Atomic-Approach to Predict the Energetically Favored Composition Region and to Characterize the Short-, Medium-, and Extended-Range Structures of the Ti-Nb-Al Ternary Metallic Glasses


Abstract
1. Introduction
2. Construction of Ti-Nb-Al Interatomic Potential
3. Metallic Glass Formation of Ti-Nb-Al System
3.1. Atomic Simulation Methods
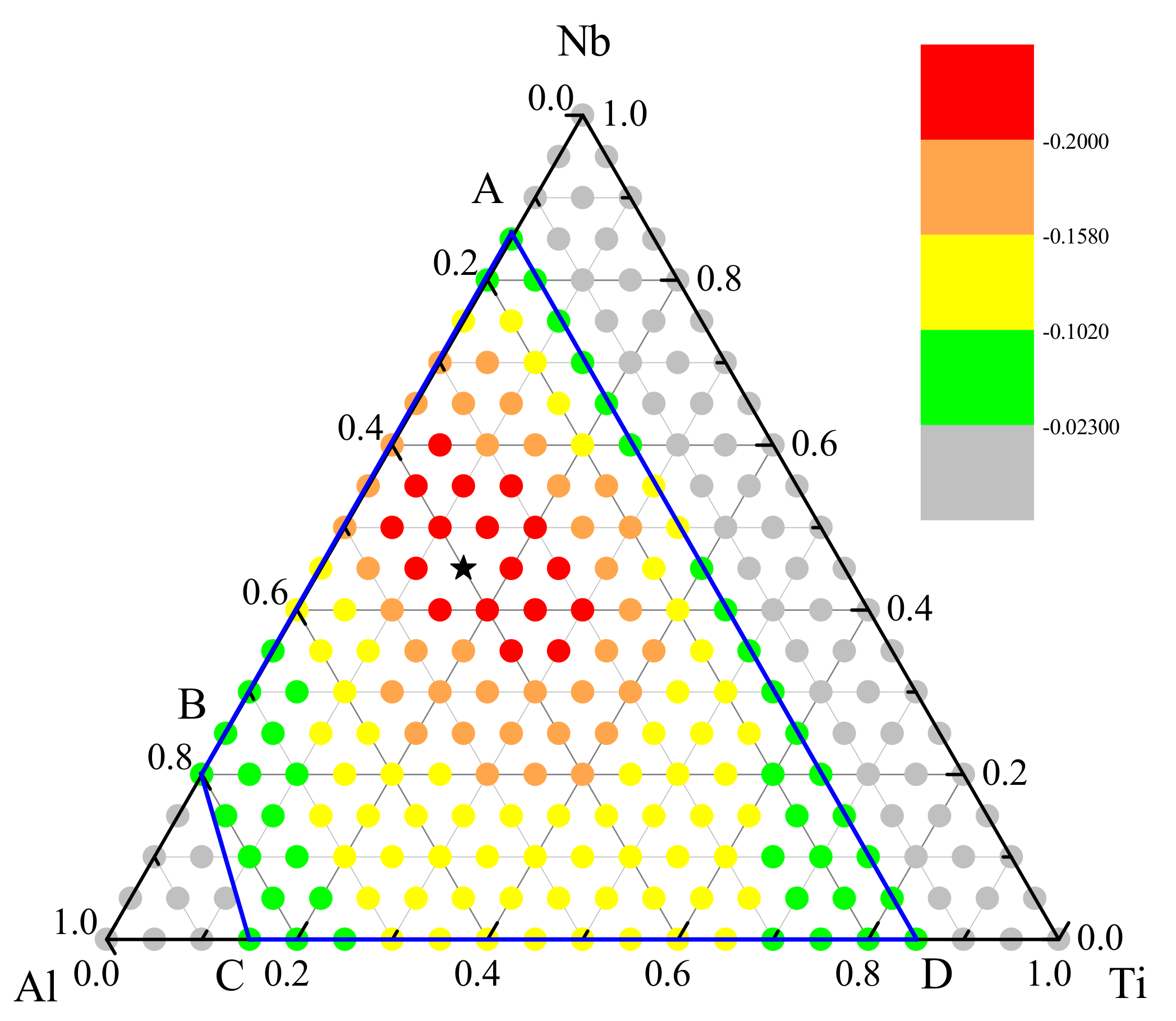
3.2. Glass Formation Region of Ti-Nb-Al System
3.3. Optimization of Glass—Formation Compositions
4. Atomic Structure of Ti-Nb-Al Metallic Glass
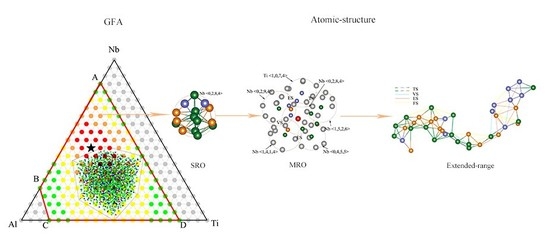
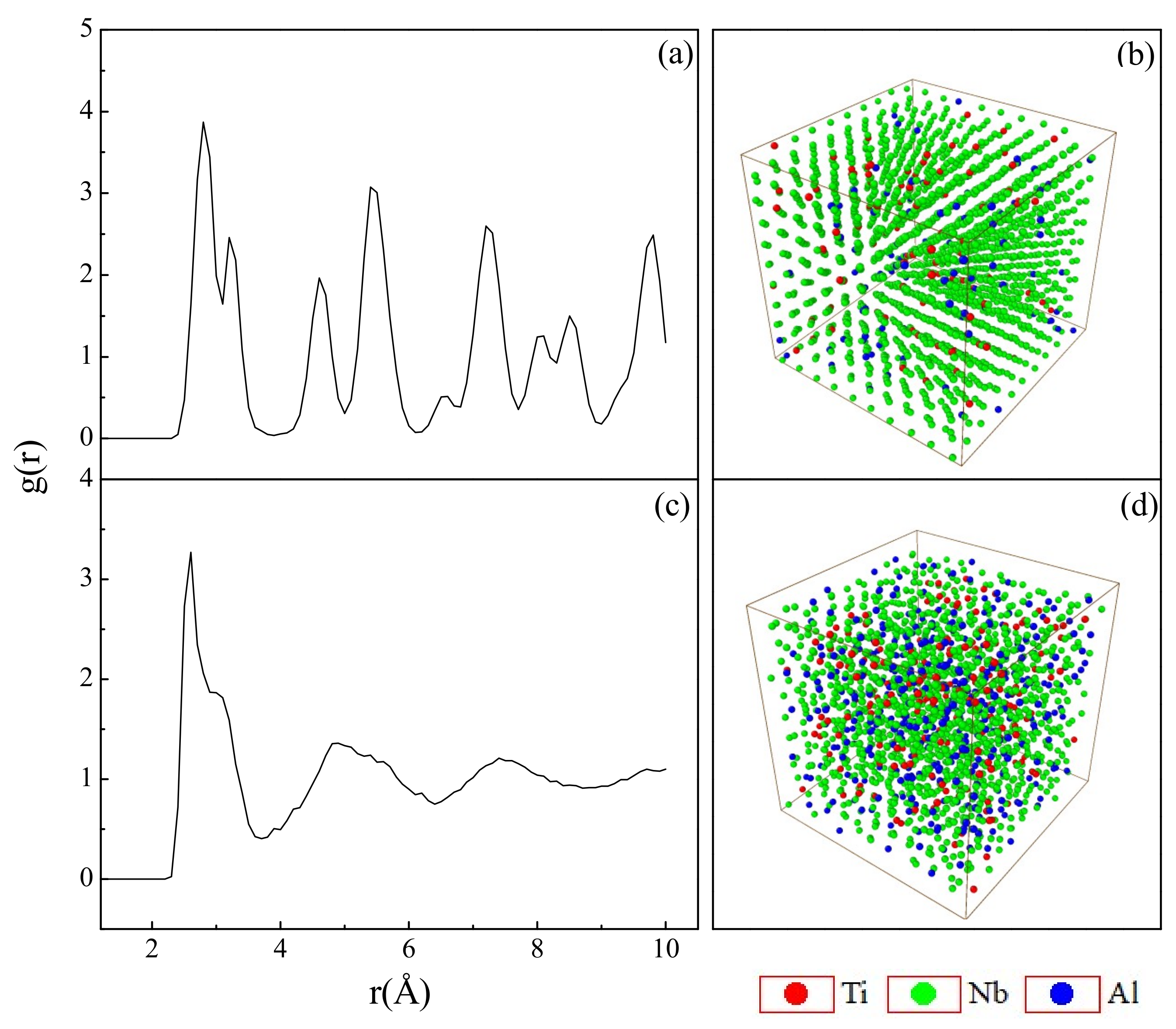
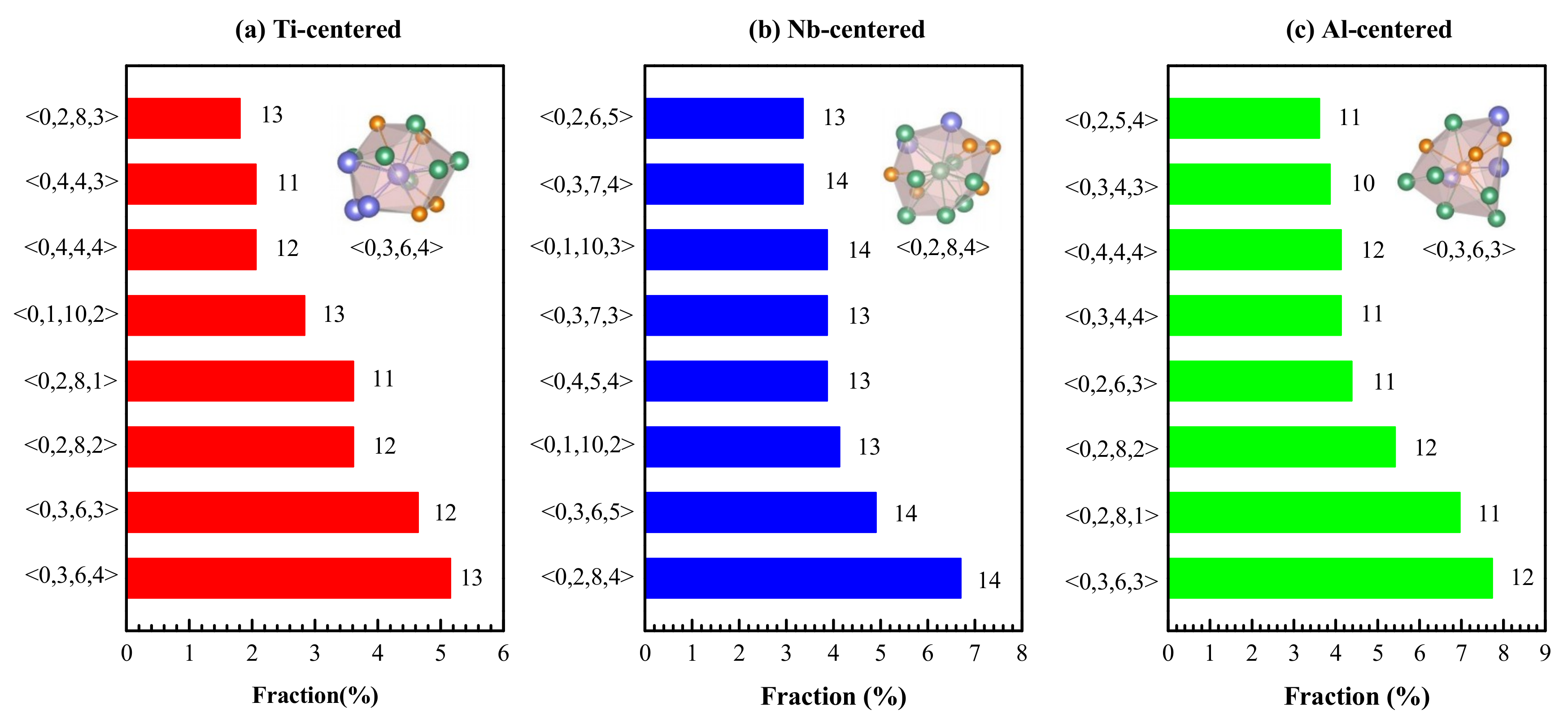
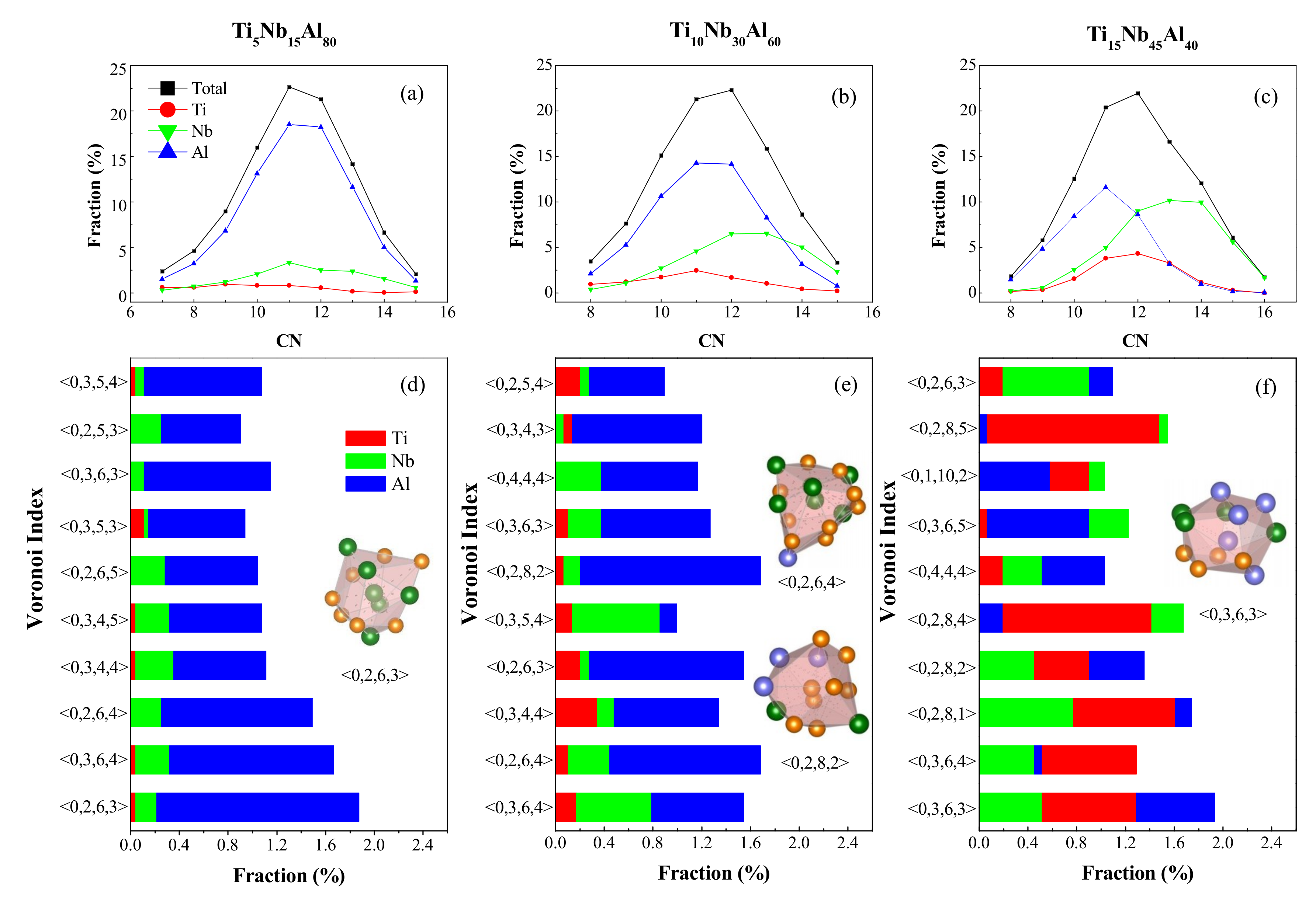
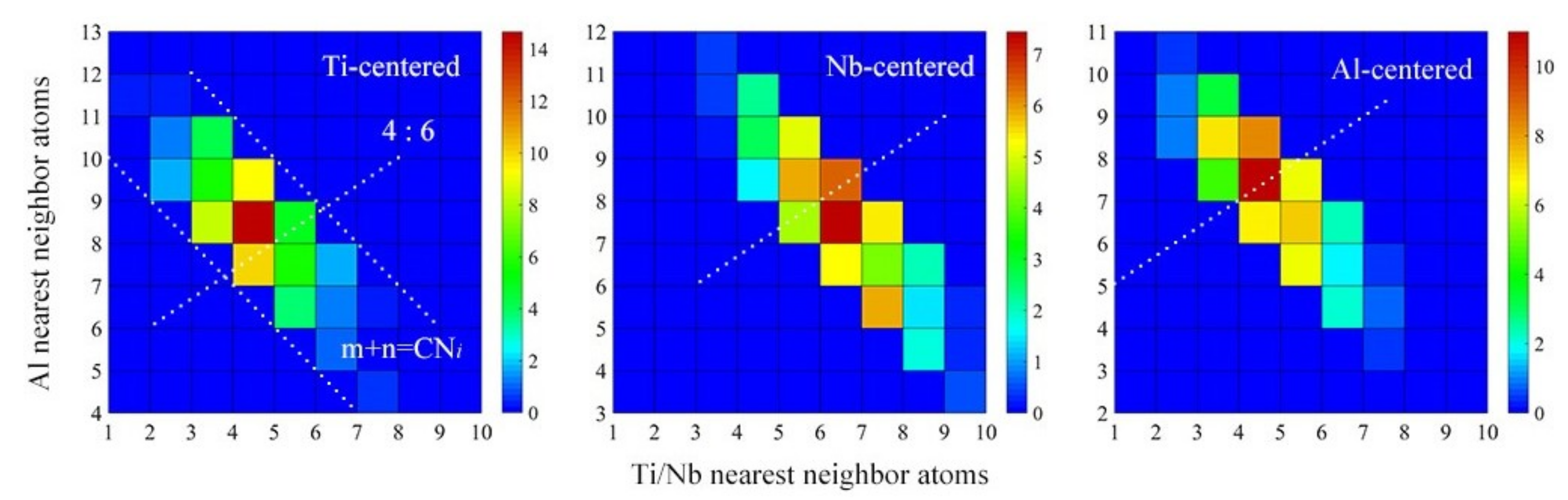
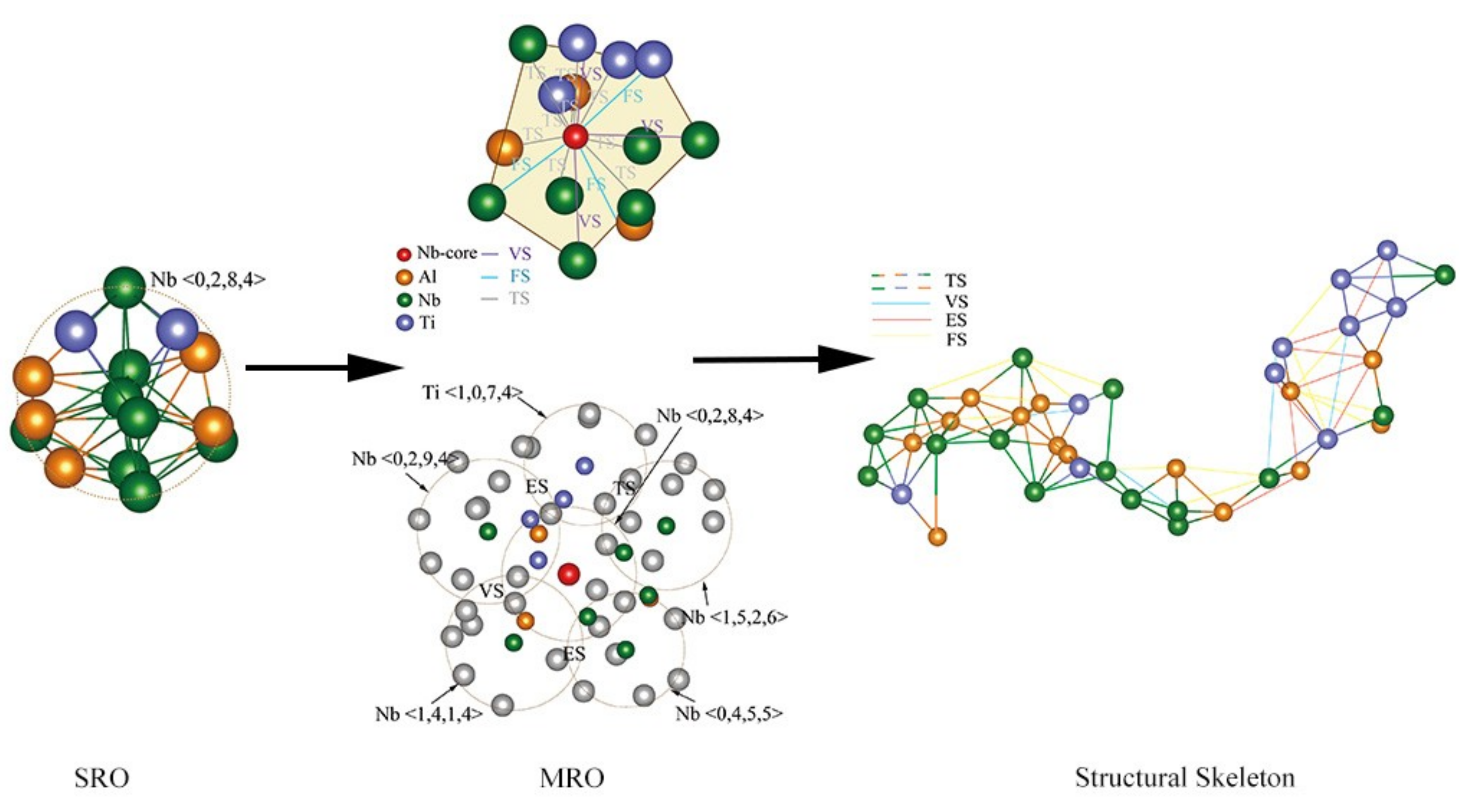
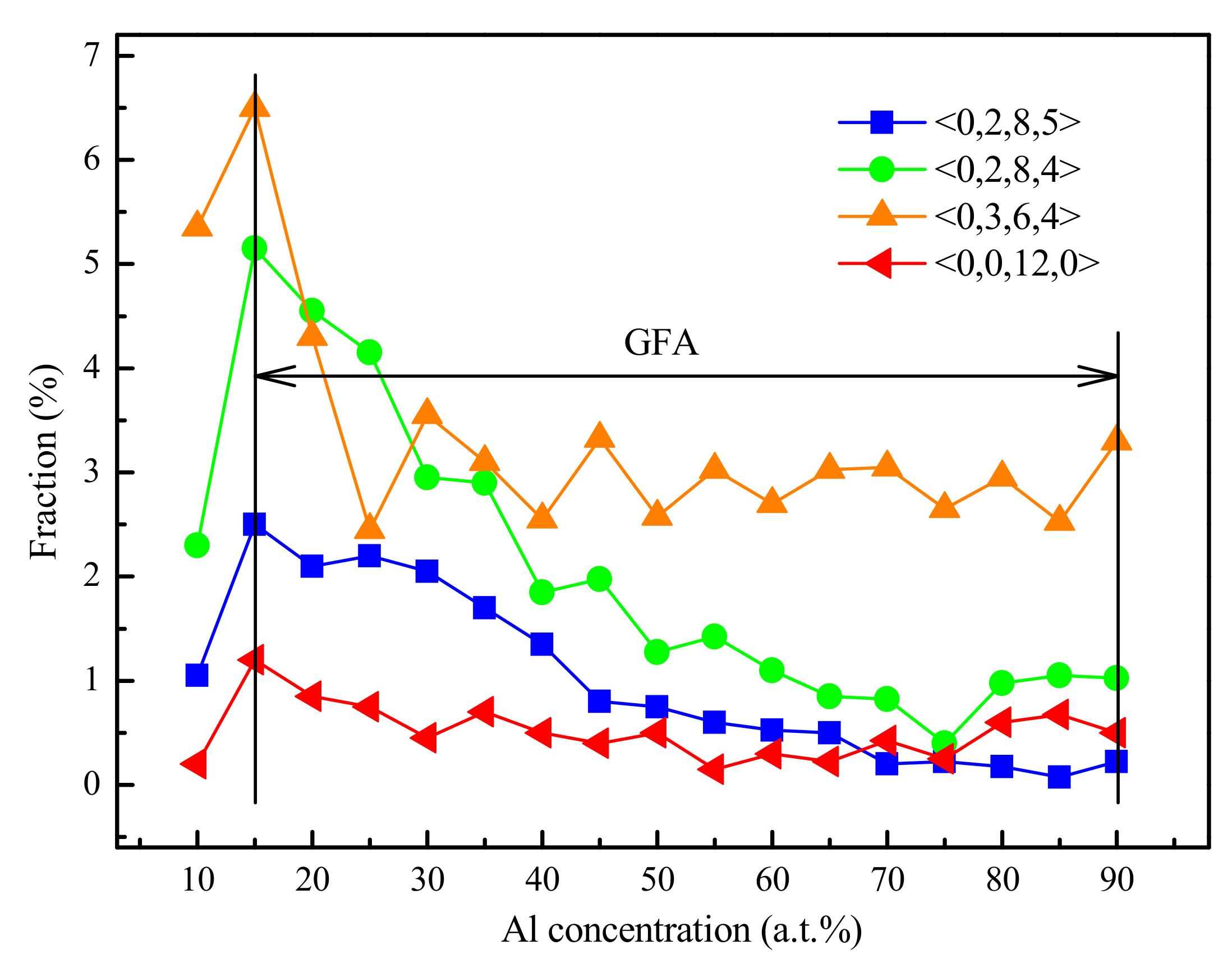
4.1. Short-Range Order and Chemical Microscopic Heterogeneity in Metallic Glass
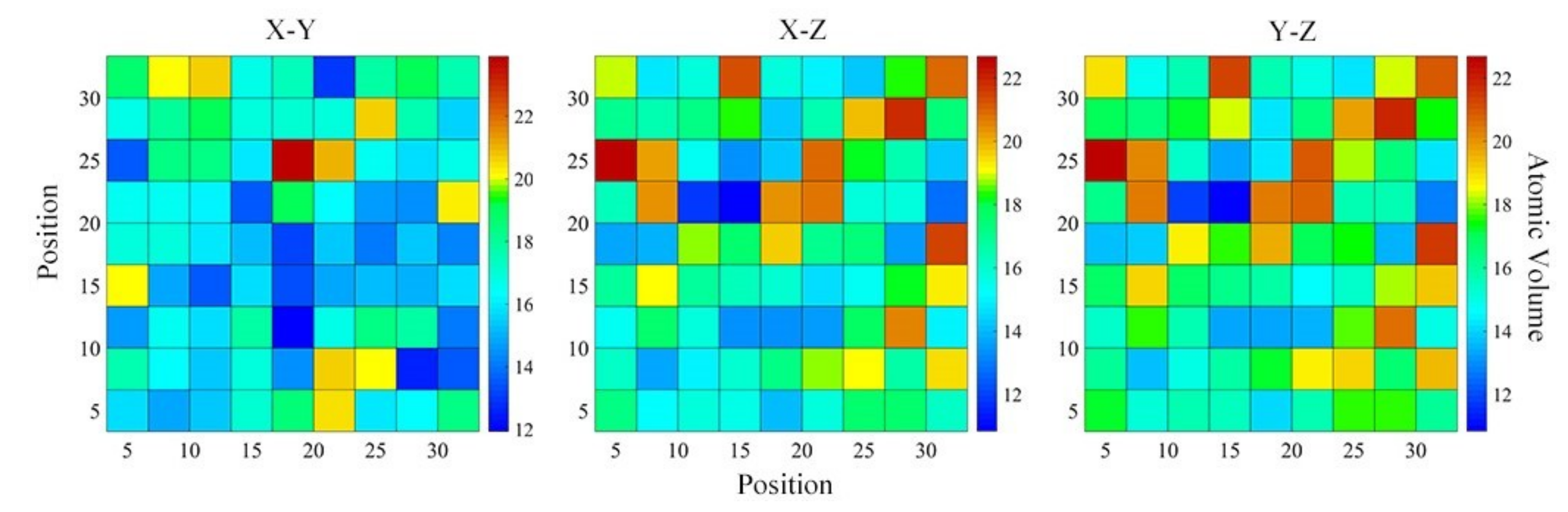
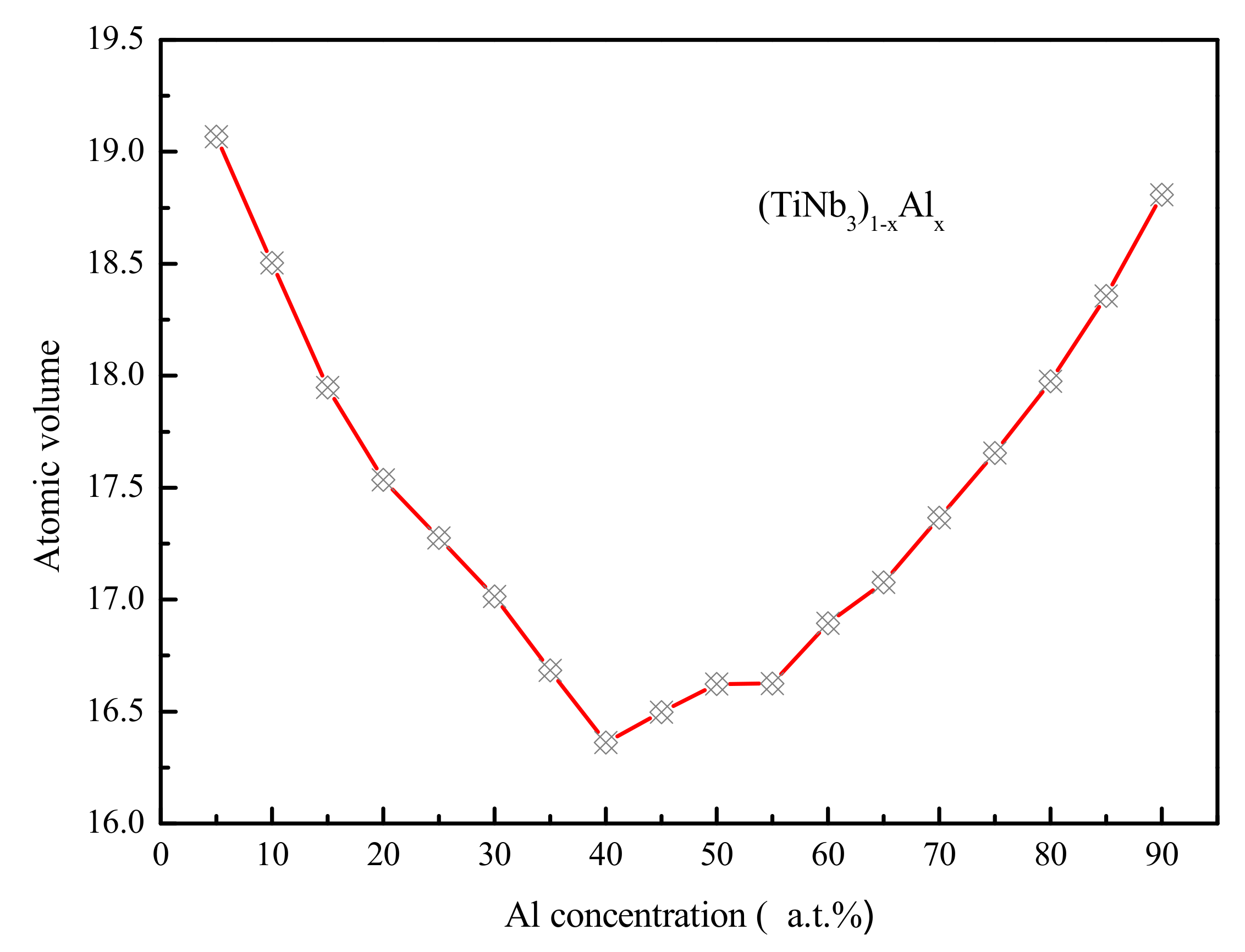
4.2. Medium-Range Order, Cluster Correlation Heterogeneity and Atomic Volume
4.3. Structural Skeleton in the Extended Scale
5. Further Discussion about the GFA and Atomic Structure
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Klement, W.; Willens, R.H.; Duwez, P. Non-crystalline Structure in Solidified Gold–Silicon Alloys. Nature 1960, 187, 869–870. [Google Scholar] [CrossRef]
- Greer, A.L. Metallic glasses. Science 1995, 267, 1947–1953. [Google Scholar] [CrossRef]
- Inoue, A. Stabilization of metallic super-cooled liquid and bulk amorphous alloys. Acta Mater. 2000, 48, 279–306. [Google Scholar] [CrossRef]
- Han, F.F.; Inoue, A.; Han, Y. Novel heating-induced reversion during crystallization of Al-based glassy alloys. Sci. Rep. 2017, 7, 46113. [Google Scholar] [CrossRef]
- Inoue, A. Amorphous, nano-quasi-crystalline and nano-crystalline alloys in Al-based systems. Prog. Mater. Sci. 1998, 43, 365–520. [Google Scholar] [CrossRef]
- Inoue, A.; Kimura, H.M.; Masumoto, T.; Tsai, A.P.; Bizen, Y. Al-Ge-(Cr or Mn) and Al-Si-(Cr or Mn) quasi-crystals with high metalloid concentration prepared by rapid quenching. J. Mater. Sci. Lett. 1987, 6, 771–774. [Google Scholar] [CrossRef]
