Fabrication and Histological Evaluation of Porous Carbonate Apatite Block from Gypsum Block Containing Spherical Phenol Resin as a Porogen

Abstract
1. Introduction
2. Materials and Methods
2.1. Fabrication of Cylindrical CO3Ap and HAp Blocks
2.2. Characterization of Samples
2.3. Surgical Procedure
2.4. Image Analysis
2.5. Histological Procedures
2.6. Statistical Analysis
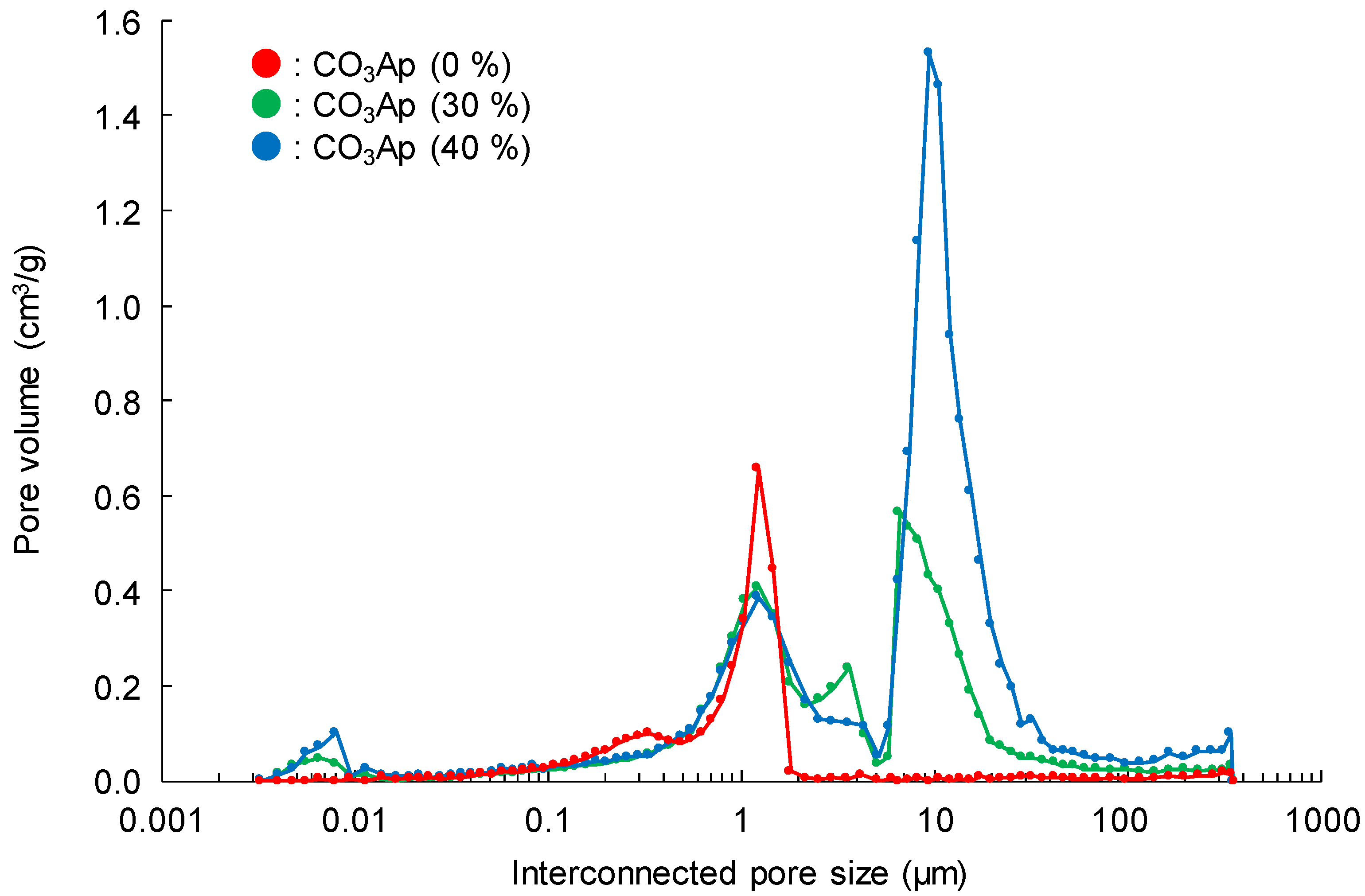
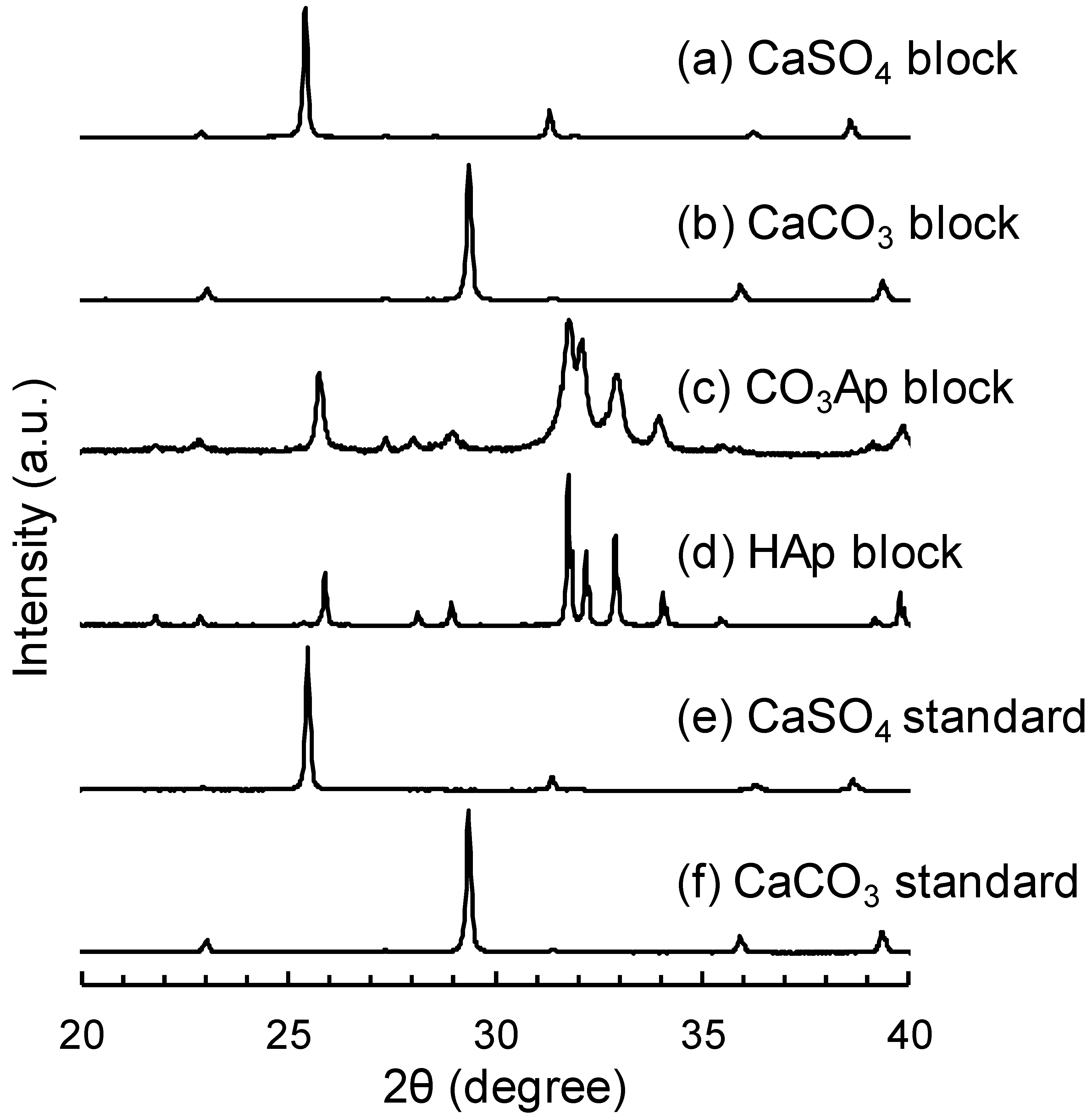
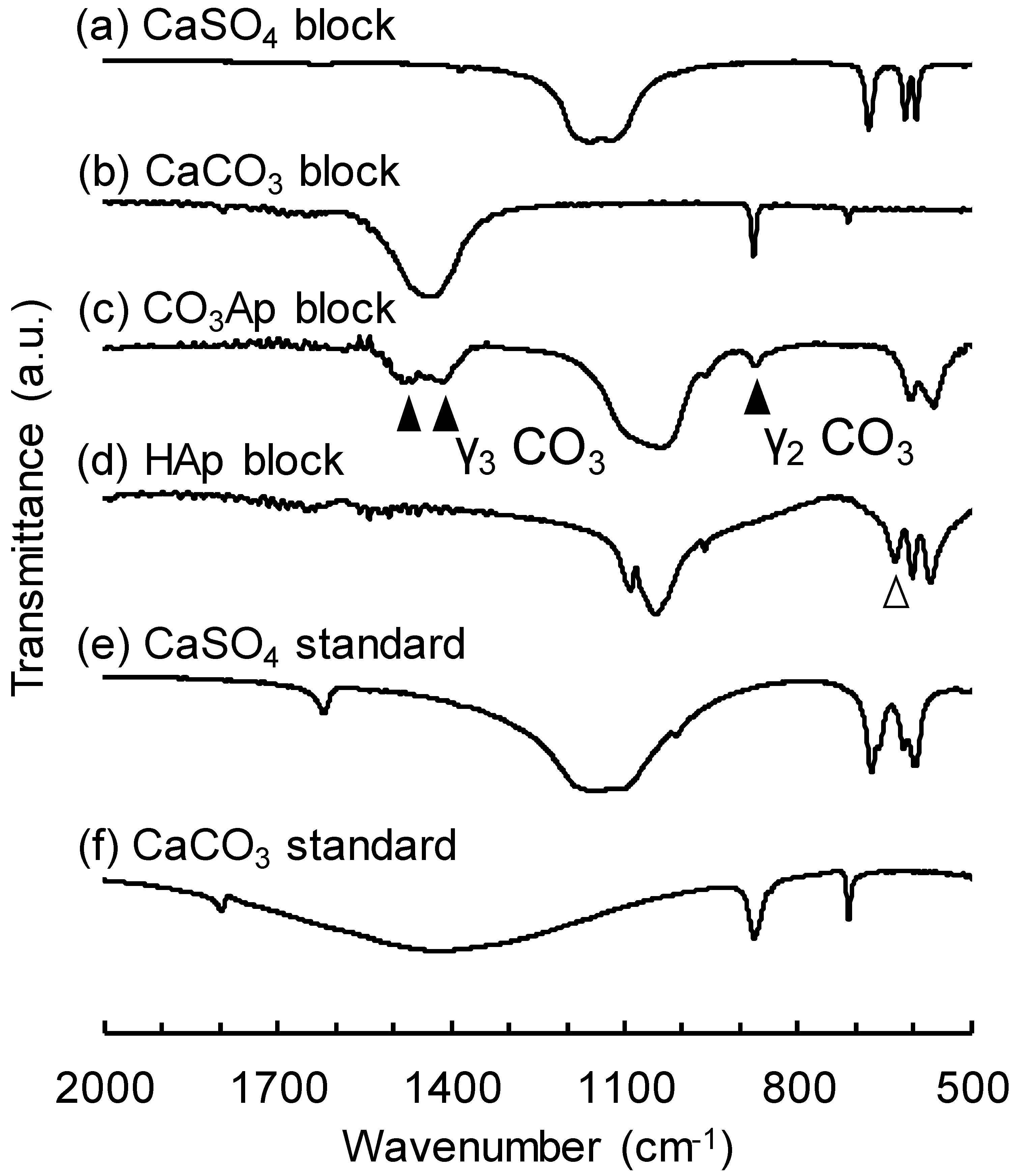
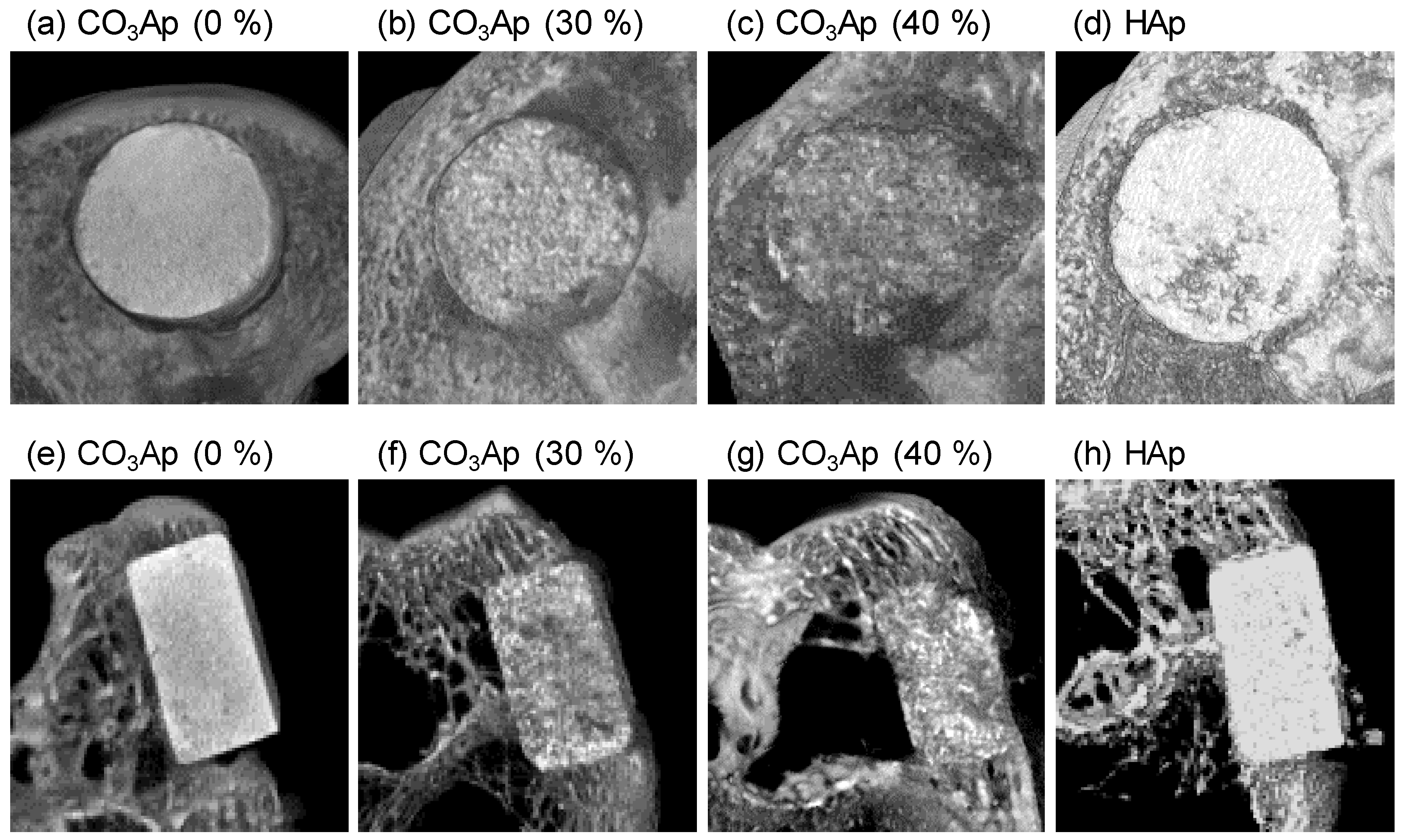
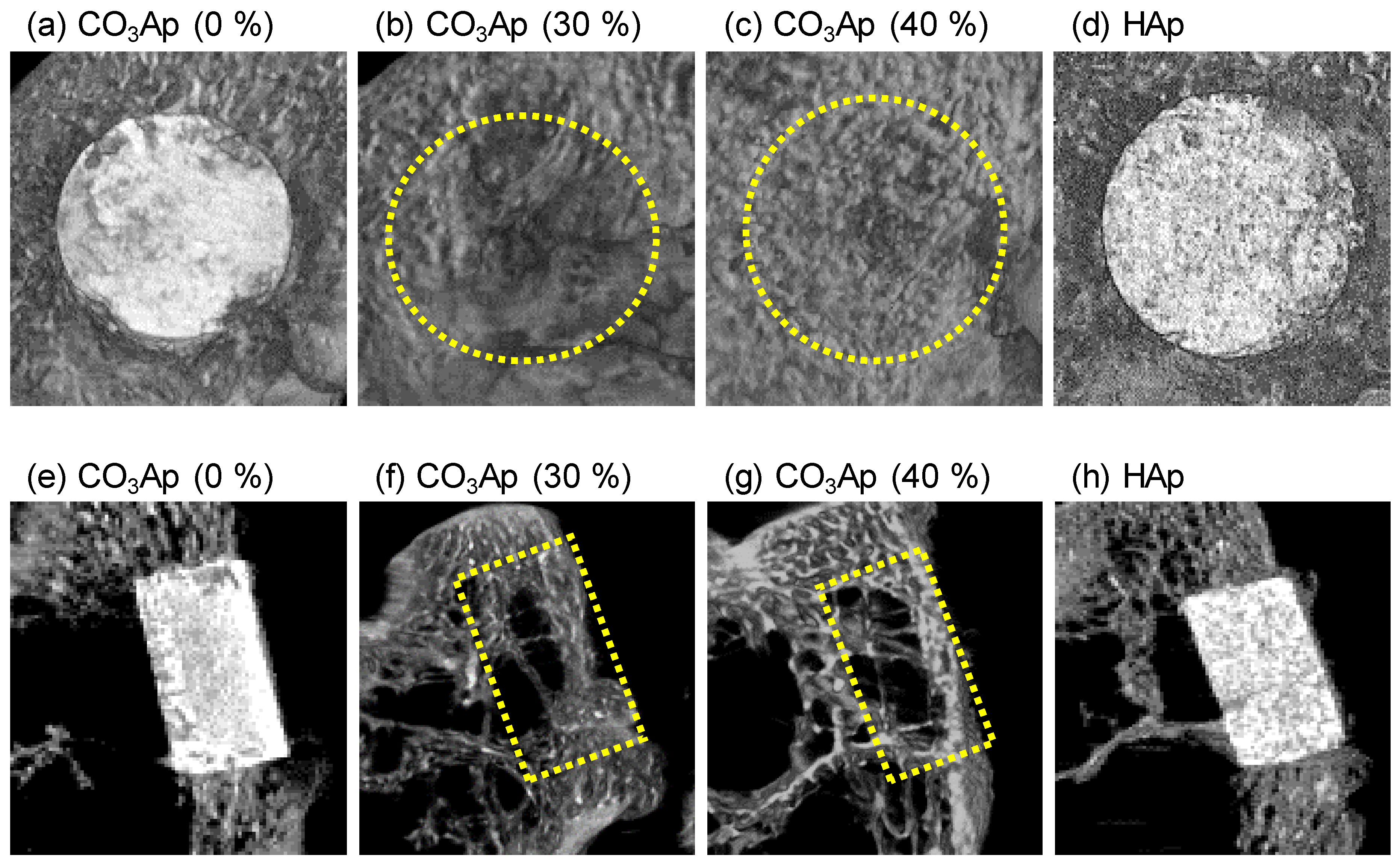
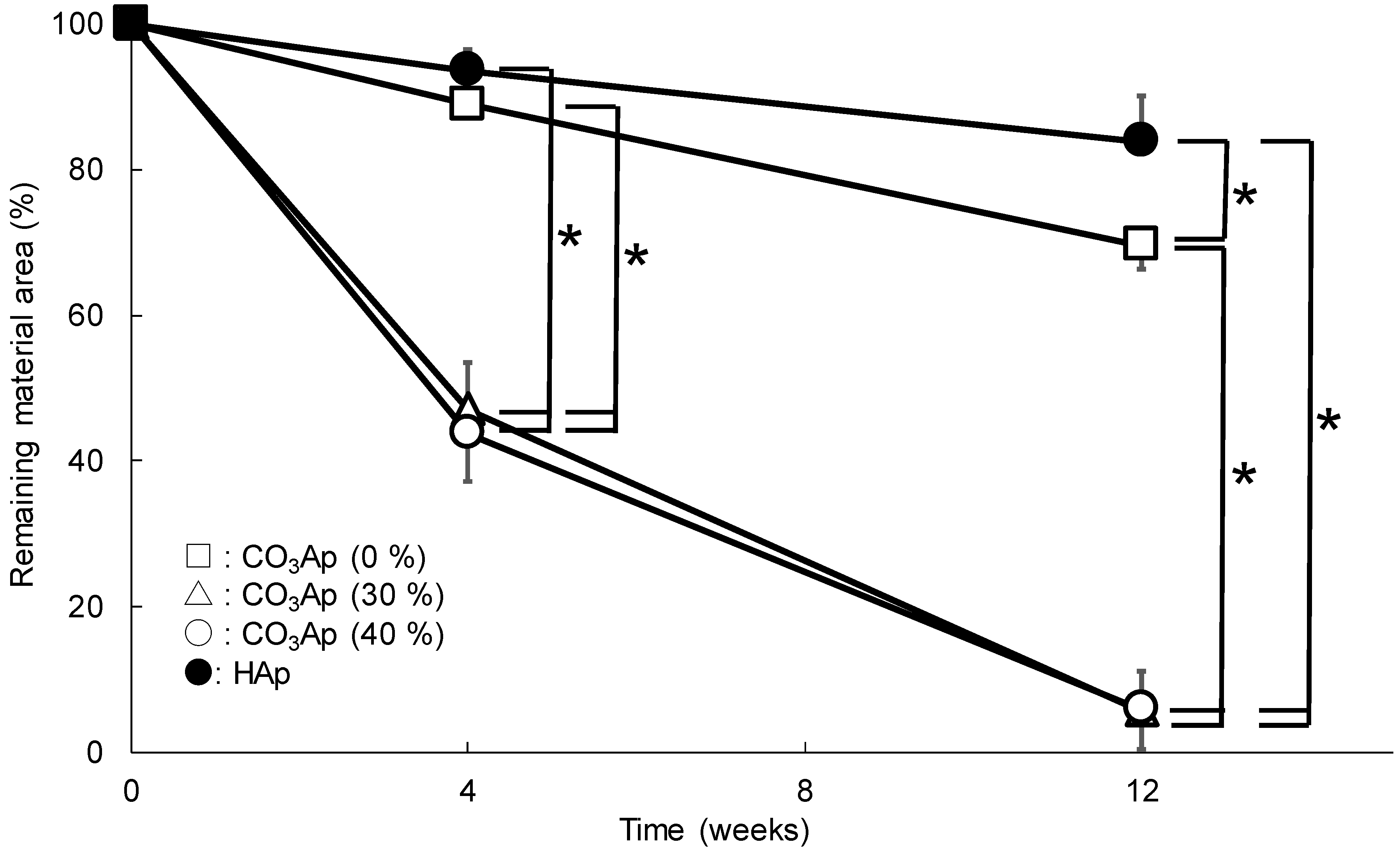
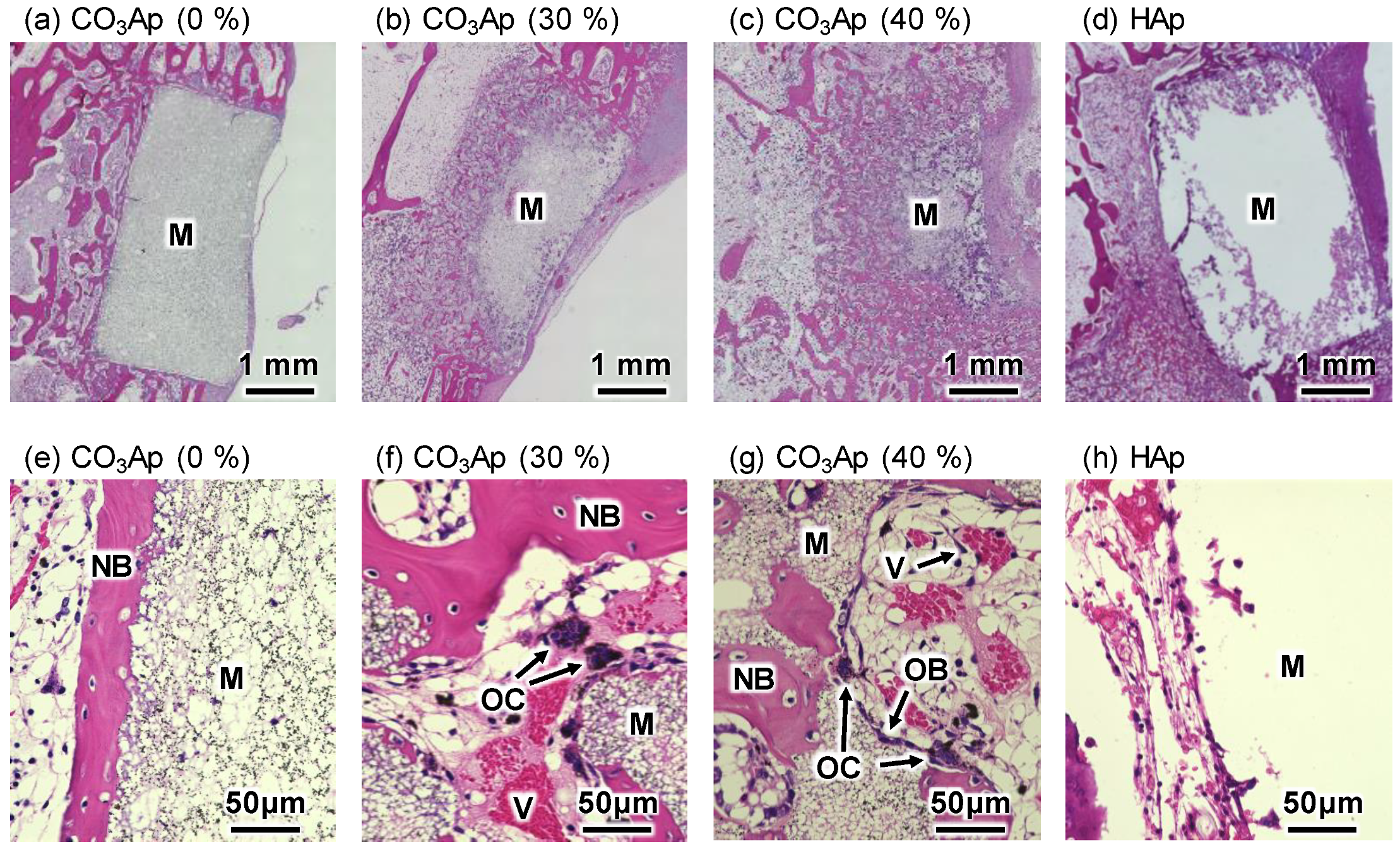
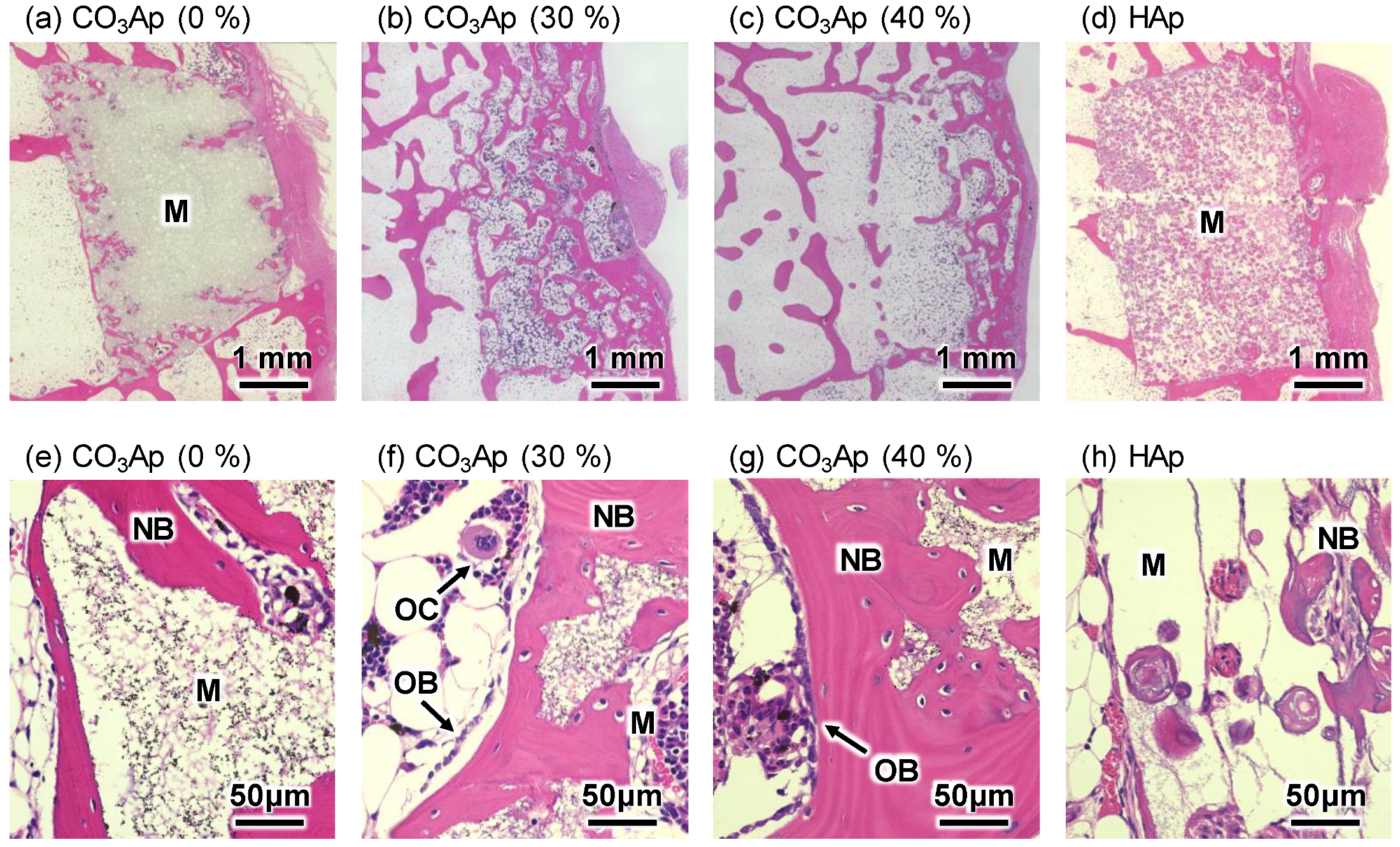
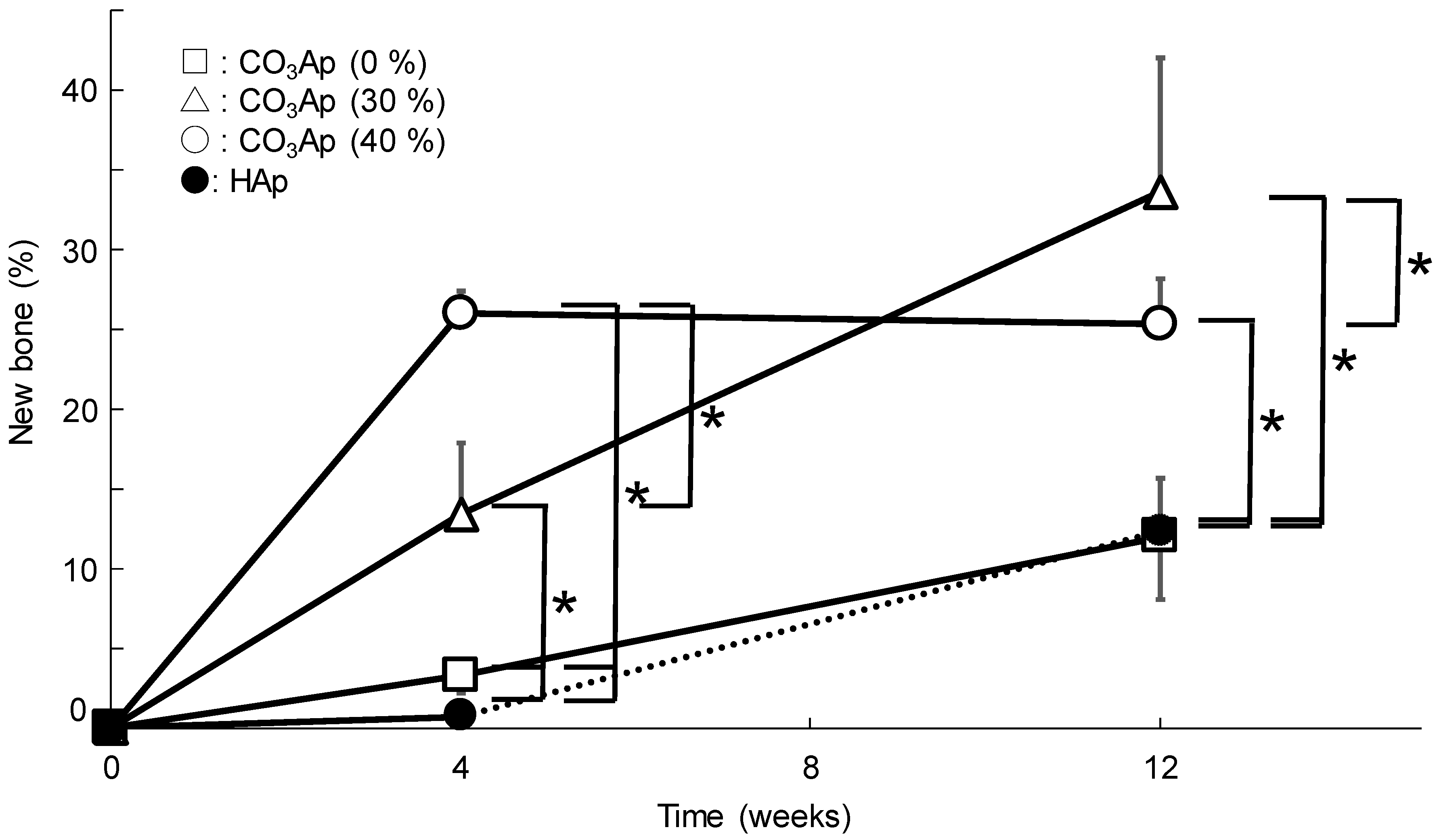
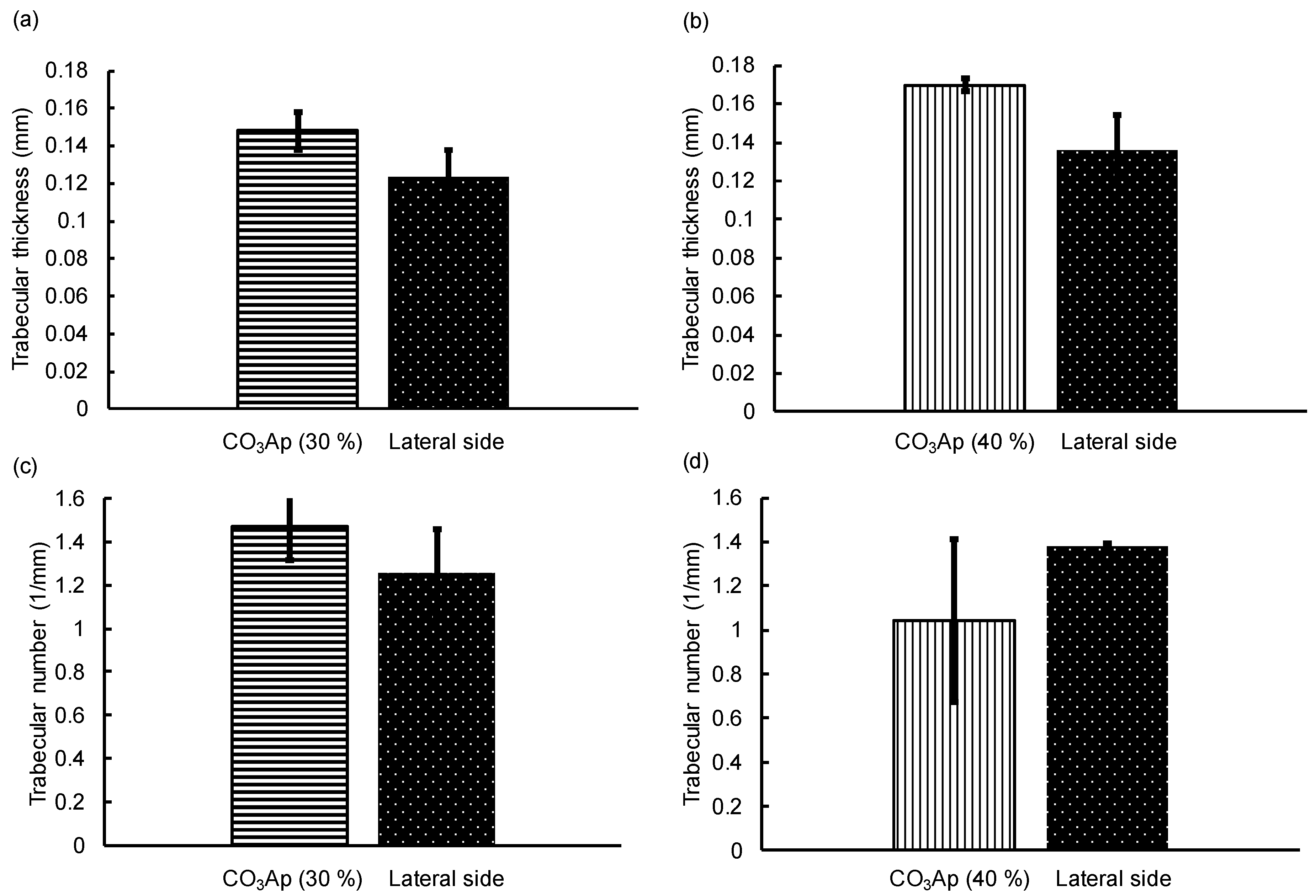
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moore, W.R.; Graves, S.E.; Bain, G.I. Synthetic bone graft substitutes. ANZ J. Surg. 2001, 71, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Brekke, J.H.; Toth, J.M. Principles of tissue engineering applied to programmable osteogenesis. J. Biomed. Mater. Res. 1998, 43, 380–398. [Google Scholar] [CrossRef]
- Nasr, H.F.; Aichelmann-Reidy, M.E.; Yukna, R.A. Bone and bone substitutes. Periodontol. 2000 1999, 19, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Kurz, L.T.; Garfin, S.R.; Booth, R.E.J. Harvesting autogenous iliac bone grafts: A review of complications and techniques. Spine 1989, 14, 1324. [Google Scholar] [CrossRef]
- Uchida, A.; Araki, N.; Shinto, Y.; Yoshikawa, H.; Kurisaki, E.; Ono, K. The use of calcium hydroxyapatite ceramic in bone tumour surgery. J. Bone Joint Surg. 1990, 72, 298–302. [Google Scholar] [CrossRef]
- Termine, J.D.; Eanes, E.D.; Greenfield, D.J.; Nylen, M.U.; Harper, R.A. Hydrazine-deproteinated bone mineral. Phys. Chem. Prop. Calcif. Tissue Res. 1973, 12, 73–90. [Google Scholar] [CrossRef]
