Solid-State Nonlinear Optical Properties of Mononuclear Copper(II) Complexes with Chiral Tridentate and Tetradentate Schiff Base Ligands

 ,
,  ,
,  ,
, 
Abstract

1. Introduction
2. Materials and Methods
2.1. General Information
2.2. Synthesis of [Cu(La)(py)](ClO4) (1a)
2.3. Synthesis of [Cu(Lb)(py)](ClO4) (1b) and Isolation of [Cu(Lb)(H2O)](ClO4) (1b’) as Intermediate
2.4. Synthesis of [Cu(Lc)(py)](ClO4) (1c)
2.5. Synthesis of [Cu(5-NO2-5′-H-sal-(1R,2R)-dpen] (2a)
2.6. Synthesis of [Cu(5-NO2-5′-OMe-sal-(1R,2R)-dpen] (2b)
2.7. Synthesis of [Cu(5-NO2-5′-OMe-sal-(1S,2S)-chxn] (2c)
2.8. X-ray Data Collection and Structure Determination
2.9. Computational Details
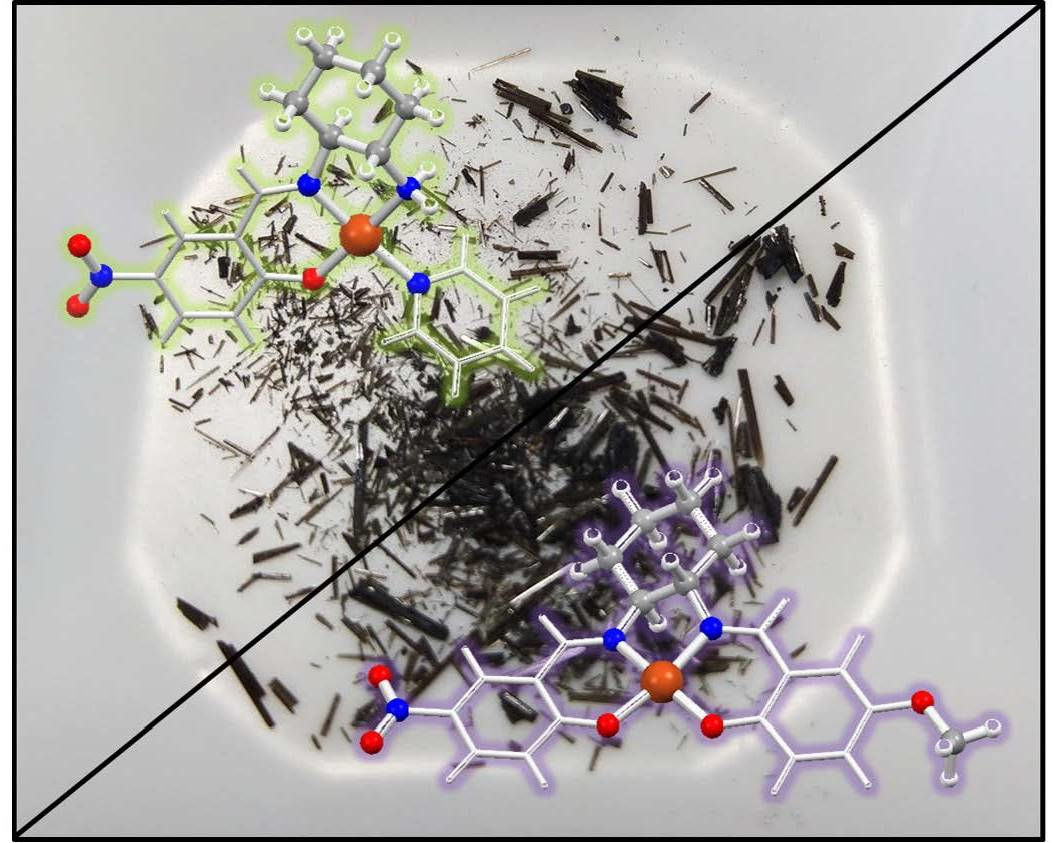
2.10. Kurtz–Perry Powder Measurements
3. Results and Discussion
3.1. Synthesis and IR/MS Characterization
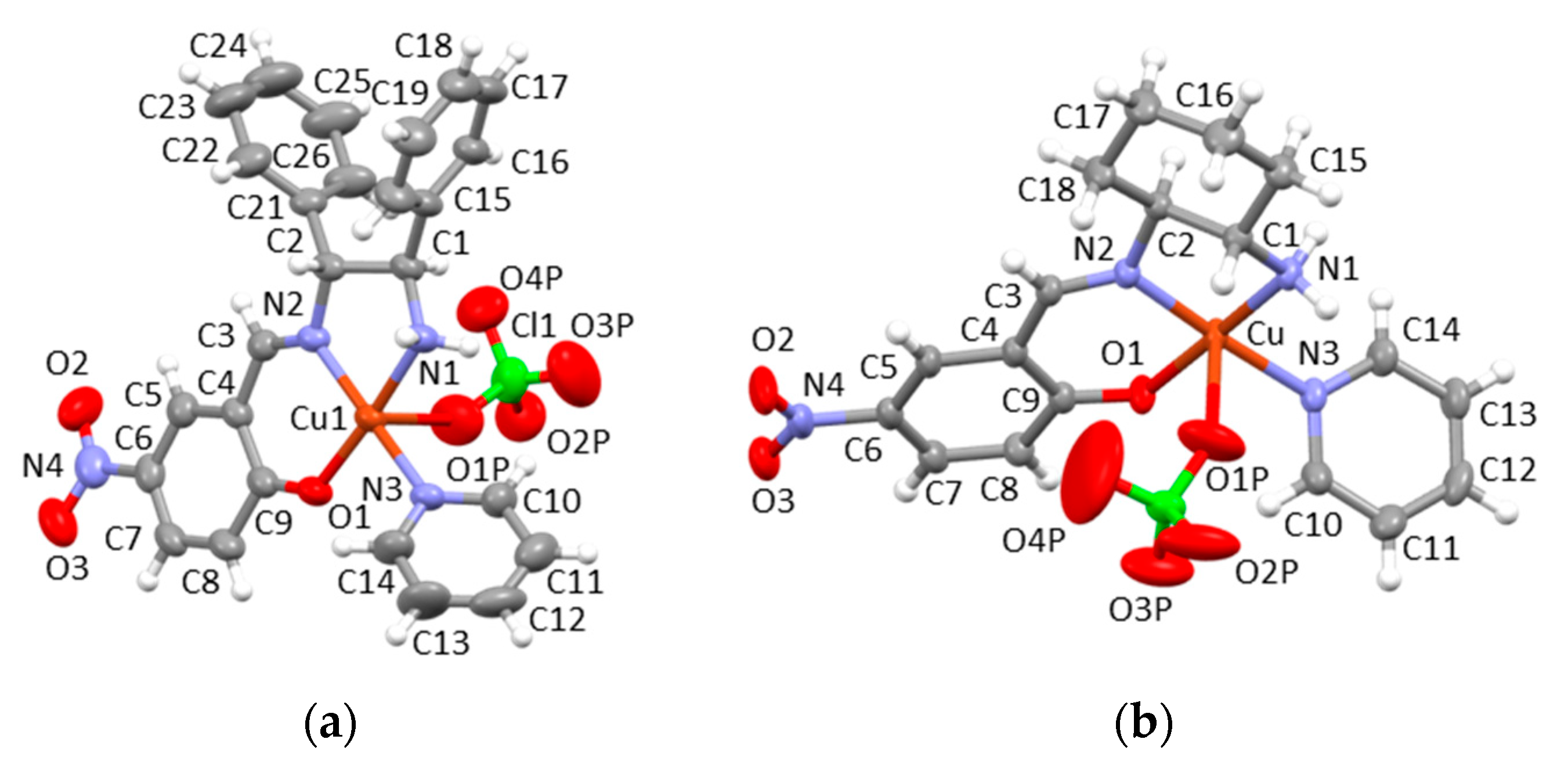
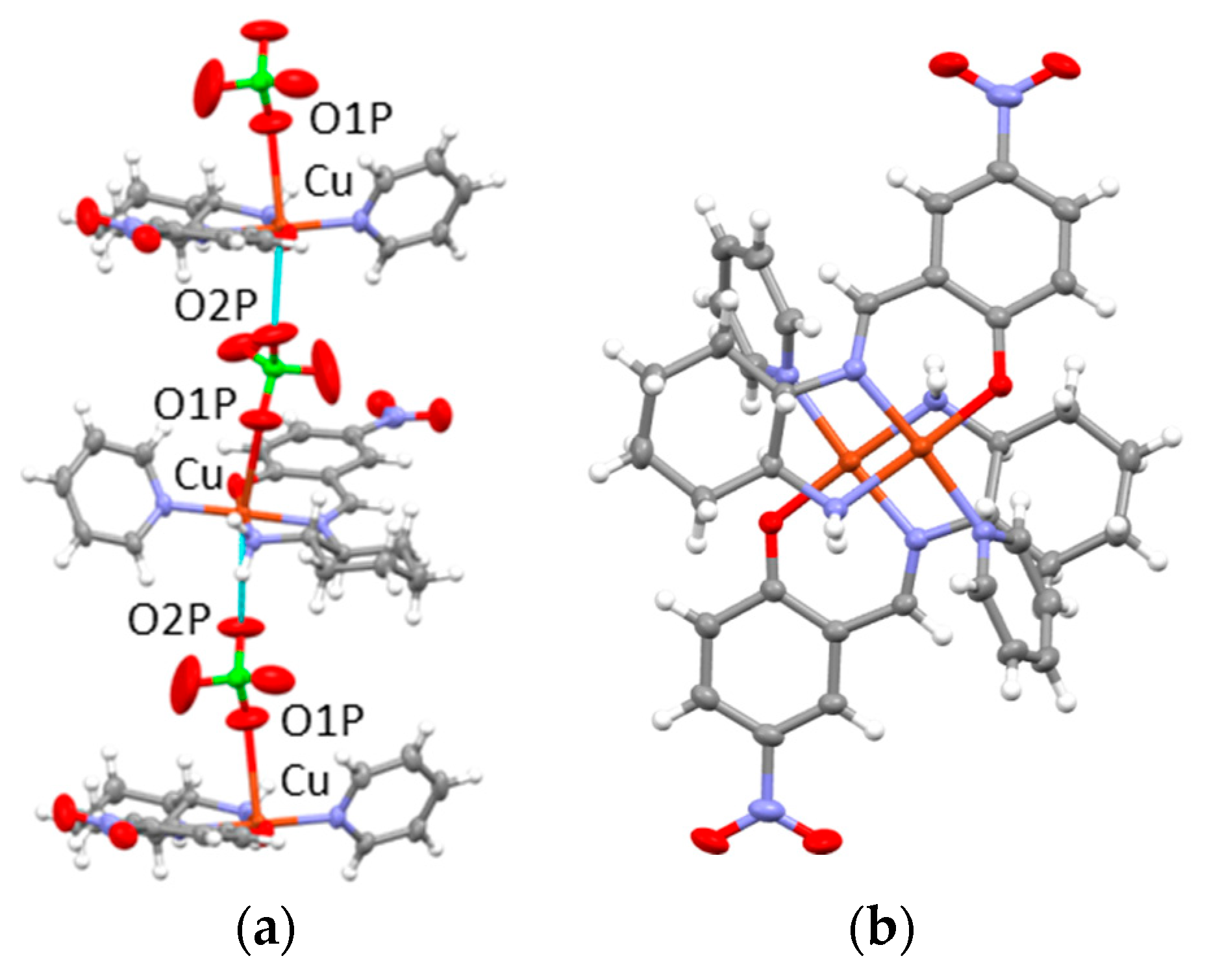
3.2. X-ray Structures of 1a∙0.5H2O, 1b, and 1c
3.3. Absorption Spectroscopy
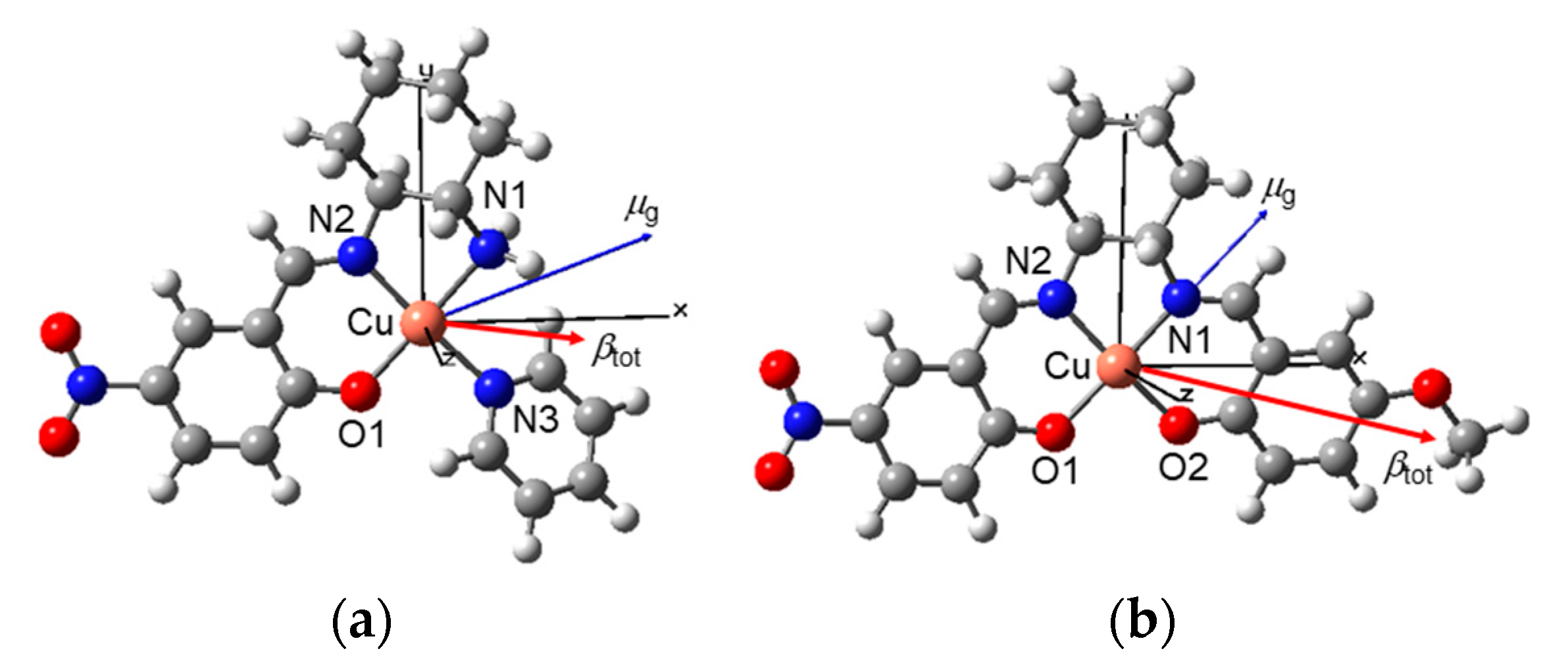
3.4. DFT Structural, Electronic, and NLO Properties
3.5. Solid-State NLO Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Collins, R.J.; Nelson, D.F.; Schawlow, A.L.; Bond, W.; Garrett, C.G.B.; Kaiser, W. Coherence, Narrowing, Directionality, and Relaxation Oscillations in the Light Emission from Ruby. Phys. Rev. Lett. 1960, 5, 303–305. [Google Scholar] [CrossRef]
- Franken, P.A.; Hill, A.E.; Peters, C.W.; Weinreich, G. Generation of Optical Harmonics. Phys. Rev. Lett. 1961, 7, 118–119. [Google Scholar] [CrossRef]
- Boyd, R.W. Nonlinear Optics, 3rd ed.; Academic Press: Amsterdam, The Netherlands; Boston, MA, USA, 2008; ISBN 978-0-12-369470-6. [Google Scholar]
- Prasad, P.N.; Williams, D.J. Introduction to Nonlinear Optical Effects in Molecules and Polymers; Wiley: New York, NY, USA, 1991; ISBN 978-0-471-51562-3. [Google Scholar]
