C.; Feng, C.W.; Wen, Z.H. Bone formation using cross-linked chitosan scaffolds in rat calvarial defects. Implant Dent. 2018, 27, 15–21. [Google Scholar] [CrossRef]
- Balagangadharan, K.; Dhivya, S.; Selvamurugan, N. Chitosan based nanofibers in bone tissue engineering. Int. J. Biol. Macromol. 2017, 104, 1372–1382. [Google Scholar] [CrossRef]
- LogithKumar, R.; KeshavNarayan, A.; Dhivya, S.; Chawla, A.; Saravanan, S.; Selvamurugan, N. A review of chitosan and its derivatives in bone tissue engineering. Carbohydr. Polym. 2016, 151, 172–188. [Google Scholar] [CrossRef]
- Yang, X.; Chen, X.; Wang, H. Acceleration of osteogenic differentiation of preosteoblastic cells by chitosan containing nanofibrous scaffolds. Biomacromolecules 2009, 10, 2772–2778. [Google Scholar] [CrossRef]
- Di Martino, A.; Sittinger, M.; Risbud, M.V. Chitosan: A versatile biopolymer for orthopaedic tissue-engineering. Biomaterials 2005, 26, 5983–5990. [Google Scholar] [CrossRef]
- Palma, P.J.; Ramos, J.C.; Martins, J.B.; Diogenes, A.; Figueiredo, M.H.; Ferreira, P.; Viegas, C.; Santos, J.M. Histologic evaluation of regenerative endodontic procedures with the use of chitosan scaffolds in immature dog teeth with apical periodontitis. J. Endod. 2017, 43, 1279–1287. [Google Scholar] [CrossRef]
- Stępniewski, M.; Martynkiewicz, J.; Gosk, J. Chitosan and its composites: Properties for use in bone substitution. Polim. Med. 2017, 47, 49–53. [Google Scholar] [CrossRef]
- Bi, L.; Cheng, W.; Fan, H.; Pei, G. Reconstruction of goat tibial defects using an injectable tricalcium phosphate/chitosan in combination with autologous platelet-rich plasma. Biomaterials 2010, 31, 3201–3211. [Google Scholar] [CrossRef]
- Zhou, D.; Qi, C.; Chen, Y.X.; Zhu, Y.J.; Sun, T.W.; Chen, F.; Zhang, C.Q. Comparative study of porous hydroxyapatite/chitosan and whitlockite/chitosan scaffolds for bone regeneration in calvarial defects. Int. J. Nanomed. 2017, 12, 2673–2687. [Google Scholar] [CrossRef]
- Keller, L.; Regiel-Futyra, A.; Gimeno, M.; Eap, S.; Mendoza, G.; Andreu, V.; Wagner, Q.; Kyzioł, A.; Sebastian, V.; Stochel, G.; et al. Chitosan-based nanocomposites for the repair of bone defects. Nanomedicine 2017, 13, 2231–2240. [Google Scholar] [CrossRef]
- Shamekhi, M.A.; Mirzadeh, H.; Mahdavi, H.; Rabiee, A.; Mohebbi-Kalhori, D.; Baghaban Eslaminejad, M. Graphene oxide containing chitosan scaffolds for cartilage tissue engineering. Int. J. Biol. Macromol. 2019, 127, 396–405. [Google Scholar] [CrossRef]
- Valencia, C.; Valencia, C.H.; Zuluaga, F.; Valencia, M.E.; Mina, J.H.; Grande-Tovar, C.D. Synthesis and application of scaffolds of chitosan-graphene oxide by the freeze-drying method for tissue regeneration. Molecules 2018, 23, 2651. [Google Scholar] [CrossRef]
- Hermenean, A.; Codreanu, A.; Herman, H.; Balta, C.; Rosu, M.; Mihali, C.V.; Ivan, A.; Dinescu, S.; Ionita, M.; Costache, M. Chitosan-graphene oxide 3D scaffolds as promising tools for bone regeneration in critical-size mouse calvarial defects. Sci. Rep. 2017, 7, 16641. [Google Scholar] [CrossRef]
- Sivashankari, P.R.; Moorthi, A.; Abudhahir, K.M.; Prabaharan, M. Preparation and characterization of three-dimensional scaffolds based on hydroxypropyl chitosan-graft-graphene oxide. Int. J. Biol. Macromol. 2018, 110, 522–530. [Google Scholar] [CrossRef]
- Dinescu, S.; Ionita, M.; Pandele, A.M.; Galateanu, B.; Iovu, H.; Ardelean, A.; Costache, M.; Hermenean, A. In vitro cytocompatibility evaluation of chitosan/graphene oxide 3D scaffold composites designed for bone tissue engineering. Biomed. Mater. Eng. 2014, 24, 2249–2256. [Google Scholar]