- Doi, Y.; Shibutani, T.; Moriwaki, Y.; Kajimoto, T.; Iwayama, Y. Sintered carbonate apatites as bioresorbable bone substitutes. J. Biomed. Mater. Res. 1998, 39, 603–610. [Google Scholar] [CrossRef]
- Legeros, R.Z.; Trautz, O.R.; Legeros, J.P.; Klein, E.; Shirra, W.P. Apatite crystallites: Effects of carbonate on morphology. Science 1967, 155, 1409–1411. [Google Scholar] [CrossRef]
- Ishikawa, K. Bone substitute fabrication based on dissolution-precipitation reaction. Materials 2010, 3, 1138–1155. [Google Scholar] [CrossRef]
- Ishikawa, K.; Matsuya, S.; Lin, X.; Zhang, L.; Yuasa, T.; Miyamoto, Y. Fabrication of low crystalline B-type carbonate apatite block from low crystalline calcite block. J. Ceram. Soc. Jpn. 2010, 118, 341–344. [Google Scholar] [CrossRef]
- Lee, Y.; Hahm, Y.M.; Matsuya, S.; Nakagawa, M.; Ishikawa, K. Characterization of macroporous carbonate-substituted hydroxyapatite bodies prepared in different phosphate solutions. J. Mater. Sci. 2007, 42, 7843–7849. [Google Scholar] [CrossRef]
- Zaman, C.T.; Takeuchi, A.; Matsuya, S.; Zaman, Q.H.; Ishikawa, K. Fabrication of B-type carbonate apatite blocks by the phosphorylation of free-molding gypsum-calcite composite. Dent Mater. J. 2008, 27, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Daitou, F.; Maruta, M.; Kawachi, G.; Tsuru, K.; Matsuya, S.; Terada, Y.; Ishikawa, K. Fabrication of carbonate apatite block based on internal dissolution-precipitation reaction of dicalcium phosphate and calcium carbonate. Dent Mater. J. 2010, 29, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Maruta, M.; Matsuya, S.; Nakamura, S.; Ishikawa, K. Fabrication of low-crystalline carbonate apatite foam bone replacement based on phase transformation of calcite foam. Dent Mater. J. 2011, 30, 14–20. [Google Scholar] [CrossRef]