- Materials for Nonlinear Optics: Chemical Perspectives; Marder, S.R., Sohn, J.E., Stucky, G.D., Eds.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1991; ISBN 978-0-8412-1939-7. [Google Scholar]
- Papadopoulos, M.G.; Sadlej, A.J.; Leszczynski, J. (Eds.) Non-Linear Optical Properties of Matter: From Molecules to Condensed Phases; Challenges and Advances in Computational Chemistry and Physics; Springer: Dordrecht, The Netherlands, 2006; ISBN 978-1-4020-4849-4. [Google Scholar]
- Structure-Property Relationships in Non-Linear Optical Crystals I.; Wu, X.-T., Chen, L., Eds.; Structure and Bonding; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2012; Volume 144, ISBN 978-3-642-29617-8. [Google Scholar]
- Wu, X.-T.; Chen, L. (Eds.) Structure-Property Relationships in Non-Linear Optical Crystals II.; Structure and Bonding; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2012; Volume 145, ISBN 978-3-642-29620-8. [Google Scholar]
- Nonlinear Optics; Kamanina, N., Ed.; InTech Europe: Rijeka, Croatia, 2012; ISBN 978-953-51-0131-4. [Google Scholar]
- Zyss, J.; Oudar, J.L. Relations between microscopic and macroscopic lowest-order optical nonlinearities of molecular crystals with one- or two-dimensional units. Phys. Rev. A 1982, 26, 2028–2048. [Google Scholar] [CrossRef]
- Humphrey, M.G. Ruthenium Alkynyl Complexes in Non-Linear Optics. Aus. J. Chem. 2018, 71, 731–742. [Google Scholar] [CrossRef]
- Kaur, S.; Kaur, M.; Kaur, P.; Clays, K.; Singh, K. Ferrocene chromophores continue to inspire. Fine-tuning and switching of the second-order nonlinear optical response. Coord. Chem. Rev. 2017, 343, 185–219. [Google Scholar] [CrossRef]