- Hummers, W.; Offeman, R. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Park, S.; Ruoff, R.S. Chemical methods for the production of graphenes. Nat. Nanotechnol. 2009, 4, 217–224. [Google Scholar] [CrossRef]
- Lerf, A.; He, H.; Forster, M.; Klinowski, J. Structure of graphite oxide revisited. J. Phys. Chem. B 1998, 102, 4477–4482. [Google Scholar] [CrossRef]
- Wu, C.; Xia, L.; Han, P.; Xu, M.; Fang, B.; Wang, J.; Chang, J.; Xiao, Y. Graphene oxide modified beta-tricalcium phosphate bioceramics stimulate in vitro and in vivo osteogenesis. Carbon 2015, 93, 116–129. [Google Scholar] [CrossRef]
- Lee, J.H.; Shin, Y.C.; Lee, S.M.; Jin, O.; Kang, S.H.; Hong, S.W.; Jeong, C.M.; Huh, J.B.; Han, D.W. Enhanced osteogenesis by reduced graphene oxide/hydroxyapatite nanocomposites. Sci. Rep. 2015, 5, 18833. [Google Scholar] [CrossRef]
- La, W.G.; Jin, M.; Park, S.; Yoon, H.H.; Jeong, G.J.; Bhang, S.H.; Park, H.; Char, K.; Kim, B.S. Delivery of bone morphogenetic protein-2 and substance P using graphene oxide for bone regeneration. Int. J. Nanomed. 2014, 9, 107–116. [Google Scholar]
- Depan, D.; Girase, B.; Shah, J.S.; Misra, R.D. Structure-process-property relationship of the polar graphene oxide-mediated cellular response and stimulated growth of osteoblasts on hybrid chitosan network structure nanocomposite scaffolds. Acta Biomater. 2011, 7, 3432–3445. [Google Scholar] [CrossRef]
- Liu, Y.; Wen, J.; Gao, Y.; Li, T.; Wang, H.; Yan, H.; Niu, B.; Guo, R. Antibacterial graphene oxide coatings on polymer substrate. Appl. Surf. Sci. 2018, 436, 624–630. [Google Scholar] [CrossRef]
- Pandit, S.; Karunakaran, S.; Boda, S.K.; Basu, B.; De, M. High Antibacterial activity of functionalized chemically exfoliated MoS2. ACS Appl. Mater. Interfaces 2016, 8, 31567–31573. [Google Scholar] [CrossRef]
- Krishnamoorthy, K.; Veerapandian, M.; Yun, K.; Kim, S.-J. The chemical and structural analysis of graphene oxide with different degrees of oxidation. Carbon 2013, 53, 38–49. [Google Scholar] [CrossRef]
- Mattevi, C.; Eda, G.; Agnoli, S.; Miller, S.; Andre Mkhoyan, K.; Celik, O.; Mastrogiovanni, D.; Granozzi, G.; Garfunkel, E.; Chhowalla, M. Evolution of electrical, chemical, and structural properties of transparent and conducting chemically derived graphene thin films. Adv. Funct. Mater. 2009, 19, 2577–2583. [Google Scholar] [CrossRef]
- Feng, R.; Yu, Y.; Shen, C.; Jiao, Y.; Zhou, C. Impact of graphene oxide on the structure and function of important multiple blood components by a dose-dependent pattern. J. Biomed. Mater. Res. A 2015, 103, 2006–2014. [Google Scholar] [CrossRef]
- Kucki, M.; Rupper, P.; Sarrieu, C.; Melucci, M.; Treossi, E.; Schwarz, A.; León, V.; Kraegeloh, A.; Flahaut, E.; Vázquez, E.; et al. Interaction of graphene-related materials with human intestinal cells: An in vitro approach. Nanoscale 2016, 8, 8749–8760. [Google Scholar] [CrossRef]
- Wei, X.Q.; Hao, L.Y.; Shao, X.R.; Zhang, Q.; Jia, X.Q.; Zhang, Z.R.; Lin, Y.F.; Peng, Q. Insight into the interaction of graphene oxide with serum proteins and the impact of the degree of reduction and concentration. ACS Appl. Mater. Interfaces 2015, 7, 13367–13374. [Google Scholar] [CrossRef]