- Inoue, A.; Yamamoto, M.; Kimura, H.M.; Masumoto, T. Ductile aluminium-base amorphous alloys with two separate phases. J. Mater. Sci. Lett. 1987, 6, 194–196. [Google Scholar] [CrossRef]
- Inoue, A.; Ohtera, K.; Tsai, A.P.; Masumoto, T. Aluminum-based amorphous alloys with tensile strength above 980 MPa (100 kg/mm2). Jpn. J. Appl. Phys. 1988, 27, 479–482. [Google Scholar] [CrossRef]
- Inoue, A.; Sobu, S.; Louzguine, D.V.; Kimura, H.; Sasamori, K. Ultrahigh strength Al-based amorphous alloys containing Sc. J. Mater. Res. 2004, 19, 1539–1543. [Google Scholar] [CrossRef]
- He, Y.; Poon, S.J.; Shiflet, G.J. Synthesis and properties of metallic glasses that contain aluminum. Science 1988, 241, 1640–1642. [Google Scholar] [CrossRef]
- Inoue, A.; Matsumoto, N.; Masumoto, T. Al-Ni-Y-Co amorphous alloys with high mechanical strengths, wide supercooled liquid region and large glass-forming capacity. Mater. Trans. JIM 2007, 31, 493–500. [Google Scholar] [CrossRef]
- Huang, Y.; Shen, J.; Sun, J.; Zhang, Z. Enhanced strength and plasticity of a Ti-based metallic glass at cryogenic temperatures. Mater. Sci. Eng. A 2008, 498, 203–207. [Google Scholar] [CrossRef]
- Masumoto, T.; Inoue, A.; Sakai, S.; Kimura, H.; Hoshi, A. Superconductivity of Ductile Nb-Based Amorphous Alloys. Trans. Jpn. Inst. Met. 1980, 21, 115–122. [Google Scholar] [CrossRef]
- Inoue, A.; Zhang, T.; Masumoto, T. Glass-forming ability of alloys. J. Non-Cryst. Solids 1993, 156, 473–480. [Google Scholar] [CrossRef]
- Turnbull, D. Kinetics of solidification of super-cooled liquid mercury droplets. J. Chem. Phys. 1952, 20, 411–424. [Google Scholar] [CrossRef]
- Xia, L.; Ding, D.; Shan, S.T.; Dong, Y.D. The glass forming ability of Cu-rich Cu-Hf binary alloys. J. Phys. Condens. Mater. 2006, 18, 3543–3548. [Google Scholar] [CrossRef]
- Guo, F.Q.; Enouf, S.; Shiflet, G.; Poon, J. Role of atomic size on glass forming ability and thermal stability of Al-based amorphous alloys. Mater. Trans. JIM 2000, 41, 1406–1409. [Google Scholar] [CrossRef]