- Kodikara, M.S.; Stranger, R.; Humphrey, M.G. Computational studies of the nonlinear optical properties of organometallic complexes. Coord. Chem. Rev. 2018, 375, 389–409. [Google Scholar] [CrossRef]
- Humphrey, M.G.; Schwich, T.; West, P.J.; Cifuentes, M.P.; Samoc, M. Nonlinear Optical Properties of Coordination and Organometallic Complexes. In Comprehensive Inorganic Chemistry II.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 781–835. ISBN 978-0-08-096529-1. [Google Scholar]
- Di Bella, S.; Dragonetti, C.; Pizzotti, M.; Roberto, D.; Tessore, F.; Ugo, R. Coordination and Organometallic Complexes as Second-Order Nonlinear Optical Molecular Materials. In Molecular Organometallic Materials for Optics; Bozec, H., Guerchais, V., Eds.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2010; Volume 28, pp. 1–55. ISBN 978-3-642-01865-7. [Google Scholar]
- Mingabudinova, L.R.; Vinogradov, V.V.; Milichko, V.A.; Hey-Hawkins, E.; Vinogradov, A.V. Metal–organic frameworks as competitive materials for non-linear optics. Chem. Soc. Rev. 2016, 45, 5408–5431. [Google Scholar] [CrossRef]
- Lacroix, P.G.; Malfant, I.; Lepetit, C. Second-order nonlinear optics in coordination chemistry: An open door towards multi-functional materials and molecular switches. Coord. Chem. Rev. 2016, 308, 381–394. [Google Scholar] [CrossRef]
- Di Bella, S. Second-order nonlinear optical properties of transition metal complexes. Chem. Soc. Rev. 2001, 30, 355–366. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Breith, E.; Lübbe, E.; Tsumaki, T. Tricyclische orthokondensierte Nebenvalenzringe. Justus Liebig’s Ann. Der Chem. 1933, 503, 84–130. [Google Scholar] [CrossRef]
- Rigamonti, L. Schiff base metal complexes for second order nonlinear optics. La Chimica e L’Industria 2010, 118, 122. [Google Scholar]