- Costa de Oliveira, M.A.; Mecheri, B.; D’Epifanio, A.; Placidi, E.; Arciprete, F.; Valentini, F.; Perandini, A.; Valentini, V.; Licoccia, S. Graphene oxide nanoplatforms to enhance catalytic performance of iron phthalocyanine for oxygen reduction reaction in bioelectrochemical systems. J. Power Sources 2017, 356, 381–388. [Google Scholar] [CrossRef]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electrone phonon coupling, doping and non adiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Mazaheri, M.; Akhavan, O.; Simchi, A. Flexible bactericidal graphene oxide-chitosan layers for stem cell proliferation. Appl. Surf. Sci. 2014, 301, 456–462. [Google Scholar] [CrossRef]
- Pakulski, D.; Czepa, W.; Witomska, S.; Aliprandi, A.; Pawluć, P.; Patroniak, V.; Ciesielski, A.; Samorì, P. Graphene oxide-branched polyethylenimine foams for efficient removal of toxic cations from water. J. Mater. Chem. A 2018, 6, 9384–9390. [Google Scholar] [CrossRef]
- Kavya, K.C.; Jayakumar, R.; Nair, S.; Chennazhi, K.P. Fabrication and characterization of chitosan/gelatin/nSiO2 composite scaffold for bone tissue engineering. Int. J. Biol. Macromol. 2013, 59, 255–263. [Google Scholar] [CrossRef]
- Unnithan, A.R.; Park, C.H.; Kim, C.S. Nanoengineered bioactive 3D composite scaffold: A unique combination of graphene oxide and nanotopography for tissue engineering applications. Compos. Part B 2016, 90, 503–511. [Google Scholar] [CrossRef]
- Kundie, F.; Azhari, C.H.; Muchtar, A.; Ahmad, Z.A. Effects of filler size on the mechanical properties of polymer-filled dental composites: A review of recent developments. J. Phys. Sci. 2018, 29, 141–165. [Google Scholar] [CrossRef]
- Kim, J.; Kim, S.W.; Yun, H.; Kim, B.J. Impact of size control of graphene oxide nanosheets for enhancing electrical and mechanical properties of carbon nanotube–polymer composites. RSC Adv. 2017, 7, 30221–30228. [Google Scholar] [CrossRef]
- Medhekar, N.V.; Ramasubramaniam, A.; Ruoff, R.S.; Shenoy, V.B. Hydrogen bond networks in graphene oxide composite paper: Structure and mechanical properties. ACS Nano 2010, 4, 2300–2306. [Google Scholar] [CrossRef]
- Compton, O.C.; Cranford, S.W.; Putz, K.W.; An, Z.; Brinson, L.C.; Buehler, M.J.; Nguyen, S.T. Tuning the mechanical properties of graphene oxide paper and its associated polymer nanocomposites by controlling cooperative intersheet hydrogen bonding. ACS Nano 2012, 6, 2008–2019. [Google Scholar] [CrossRef]
- Dikin, D.A.; Stankovich, S.; Zimney, E.J.; Piner, R.D.; Dommett, G.H.B.; Evmenenko, G.; Nguyen, S.T.; Ruoff, R.S. Preparation and Characterization of Graphene Oxide Paper. Nature 2007, 448, 457–460. [Google Scholar] [CrossRef]
- Liao, K.H.; Lin, Y.S.; Macosko, C.W.; Haynes, C.L. Cytotoxicity of graphene oxide and graphene in human erythrocytes and skin fibroblasts. ACS Appl. Mater. Interfaces 2011, 3, 2607–2615. [Google Scholar] [CrossRef]
- Bengtson, S.; Kling, K.; Madsen, A.M.; Noergaard, A.W.; Jacobsen, N.R.; Clausen, P.A.; Alonso, B.; Pesquera, A.; Zurutuza, A.; Ramos, R.; et al. No cytotoxicity or genotoxicity of graphene and graphene oxide in murine lung epithelial FE1 cells in vitro. Environ. Mol. Mutagen. 2016, 57, 469–482. [Google Scholar] [CrossRef]
- Zhi, X.; Fang, H.; Bao, C.; Shen, G.; Zhang, J.; Wang, K.; Guo, S.; Wan, T.; Cui, D. The immunotoxicity of graphene oxides and the effect of PVP-coating. Biomaterials 2013, 34, 5254–5261. [Google Scholar] [CrossRef]
- De Marco, P.; Zara, S.; De Colli, M.; Radunovic, M.; Lazović, V.; Ettorre, V.; Di Crescenzo, A.; Piattelli, A.; Cataldi, A.; Fontana, A. Graphene oxide improves the biocompatibility of collagen membranes in an in vitro model of human primary gingival fibroblasts. Biomed. Mater. 2017, 12, 055005. [Google Scholar] [CrossRef]