- He, Y.; Dougherty, G.M.; Shiflet, G.J.; Poon, S.J. Unique metallic glass forming ability and ultra-high tensile strength in Al-Ni-Fe-Gd alloys. Acta Metall. Mater. 1993, 41, 337–343. [Google Scholar] [CrossRef]
- Li, J.H.; Dai, Y.; Cui, Y.Y. Atomic theory for predicting the binary metallic glass formation. Mater. Sci. Eng. R 2011, 72, 1–28. [Google Scholar] [CrossRef]
- Cui, Y.Y.; Wang, T.L.; Li, J.H.; Dai, Y.; Liu, B.X. Thermodynamic calculation and inter-atomic potential to predict the favored composition region for the Cu-Zr-Al metallic glass formation. Phys. Chem. Chem. Phys. 2011, 13, 4103–4108. [Google Scholar] [CrossRef]
- Yang, M.H.; Li, J.H.; Liu, B.X. Proposed correlation of structure network inherited from producing techniques and deformation behavior for Ni-Ti-Mo metallic glasses via atomistic simulations. Sci. Rep. 2016, 6, 29722. [Google Scholar] [CrossRef] [PubMed]
- Yavari, A.R. Materials science: A new order for metallic glasses. Nature 2006, 439, 405–406. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Q.; Ma, E. Atomic-level structure and structure-property relationship in metallic glasses. Prog. Mater. Sci. 2011, 56, 379–473. [Google Scholar] [CrossRef]
- Xing, L.Q.; Hufnagel, T.C.; Eckert, J.; Loser, W. Relation between short-range order and crystallization behavior in Zr-based amorphous alloys. Appl. Phys. Lett. 2000, 77, 1970–1972. [Google Scholar] [CrossRef]
- Peng, H.L.; Li, M.Z.; Wang, W.H.; Wang, C.Z.; Ho, K.M. Effect of local structures and atomic packing on glass forming ability in CuxZr100−x metallic glasse. Appl. Phys. Lett. 2010, 96, 021901. [Google Scholar] [CrossRef]
- Hirata, A.; Kang, L.J.; Fujita, T.; Klumov, B.; Matsue, K. Geometric frustration of icosahedron in metallic glasses. Science 2013, 341, 376–379. [Google Scholar] [CrossRef]
- Kartouzian, A. Cluster-assembled metallic glasses. Nanoscale Res. Lett. 2013, 8, 339. [Google Scholar] [CrossRef]
- Bednarcik, J.; Michalik, S.; Kolesar, V. In situ XRD studies of nanocrystallization of Fe-based metallic glass: A comparative study by reciprocal and direct space methods. Phys. Chem. Chem. Phys. 2013, 15, 8470–8479. [Google Scholar] [CrossRef]