- Liu, X.; Manzur, C.; Novoa, N.; Celedón, S.; Carrillo, D.; Hamon, J.-R. Multidentate unsymmetrically-substituted Schiff bases and their metal complexes: Synthesis, functional materials properties, and applications to catalysis. Coord. Chem. Rev. 2018, 357, 144–172. [Google Scholar] [CrossRef]
- Nayar, C.R.; Ravikumar, R. Review: Second order nonlinearities of Schiff bases derived from salicylaldehyde and their metal complexes. J. Coord. Chem. 2014, 67, 1–16. [Google Scholar] [CrossRef]
- Lacroix, P.G. Second-Order Optical Nonlinearities in Coordination Chemistry: The Case of Bis(salicylaldiminato)metal Schiff Base Complexes. Eur. J. Inorg. Chem. 2001, 2001, 339–348. [Google Scholar] [CrossRef]
- Di Bella, S.; Fragala, I.; Ledoux, I.; Diaz-Garcia, M.A.; Lacroix, P.G.; Marks, T.J. Sizable Second-Order Nonlinear Optical Response of Donor-Acceptor Bis(salicylaldiminato)nickel(II) Schiff Base Complexes. Chem. Mater. 1994, 6, 881–883. [Google Scholar] [CrossRef]
- Di Bella, S.; Fragala, I.; Ledoux, I.; Marks, T.J. Role of Metal Electronic Properties in Tuning the Second-Order Nonlinear Optical Response of Coordination Complexes. A Combined Experimental and Theoretical Investigation of a Homologous Series of (N,N′-Disalicylidene-1,2-phenylenediaminato)M(II) (M = Co, Ni, Cu) Complexes. J. Am. Chem. Soc. 1995, 117, 9481–9485. [Google Scholar]
- Lacroix, P.G.; Di Bella, S.; Ledoux, I. Synthesis and Second-Order Nonlinear Optical Properties of New Copper(II), Nickel(II), and Zinc(II) Schiff-Base Complexes. Toward a Role of Inorganic Chromophores for Second Harmonic Generation. Chem. Mater. 1996, 8, 541–545. [Google Scholar] [CrossRef]
- Di Bella, S.; Fragalà, I.; Ledoux, I.; Diaz-Garcia, M.A.; Marks, T.J. Synthesis, Characterization, Optical Spectroscopic, Electronic Structure, and Second-Order Nonlinear Optical (NLO) Properties of a Novel Class of Donor−Acceptor Bis(salicylaldiminato)nickel(II) Schiff Base NLO Chromophores. J. Am. Chem. Soc. 1997, 119, 9550–9557. [Google Scholar] [CrossRef]