- Sunouchi, K.; Tsuru, K.; Maruta, M.; Kawachi, G.; Matsuya, S.; Terada, Y.; Ishikawa, K. Fabrication of solid and hollow carbonate apatite microspheres as bone substitutes using calcite microspheres as a precursor. Dent. Mater. J. 2012, 31, 549–557. [Google Scholar] [CrossRef]
- Ishikawa, K.; Munar, M.L.; Tsuru, K.; Miyamoto, Y. Fabrication of carbonate apatite honeycomb and its tissue response. J. Biomed. Mater. Res. Part. A 2019, 107A, 1014–1020. [Google Scholar] [CrossRef]
- Tsuru, K.; Kanazawa, M.; Yoshimoto, A.; Nakashima, Y.; Ishikawa, K. Fabrication of carbonate apatite block through a dissolution–precipitation reaction using calcium hydrogen phosphate dihydrate block as a precursor. Materials 2017, 10, 374. [Google Scholar] [CrossRef]
- Kanazawa, M.; Tsuru, K.; Fukuda, N.; Sakemi, Y.; Nakashima, Y.; Ishikawa, K. Evaluation of carbonate apatite blocks fabricated from dicalcium phosphate dihydrate blocks for reconstruction of rabbit femoral and tibial defects. J. Mater. Sci. Mater. Med. 2017, 28, 85–96. [Google Scholar] [CrossRef]
- Wakae, H.; Takeuchi, A.; Udoh, K.; Matsuya, S.; Munar, M.; LeGeros, R.Z.; Nakasima, A.; Ishikawa, K. Fabrication of macroporous carbonate apatite foam by hydrothermal conversion of α-tricalcium phosphate in carbonate solutions. J. Biomed. Mater. Res. A. 2008, 87, 957–963. [Google Scholar] [CrossRef]
- Takeuchi, A.; Munar, M.L.; Wakae, H.; Maruta, M.; Matsuya, S.; Tsuru, K.; Ishikawa, K. Effect of temperature on crystallinity of carbonate apatite foam prepared from alpha-tricalcium phosphate by hydrothermal treatment. Bio-Med. Mater. Eng. 2009, 19, 205–211. [Google Scholar]