- Jin, Z.H.; Lu, K.; Gong, Y.D.; Hu, Z.Q. Glass transition and atomic structures in supercooled Ga0.15Zn0.15Mg0.7 metallic liquids: A constant pressure molecular dynamics study. J. Chem. Phys. 1997, 106, 8830–8840. [Google Scholar] [CrossRef]
- Fan, C.; Liaw, P.K.; Wilson, T.W. Structural model for bulk amorphous alloys. Appl. Phys. Lett. 2006, 89, 111905. [Google Scholar] [CrossRef]
- Gonçalves, A.P.; Lopes, E.B.; Rouleau, O. Conducting glasses as new potential thermoelectric materials: The Cu–Ge–Te case. J. Mater. Chem. 2010, 20, 1516–1521. [Google Scholar] [CrossRef]
- Liu, X.J.; Xu, Y.; Hui, X. Metallic liquids and glasses: Atomic order and global packing. Phys. Rev. Lett. 2010, 105, 155501. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.W.; Luo, W.K.; Alamgir, F.M. Atomic packing and short-to-medium-range order in metallic glasses. Nature 2006, 439, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Guan, P.; Fujita, T. Direct observation of local atomic order in a metallic glass. Nat. Mater. 2011, 10, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.F.; Fujita, T.; Hirata, A. Structural origins of the excellent glass forming ability of Pd40Ni40P20. Phys. Rev. Lett. 2012, 108, 175501. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Q.; Ma, E.; Sheng, H.W. Atomic level structure in multicomponent bulk metallic glass. Phys. Rev. Lett. 2009, 102, 245501. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Stoica, A.D.; Wang, X.L. Power-law scaling and fractal nature of medium-range order in metallic glasses. Nat. Mater. 2009, 8, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.D.; Yin, S.; Cao, Q.P. Atomic structure of binary Cu64.5Zr35.5 bulk metallic glass. Appl. Phys. Lett. 2008, 92, 011902. [Google Scholar] [CrossRef]
- Lee, M.; Lee, C.M.; Lee, K.R. Networked interpenetrating connections of icosahedra: Effects on shear transformations in metallic glass. Acta Mater. 2011, 59, 159–170. [Google Scholar] [CrossRef]
- Ichitsubo, T.; Matsubara, E.; Yamamoto, T. Microstructure of fragile metallic glasses inferred from ultrasound-accelerated crystallization in Pd-based metallic glasses. Phys. Rev. Lett. 2005, 95, 245501. [Google Scholar] [CrossRef]