- Averseng, F.; Lacroix, P.G.; Malfant, I.; Lenoble, G.; Cassoux, P.; Nakatani, K.; Maltey-Fanton, I.; Delaire, J.A.; Aukauloo, A. Synthesis, Crystal Structure, and Second-Order Nonlinear Optical Properties of a New Bis(salicylaldiminato)nickel(II) Metal Complex. Chem. Mater. 1999, 11, 995–1002. [Google Scholar] [CrossRef]
- Di Bella, S.; Fragalà, I. Synthesis and second-order nonlinear optical properties of bis(salicylaldiminato)M(II) metalloorganic materials. Synth. Metals 2000, 115, 191–196. [Google Scholar] [CrossRef]
- Averseng, F.; Lacroix, P.G.; Malfant, I.; Périssé, N.; Lepetit, C.; Nakatani, K. Enhanced Second Harmonic Generation on Passing from a Mono- to a Dicopper(II) Bis(salicylaldiminato) Schiff Base Complex. Inorg. Chem. 2001, 40, 3797–3804. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, S.; Fragalà, I.; Ledoux, I.; Zyss, J. Dipolar Donor-Acceptor-Substituted Schiff Base Complexes with Large Off-Diagonal Second-Order Nonlinear Optical Tensor Components. Chem. Eur. J. 2001, 7, 3738–3743. [Google Scholar] [CrossRef]
- Costes, J.P.; Lamère, J.F.; Lepetit, C.; Lacroix, P.G.; Dahan, F.; Nakatani, K. Synthesis, Crystal Structures, and Nonlinear Optical (NLO) Properties of New Schiff-Base Nickel(II) Complexes. Toward a New Type of Molecular Switch? Inorg. Chem. 2005, 44, 1973–1982. [Google Scholar] [CrossRef]
- Di Bella, S.; Oliveri, I.P.; Colombo, A.; Dragonetti, C.; Righetto, S.; Roberto, D. An unprecedented switching of the second-order nonlinear optical response in aggregate bis(salicylaldiminato)zinc(II) Schiff-base complexes. Dalton Trans. 2012, 41, 7013–7016. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, S.; Fragalà, I.; Marks, T.J.; Ratner, M.A. Large Second-Order Optical Nonlinearities in Open-Shell Chromophores. Planar Metal Complexes and Organic Radical Ion Aggregates. J. Am. Chem. Soc. 1996, 118, 12747–12751. [Google Scholar] [CrossRef]