- Karashima, S.; Takeuchi, A.; Matsuya, S.; Udoh, K.; Koyano, K.; Ishikawa, K. Fabrication of low-crystallinity hydroxyapatite foam based on the setting reaction of alpha-tricalcium phosphate foam. J. Biomed. Mater. Res. A 2009, 88, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, Y.; Tsuru, K.; Ishikawa, K. Fabrication of carbonate apatite foam based on the setting reaction of α-tricalcium phosphate foam granules. Ceram. Int. 2016, 42, 204–210. [Google Scholar] [CrossRef]
- Arifta, T.I.; Munar, M.L.; Tsuru, K.; Ishikawa, K. Fabrication of interconnected porous calcium-deficient hydroxyapatite using the setting reaction of α tricalcium phosphate spherical granules. Cerams Int. 2017, 43, 11149–11155. [Google Scholar] [CrossRef]
- Ishikawa, K.; Arifta, T.A.; Hayashi, K.; Tsuru, K. Fabrication and evaluation of interconnected porous carbonate apatite from alpha tricalcium phosphate spheres. J. Biomed. Mater. Res. Part. B Appl. Biomater. 2019, 107, 269–277. [Google Scholar] [CrossRef]
- Lowmunkong, R.; Sohmura, T.; Takahashi, J.; Suzuki, Y.; Matsuya, S.; Ishikawa, K. Transformation of 3DP gypsum model to HA by treating in ammonium phosphate solution. J. Biomed. Mater. Res. Part B App. Biomater. 2007, 80B, 386–393. [Google Scholar] [CrossRef]
- Lowmunkong, R.; Sohmura, T.; Suzuki, Y.; Matsuya, S.; Ishikawa, K. Fabrication of freeform bone-filling calcium phosphate ceramics by gypsum 3D printing method. J. Biomed. Mater. Res. B Appl. Biomater. 2009, 90, 531–539. [Google Scholar] [CrossRef]
- Nomura, S.; Tsuru, K.; Matsuya, S.; Takahashi, I.; Ishikawa, K. Fabrication of carbonate apatite block from set gypsum based on dissolution-precipitation reaction in phosphate-carbonate mixed solution. Dent. Mater. J. 2014, 33, 166–172. [Google Scholar] [CrossRef]