- Wu, Z.W.; Li, M.Z.; Wang, W.H. Correlation between structural relaxation and connectivity of icosahedral cluster in Cu-Zr metallic glass-forming liquids. Phys. Rev. B 2013, 88, 054202. [Google Scholar] [CrossRef]
- Luo, W.K.; Sheng, H.W.; Alamgir, F.M. Icosahedral short-range order in amorphous alloys. Phys. Rev. Lett. 2004, 92, 145502. [Google Scholar] [CrossRef] [PubMed]
- Soklaski, R.; Nussinov, Z.; Markow, Z. Connectivity of icosahedral network and a dramatically growing static length scale in Cu-Zr binary metallic glasses. Phys. Rev. B 2013, 87, 184203. [Google Scholar] [CrossRef]
- Li, M.; Wang, C.Z.; Hao, S.G. Structural heterogeneity and medium-range order in ZrxCu100−x metallic glasses. Phys. Rev. B 2009, 80, 184201. [Google Scholar] [CrossRef]
- Dai, X.D.; Kong, Y.; Li, J.H. Long-range empirical potential model: Application to fcc transition metals and alloys. Phys. Rev. B 2007, 75, 104101. [Google Scholar] [CrossRef]
- Dai, X.D.; Li, J.H.; Kong, Y. Long-range empirical potential for the bcc structured transition metals. Phys. Rev. B 2007, 75, 052102. [Google Scholar] [CrossRef]
- Dai, Y.; Li, J.H.; Liu, B.X. Long-range empirical potential model: Extension to hexagonal close-packed metals. J. Phys. Condens. Matter. 2009, 21, 385402. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Yang, M.H.; Liu, J.B. Atomistic simulation study of favored compositions of Ni-Nb-Al metallic glasses. Sci. China. Technol. Sci. 2018, 61, 1829–1838. [Google Scholar] [CrossRef]
- Sabeena, M.; Murugesan, S.; Anees, P. Crystal structure and bonding characteristics of transformation products of bcc beta in Ti-Mo alloys. J. Alloys Compd. 2017, 705, 769–781. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J. First-principles simulation: Ideas. illustrations and the CASTEP code. J. Phys. Condens. Mater. 2002, 14, 2717. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J. First principles methods using CASTEP. Zeitschrift für Kristallographie 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244. [Google Scholar] [CrossRef]