- Averseng, F.; Lepetit, C.; Lacroix, P.G.; Tuchagues, J.P. Theoretical Investigation of the Effect of a Spin Transition on the Second-Order Molecular Hyperpolarizability of a Bis(salicylaldiminato)Fe II Schiff Base Complex. Chem. Mater. 2000, 12, 2225–2229. [Google Scholar] [CrossRef]
- Di Bella, S.; Fragalà, I. Two-dimensional characteristics of the second-order nonlinear optical response in dipolar donor–acceptor coordination complexes. New. J. Chem. 2002, 26, 285–290. [Google Scholar]
- Di Bella, S.; Fragalà, I. Second-Order Nonlinear Optical Properties of Tetraaza-Coordinated Nickel(II) Complexes. Eur. J. Inorg. Chem. 2003, 2003, 2606–2611. [Google Scholar] [CrossRef]
- Rigamonti, L.; Demartin, F.; Forni, A.; Righetto, S.; Pasini, A. Copper(II) Complexes of salen Analogues with Two Differently Substituted (Push−Pull) Salicylaldehyde Moieties. A Study on the Modulation of Electronic Asymmetry and Nonlinear Optical Properties. Inorg. Chem. 2006, 45, 10976–10989. [Google Scholar] [CrossRef]
- Rigamonti, L.; Forni, A.; Righetto, S.; Pasini, A. Push-pull unsymmetrical substitution in nickel(II) complexes with tetradentate N2O2 Schiff base ligands: Synthesis, structures and linear-nonlinear optical studies. Dalton Trans. 2019, 48, 11217–11234. [Google Scholar] [CrossRef]
- Gradinaru, J.; Forni, A.; Druta, V.; Tessore, F.; Zecchin, S.; Quici, S.; Garbalau, N. Structural, Spectral, Electric-Field-Induced Second Harmonic, and Theoretical Study of Ni(II), Cu(II), Zn(II), and VO(II) Complexes with [N2O2] Unsymmetrical Schiff Bases of S-Methylisothiosemicarbazide Derivatives. Inorg. Chem. 2007, 46, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, A.; Fuentealba, M.; Carrillo, D.; Manzur, C.; Ledoux-Rak, I.; Hamon, J.