- Ayukawa, Y.; Suzuki, Y.; Koyano, K.; Ishikawa, K.; Tsuru, K. Histological comparison in rats between carbonate apatite fabricated from gypsum and sintered hydroxyapatite on bone remodeling. BioMed Res. Int. 2015, 2015, 579541. [Google Scholar] [CrossRef]
- Ishikawa, K. Carbonate apatite bone replacement. In Handbook of Bioceeramics and Biocomposite; Antoniac, I., Ed.; Springer: Berlin, Germany, 2018; pp. 213–232. [Google Scholar]
- Ishikawa, K. Carbonate apatite scaffold for regenerative medicine. In Handbook of Intelligent Scaffold for Tissue Engineering and Regenerative Medicine, 2nd ed.; Khang, G., Ed.; Pan Stanford Publishing: Temasek Avenue, Singapore, 2017; pp. 141–160. [Google Scholar]
- Ishikawa, K. Carbonate apatite bone replacement: Learn from the bone. J. Ceramic Soc. Jpn. in press. [CrossRef]
- Nagai, H.; Fujioka-Kobayashi, M.; Fujisawa, K.; Ohe, G.; Takamaru, N.; Hara, K.; Uchida, D.; Tamatani, T.; Ishikawa, K. Miyamoto Y: Effects of low crystalline carbonate apatite on proliferation and osteoblastic differentiation of human bone marrow cells. J. Mater. Sci. Mater. Med. 2015, 26, 99–107. [Google Scholar] [CrossRef]
- You, C.; Lee, M.H.; Lee, H.J.; Han, M.H.; Kwon, T.Y.; Kim, K.H.; Oh, D.S. The effect of macro/micro combination pore structure of biphasic calcium phosphate scaffold on bioactivity. Ceram. Int. 2016, 43, 3540–3546. [Google Scholar] [CrossRef]
- Huang, H.B.; Yang, Y.; Chen, L.H.; Wang, Y.; Huang, S.Z.; Tao, J.W.; Ma, X.T.; Hasan, T.; Li, Y.; Xu, Y. Hierarchical TiO/C nanocomposite monoliths with a robust scaffolding architecture, mesopore-macropore network and TiO-C heterostructure for high-performance lithium ion batteries. Nanoscale 2016, 8, 10928–10937. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Gerges, I.; Tocchio, A.; Tamplenizza, M.; Aprile, P.; Recordati, C.; Martello, F.; Martin, I.; Milani, P.; Lenardi, C. Biologically and mechanically driven design of an RGD-mimetic macroporous foam for adipose tissue engineering applications. Biomaterials 2016, 104, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Collins, B.; Goehl, T.; Dickson, I.R.; Glimcher, M.J. The carbonate environment in bone mineral: A resolution-enhanced Fourier transform infrared spectroscopy study. Calcif. Tissue Int. 1989, 45, 157–164. [Google Scholar] [CrossRef]
- Elliott, J.C.; Hobomb, D.W.; Young, R.A. Infrared determination of the degree of substitution of hydroxyl bicarbonate ions in human dental enamel. Calcif. Tissue Int. 1985, 37, 372–375. [Google Scholar] [CrossRef]
- Kopperdahl, D.L.; Keaveny, T.M. Yield strain behavior of trabecular bone. J. Biomech. 1998, 31, 601–608. [Google Scholar] [CrossRef]
- Kuboki, Y.; Jin, Q.; Takita, H. Geometry of carriers controlling phenotypic expression in bmp-induced osteogenesis and chondrogenesis. J. Bone Joint Surg. Am. 2001, 83, S05–S115. [Google Scholar] [CrossRef]
- LeGeros, R.Z. Properties of osteoconductive biomaterials: Calcium phosphates. Clin. Orthop. Relat. Res. 2002, 395, 81–98. [Google Scholar] [CrossRef]
- Tsuruga, E.; Takita, H.; Itoh, H.; Wakisaka, Y.; Kuboki, Y. Pore size of porous hydroxyapatite as the cell-substratum controls bmp-induced osteogenesis. J. Biochem. 1997, 121, 317–324. [Google Scholar] [CrossRef]
- Daculsi, G.; Passuti, N. Effect of the macroporosity for osseous substitution of calcium phosphate ceramics. Biomaterials 1990, 11, 86–87. [Google Scholar]