- Rose, J.H.; Smith, J.R.; Guinea, F. Universal features of the equation of state of metals. Phys. Rev. B 1984, 29, 2963. [Google Scholar] [CrossRef]
- Sheng, H.W.; Wilde, G.; Ma, E. The competing crystalline and amorphous solid solutions in the Ag-Cu system. Acta Mater. 2002, 50, 475–488. [Google Scholar] [CrossRef]
- LAMMPS Package. Available online: http://lammps.sandia.gov (accessed on 30 January 2019).
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Panagiotopoulos, A.Z.; Quirke, N.; Stapleton, M. Phase equilibria by simulation in the Gibbs ensemble: Alternative derivation, generalization and application to mixture and membrane equilibria. Mol. Phys. 1988, 63, 527–545. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: London, UK, 1987. [Google Scholar]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Dai, Y.; Li, J.H.; Che, X.L. Glass-forming region of the Ni-Nb-Ta ternary metal system determined directly from n-body potential through molecular dynamics simulations. J. Mater. Res. 2009, 24, 1815–1819. [Google Scholar] [CrossRef]
- Finney, J.L. Random Packings and the Structure of Simple Liquids. I. The Geometry of Random Close Packing. Proc. R. Soc. Lond. A 1970, 319, 479–493. [Google Scholar] [CrossRef]
- Hui, X.; Fang, H.Z.; Chen, G.L. Atomic structure of Zr41.2Ti13.8Cu12.5Ni10Be22.5 bulk metallic glass alloy. Acta Mater. 2009, 57, 376–391. [Google Scholar] [CrossRef]
- Wang, Q.; Li, J.H.; Cui, Y.Y. Calculation of driving force and local order to predict the favored and optimized compositions for Mg-Cu-Ni metallic glass formation. J. Appl. Phys. 2013, 114, 153503. [Google Scholar] [CrossRef]
- Liu, B.X.; Cheng, G.A. Glass Forming ability of the Al-Ti System under Ion Beam Mixing. Phys. Stat. Sol. A 1991, 125, 93–96. [Google Scholar] [CrossRef]
- Akiyama, E.; Yoshioka, H.; Kim, J.H. The effect of magnesium on the corrosion behavior of sputter-deposited amorphous Al-Mg-Ti ternary alloys in a neutral chloride solution. Corros. Sci. 1993, 34, 27–40. [Google Scholar] [CrossRef]
- Itsukaichi, T.; Masuyama, K.; Umemoto, M. Mechanical alloying of Al-Ti powder mixtures and their subsequent consolidation. J. Mater. Res. 1993, 8, 1817–1828. [Google Scholar] [CrossRef]