- Loh, Q.L.; Choong, C. Three-dimensional scaffolds for tissue engineering applications: Role of porosity and pore size. Tissue Eng. Part B Rev. 2013, 19, 485–502. [Google Scholar] [CrossRef]
- Han, J.; Zhou, Z.; Yin, R.; Yang, D.; Nie, J. Alginate-chitosan/hydroxyapatite polyelectrolyte complex porous scaffolds: Preparation and characterization. Int. J. Biol. Macromol. 2010, 46, 199–205. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Frequency (cm−1) | Assigned Bands | ID/IG |
|---|---|---|---|
| GOsigma | 2706 | 2D | 0.06 |
| 1565 | G | ||
| 1350 | D | ||
| GOexfoliated | 2713 | 2D | 0.27 |
| 1582 | G | ||
| 1357 | D |
| Peak BE (eV) | Species | GOsigma At (%) | GOexfoliated At (%) |
|---|---|---|---|
| 283.8 | C–C | 60.7 | 50.0 |
| 284.7 | C–OH | 21.7 | 18.0 |
| 285.9 | C–O | 9.4 | 16.0 |
| 287.0 | C=O | 8.2 | 9.0 |
| 289.0 | C(=O)O | 4.3 | 7.0 |
| Sample | CS Conc. (%, w/v) | A1644/A1560 |
|---|---|---|
| CS | – | 0.82 |
| GOsigma/CS 0.3% | 1 | 0.93 |
| GOsigma/CS 1% | 1 | 1.02 |
| GOexfoliated/CS 0.3% | 1 | 0.88 |
| GOexfoliated/CS 1% | 1 | 0.91 |
| Acronym | Method for Scaffold Preparation | GO/CS % (w/w) |
|---|---|---|
| CSSL | SL * | – |
| GOsigma/CSSL 0.3% | SL | 0.3 |
| GOsigma/CSSL 1% | SL | 1 |
| GOsigma/CSFD 1% | FD * | 1 |
| GOexfoliated/CSSL 0.3% | SL | 0.3 |
| GOexfoliated/CSSL 1% | SL | 1 |
| GOexfoliated/CSFD 1% | FD | 1 |
| Sample | Equilibrium Swelling Ratio (SR) | Water Retention (WR) | Td (°C) | Tg (°C) | Compressive Modulus (KPa) |
|---|---|---|---|---|---|
| CSSL | 19 ± 2 | 10.1 ± 0.5 | 280 | 72 | 12 ± 3 |
| GOsigma/CSSL 0.3% | 14 ± 1 | 7.1 ± 0.5 | 296 | 84 | 22 ± 2 |
| GOsigma/CSSL 1% | 9 ± 2 | 5.9 ± 0.5 | 304 | 82 | 108 ± 5 |
| GOsigma/CSFD 1% | 8 ± 1 | 4.2 ± 0.5 | ND * | ND | ND |
| GOexfoliated/CSSL 0.3% | 28 ± 4 | 7.6 ± 0.5 | 283 | 86 | 10 ± 3 |
| GOexfoliated/CSSL 1% | 16 ± 3 | 5.5 ± 0.5 | 286 | 84 | 31 ± 4 |
| GOexfoliated/CSFD 1% | 10 ± 2 | 4.0 ± 0.5 | ND | ND | ND |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francolini, I.; Perugini, E.; Silvestro, I.; Lopreiato, M.; Scotto d’Abusco, A.; Valentini, F.; Placidi, E.; Arciprete, F.; Martinelli, A.; Piozzi, A. Graphene Oxide Oxygen Content Affects Physical and Biological Properties of Scaffolds Based on Chitosan/Graphene Oxide Conjugates. Materials 2019, 12, 1142. https://doi.org/10.3390/ma12071142
Francolini I, Perugini E, Silvestro I, Lopreiato M, Scotto d’Abusco A, Valentini F, Placidi E, Arciprete F, Martinelli A, Piozzi A. Graphene Oxide Oxygen Content Affects Physical and Biological Properties of Scaffolds Based on Chitosan/Graphene Oxide Conjugates. Materials. 2019; 12(7):1142. https://doi.org/10.3390/ma12071142
Chicago/Turabian StyleFrancolini, Iolanda, Elena Perugini, Ilaria Silvestro, Mariangela Lopreiato, Anna Scotto d’Abusco, Federica Valentini, Ernesto Placidi, Fabrizio Arciprete, Andrea Martinelli, and Antonella Piozzi. 2019. "Graphene Oxide Oxygen Content Affects Physical and Biological Properties of Scaffolds Based on Chitosan/Graphene Oxide Conjugates" Materials 12, no. 7: 1142. https://doi.org/10.3390/ma12071142
APA StyleFrancolini, I., Perugini, E., Silvestro, I., Lopreiato, M., Scotto d’Abusco, A., Valentini, F., Placidi, E., Arciprete, F., Martinelli, A., & Piozzi, A. (2019). Graphene Oxide Oxygen Content Affects Physical and Biological Properties of Scaffolds Based on Chitosan/Graphene Oxide Conjugates. Materials, 12(7), 1142. https://doi.org/10.3390/ma12071142