- Shimazaki, K.; Mooney, V. Comparative study of porous hydroxyapatite and tricalcium phosphate as bone substitute. J. Orthop. Res. 1985, 3, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Uchida, A.; Nade, S.; McCartney, E.; Ching, W. Bone ingrowth into three different porous ceramics implanted into the tibia of rats and rabbits. J. Orthop. Res. 1985, 3, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Li, K.; Zhu, Y.F.; Zhang, J.; Ye, X. 3D-printed hierarchical scaffold for localized isoniazid/rifampin drug delivery and osteoarticular tuberculosis therapy. Acta Biomater. 2015, 16, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.M. Biomaterials for bone tissue engineering. Mater. Today 2008, 11, 18–25. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, S.; Zhu, Y.; Huang, Y.; Zhu, M.; Tao, C.; Zhang, C. Three-dimensional printing of strontium-containing mesoporous bioactive glass scaffolds for bone regeneration. Acta Biomater. 2014, 10, 2269–2281. [Google Scholar] [CrossRef]
- Domingos, M.; Gloria, A.; Coelho, J.; Bartolo, P.; Ciurana, J. Three-dimensional printed bone scaffolds: The role of nano/micro-hydroxyapatite particles on the adhesion and differentiation of human mesenchymal stem cells. Proc. Inst. Mech. Eng. Part. H J. Eng. Med. 2017, 231, 555–564. [Google Scholar] [CrossRef]
- Lindner, M.; Bergmann, C.; Telle, R.; Fischer, H. Calcium phosphate scaffolds mimicking the gradient architecture of native long bones. J. Biomed. Mater. Res. Part. A 2014, 102A, 3677–3684. [Google Scholar] [CrossRef]
- Adel-Khattab, D.; Giacomini, F.; Gildenhaar, R.; Berger, G.; Gomes, C.; Linow, U.; Hardt, M.; Peleska, B.; Günster, J.; Stiller, M.; et al. Development of a synthetic tissue engineered threedimensional printed bioceramic-based bone graft with homogenously distributed osteoblasts and mineralizing bone matrix in vitro. J. Tissue Eng. Regen. Med. 2018, 12, 44–58. [Google Scholar] [CrossRef]