- El-Eskandarany, M.S. Amorphization process by rod-milling TixAl100−x and the effect of annealing. J. Alloys. Compd. 1996, 234, 67–82. [Google Scholar] [CrossRef]
- Yoo, D.J.; Hwang, S.M.; Lee, S.M. Phase formation in mechanically alloyed Nb-Al powders. Appl. Phys. Lett. 2000, 19, 1327–1329. [Google Scholar]
- Karimi, H.; Ghasemi, A.; Hadi, M. Microstructure and oxidation behaviour of TiAl(Nb)/Ti2AlC composites fabricated by mechanical alloying and hot pressing. Bull. Mater. Sci. 2016, 39, 1–10. [Google Scholar] [CrossRef]
- Chen, G.H.; Suryanarayana, C.; Froes, F.H.S. Structure of mechanically alloyed Ti-Al-Nb powders. Metall. Mater. Trans. A 1995, 26, 1379–1387. [Google Scholar] [CrossRef]
- Nelson, D.R. Order, frustration, and defects in liquids and glasses. Phys. Rev. B 1983, 28, 5515–5535. [Google Scholar] [CrossRef]
- Li, F.; Liu, X.J.; Lu, Z.P. Atomic structural evolution during glass formation of a Cu–Zr binary metallic glass. Comput. Mater. Sci. 2014, 85, 147–153. [Google Scholar] [CrossRef]
- Gong, Z.Q.; Chen, Z.Y.; Chai, L.H. Thermodynamic properties of high niobium containing Ti-Al based alloy with erbium. Trans. Mater. Heat Treat. 2014, 2, 205–208. [Google Scholar]
- Miracle, D.B. A structural model for metallic glasses. Nat. Mater. 2004, 3, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, J.H.; Liu, J.B. Structural skeleton of preferentially interpenetrated clusters and correlation with shear localization in Mg-Cu-Ni ternary metallic glasses. Phys. Chem. Chem. Phys. 2014, 16, 19590–19601. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Zhao, X. Structural relaxation and its influence on the elastic properties and notch toughness of Mg-Zn-Ca bulk metallic glass. J. Alloys Compd. 2012, 515, 154–160. [Google Scholar] [CrossRef]
- Louzguine-Luzgin, D.V.; Yavari, A.R.; Fukuhara, M. Free volume and elastic properties changes in Cu-Zr-Ti-Pd bulk glassy alloy on heating. J. Alloys Compd. 2007, 431, 136–140. [Google Scholar] [CrossRef]
- Open Visualization Tool. Available online: http://www.ovito.org/ (accessed on 30 January 2019).