-R.; Saillard, J.-Y. Synthesis, Spectral, Structural, Second-Order Nonlinear Optical Properties and Theoretical Studies on New Organometallic Donor−Acceptor Substituted Nickel(II) and Copper(II) Unsymmetrical Schiff-Base Complexes. Inorg. Chem. 2010, 49, 2750–2764. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhong, C.; Zhu, X.; Tam, H.-L.; Li, K.-F.; Cheah, K.-W.; Wong, W.-Y.; Wong, W.-K.; Jones, R.A. Synthesis and two-photon absorption properties of unsymmetrical metallosalophen complexes. Polyhedron 2013, 49, 121–128. [Google Scholar] [CrossRef]
- Celedón, S.; Dorcet, V.; Roisnel, T.; Singh, A.; Ledoux-Rak, I.; Hamon, J.-R.; Carrillo, D.; Manzur, C. Main-Chain Oligomers from NiII- and CuII-Centered Unsymmetrical N2O2 Schiff-Base Complexes: Synthesis and Spectral, Structural, and Second-Order Nonlinear Optical Properties: Oligomers from Ni II and Cu II Schiff-Base Complexes. Eur. J. Inorg. Chem. 2014, 2014, 4984–4993. [Google Scholar] [CrossRef]
- Novoa, N.; Roisnel, T.; Hamon, P.; Kahlal, S.; Manzur, C.; Ngo, H.M.; Ledoux-Rak, I.; Saillard, J.-Y.; Carrillo, D.; Hamon, J.-R. Four-coordinate nickel(ii) and copper(ii) complex based ONO tridentate Schiff base ligands: Synthesis, molecular structure, electrochemical, linear and nonlinear properties, and computational study. Dalton Trans. 2015, 44, 18019–18037. [Google Scholar] [CrossRef]
- Chiang, W.; Thompson, M.E.; Vanengen, D. Synthesis and nonlinear optical properties of inorganic coordination polymers. Spec. Publ. Royal Soc. Chem. 1991, 91, 210–2016. [Google Scholar]
- Chiang, W.; Vanengen, D.; Thompson, M.E. Second-order non-linear optical properties of Fe(SALEN) complexes. Polyhedron 1996, 15, 2369–2376. [Google Scholar] [CrossRef]