- Wang, H.; Wu, G.; Zhang, J.; Zhou, K.; Yin, B.; Su, X.; Qiu, G.; Yang, G.; Zhang, X.; Zhou, G.; et al. Osteogenic effect of controlled released rhBMP-2 in 3D printed poroushydroxyapatite scaffold. Colloids Surf. B Biointerfaces 2016, 141, 491–498. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LPS (Mass %) | Porosity (%) | DTS (MPa) | ||||||
|---|---|---|---|---|---|---|---|---|
| CaSO4 | CaCO3 | CO3Ap | HAp | CaSO4 | CaCO3 | CO3Ap | HAp | |
| 0 | 37.1 ± 2.4 | 49.0 ± 1.0 | 48.8 ± 4.5 | - | 3.4 ± 0.5 | 2.3 ± 0.4 | 3.5 ± 0.3 | - |
| 30 | 66.7 ± 1.8 | 71.4 ± 1.3 | 67.5 ± 0.7 | - | 0.8 ± 0.2 | 0.3 ± 0.1 | 1.4 ± 0.2 | - |
| 40 | 73.2 ± 0.8 | 79.1 ± 1.0 | 75.8 ± 0.9 | 64.2 ± 1.0 | 0.5 ± 0.1 | 0.2 ± 0.1 | 0.7 ± 0.1 | 1.8 ± 0.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakemi, Y.; Hayashi, K.; Tsuchiya, A.; Nakashima, Y.; Ishikawa, K. Fabrication and Histological Evaluation of Porous Carbonate Apatite Block from Gypsum Block Containing Spherical Phenol Resin as a Porogen. Materials 2019, 12, 3997. https://doi.org/10.3390/ma12233997
Sakemi Y, Hayashi K, Tsuchiya A, Nakashima Y, Ishikawa K. Fabrication and Histological Evaluation of Porous Carbonate Apatite Block from Gypsum Block Containing Spherical Phenol Resin as a Porogen. Materials. 2019; 12(23):3997. https://doi.org/10.3390/ma12233997
Chicago/Turabian StyleSakemi, Yuta, Koichiro Hayashi, Akira Tsuchiya, Yasuharu Nakashima, and Kunio Ishikawa. 2019. "Fabrication and Histological Evaluation of Porous Carbonate Apatite Block from Gypsum Block Containing Spherical Phenol Resin as a Porogen" Materials 12, no. 23: 3997. https://doi.org/10.3390/ma12233997
APA StyleSakemi, Y., Hayashi, K., Tsuchiya, A., Nakashima, Y., & Ishikawa, K. (2019). Fabrication and Histological Evaluation of Porous Carbonate Apatite Block from Gypsum Block Containing Spherical Phenol Resin as a Porogen. Materials, 12(23), 3997. https://doi.org/10.3390/ma12233997