- Wang, Q.; Li, J.H.; Liu, J.B. Favored Composition Design and Atomic Structure Characterization for Ternary Al−Cu−Y Metallic Glasses via Proposed Interatomic Potential. Phys. Chem. B 2014, 118, 4442–4449. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potential Parameters | Ti-Ti | Nb-Nb | Al-Al | Ti-Al | Nb-Al | Ti-Nb |
|---|---|---|---|---|---|---|
| c0 (10−19 J/Åm) | 1.146137 | 16.721753 | 0.558355 | 0.309051 | 1.720707 | 5.543857 |
| c1 (10−19 J/Åm + 1) | −1.417844 | −22.891094 | −0.621271 | −0.338078 | −2.09683 | −8.078405 |
| c2 (10−19 J/Åm + 2) | 0.700512 | 11.808389 | 0.259702 | 0.140656 | 0.951759 | 4.703939 |
| c3 (10−19 J/Åm + 3) | −0.159982 | −2.695603 | −0.047246 | −0.025314 | −0.188004 | −1.241254 |
| c4 (10−19 J/Åm + 4) | 0.014262 | 0.228706 | 0.003148 | 0.001621 | 0.013434 | 0.124878 |
| α (10−19 J/Ån) | 0.056220 | −0.02174 | 0.035311 | 0.062566 | 0.072146 | 0.108197 |
| m | 4 | 4 | 4 | 4 | 4 | 4 |
| n | 6 | 8 | 6 | 6 | 6 | 6 |
| rc1 (Å) | 5.171296 | 4.802774 | 5.826946 | 6.551139 | 5.525992 | 4.499325 |
| rc2 (Å) | 6.935479 | 6.818573 | 7.246908 | 7.068235 | 6.452469 | 6.567841 |
| Physical Properties | hcp-Ti | fcc-Ti | bcc-Ti | |||
|---|---|---|---|---|---|---|
| Fitted | Experiments | Fitted | Ab Initio | Fitted | Ab Initio | |
| a or a, c (Å) | 2.836, 4.708 | 2.951, 4.684 | 4.008 | 4.097 | 3.217 | 3.246 |
| Ec (eV/atom) | 4.789 | 4.850 | 4.790 | 4.792 | 4.769 | 4.738 |
| C11 (Mbar) | 1.553 | 1.624 | 1.177 | 1.400 | 0.962 | 0.986 |
| C12 (Mbar) | 0.887 | 0.920 | 1.094 | 0.972 | 1.126 | 1.150 |
| C44 (Mbar) | 0.392 | 0.467 | 0.530 | 0.592 | 0.580 | 0.454 |
| C13 (Mbar) | 0.745 | 0.690 | ||||
| C33 (Mbar) | 1.862 | 1.807 | ||||
| B0 (Mbar) | 1.080 | 1.051 | 1.122 | 1.115 | 1.071 | 1.096 |
| Physical Properties | Methods | TiNb3 | TiNb | Ti3Nb |
|---|---|---|---|---|
| L12 | B2 | L12 | ||
| a or a, c (Å) | Fitted | 4.313 | 3.394 | 4.235 |
| Ab initio | 4.183 | 3.263 | 4.123 | |
| Ec (eV/atom) | Fitted | 6.696 | 6.124 | 5.447 |
| Ab initio | 6.666 | 6.135 | 5.446 | |
| B0 (Mbar) | Fitted | 1.174 | 1.506 | 1.244 |
| Ab initio | 1.346 | 1.365 | 1.236 |
| Physical Properties | Methods | AlTi3 | AlTi | Al3Ti | ||
|---|---|---|---|---|---|---|
| L12 | D019 | B2 | L12 | D022 | ||
| a or a, c (Å) | Fitted | 4.047 | 5.691, 4.701 | 3.163 | 4.030 | 3.961, 8.590 |
| Ab initio | 4.408 | 5.780, 4.647 | 3.186 | 3.983 | 3.854, 8.584 | |
| Ec (eV/atom) | Fitted | 4.764 | 4.716 | 4.397 | 4.159 | 4.105 |
| Ab initio | 4.752 | 4.738 | 4.390 | 4.137 | 4.132 | |
| B0 (Mbar) | Fitted | 1.222 | 1.114 | 1.093 | 1.090 | 0.970 |
| Ab initio | 1.214 | 1.119 | 1.098 | 1.034 | 1.030 | |
| Binary System | Enthalpy (KJ/mol) |
|---|---|
| Ti-Al | −92.23 |
| Ti-Nb | 1.30 |
| Al-Nb | −9.99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, B.; Liu, J.; Li, J.; Yang, M.; Liu, B. Atomic-Approach to Predict the Energetically Favored Composition Region and to Characterize the Short-, Medium-, and Extended-Range Structures of the Ti-Nb-Al Ternary Metallic Glasses. Materials 2019, 12, 432. https://doi.org/10.3390/ma12030432
Cai B, Liu J, Li J, Yang M, Liu B. Atomic-Approach to Predict the Energetically Favored Composition Region and to Characterize the Short-, Medium-, and Extended-Range Structures of the Ti-Nb-Al Ternary Metallic Glasses. Materials. 2019; 12(3):432. https://doi.org/10.3390/ma12030432
Chicago/Turabian StyleCai, Bei, Jianbo Liu, Jiahao Li, Menghao Yang, and Baixin Liu. 2019. "Atomic-Approach to Predict the Energetically Favored Composition Region and to Characterize the Short-, Medium-, and Extended-Range Structures of the Ti-Nb-Al Ternary Metallic Glasses" Materials 12, no. 3: 432. https://doi.org/10.3390/ma12030432
APA StyleCai, B., Liu, J., Li, J., Yang, M., & Liu, B. (2019). Atomic-Approach to Predict the Energetically Favored Composition Region and to Characterize the Short-, Medium-, and Extended-Range Structures of the Ti-Nb-Al Ternary Metallic Glasses. Materials, 12(3), 432. https://doi.org/10.3390/ma12030432