- Kurtz, S.K.; Perry, T.T. A Powder Technique for the Evaluation of Nonlinear Optical Materials. J. Appl. Phys. 1968, 39, 3798–3813. [Google Scholar] [CrossRef]
- Lenoble, G.; Lacroix, P.G.; Daran, J.C.; Di Bella, S.; Nakatani, K. Syntheses, Crystal Structures, and NLO Properties of New Chiral Inorganic Chromophores for Second-Harmonic Generation. Inorg. Chem. 1998, 37, 2158–2165. [Google Scholar] [CrossRef]
- Averseng, F.; Lacroix, P.G.; Malfant, I.; Dahan, F.; Nakatani, K. Synthesis, crystal structure and solid state NLO properties of a new chiral bis(salicylaldiminato)nickel(II) Schiff-base complex in a nearly optimized solid state environment. J. Mater. Chem. 2000, 10, 1013–1018. [Google Scholar] [CrossRef]
- Zyss, J.; Chemla, D.S.; Nicoud, J.F. Demonstration of efficient nonlinear optical crystals with vanishing molecular dipole moment: Second-harmonic generation in 3-methyl-4-nitropyridine-1-oxide. J. Chem. Phys. 1981, 74, 4800–4811. [Google Scholar] [CrossRef]
- Downing, R.S.; Urbach, F.L. Circular dichroism of square-planar, tetradentate Schiff base chelates of copper(II). J. Am. Chem. Soc. 1969, 91, 5977–5983. [Google Scholar] [CrossRef]
- Pasini, A.; Gullotti, M.; Ugo, R. Optically active complexes of Schiff bases. Part 4. An analysis of the circular-dichroism spectra of some complexes of different coordination numbers with quadridentate Schiff bases of optically active diamines. J. Chem. Soc. Dalton Trans. 1977, 4, 346–356. [Google Scholar] [CrossRef]
- Bernardo, K.; Leppard, S.; Robert, A.; Commenges, G.; Dahan, F.; Meunier, B. Synthesis and Characterization of New Chiral Schiff Base Complexes with Diiminobinaphthyl or Diiminocyclohexyl Moieties as Potential Enantioselective Epoxidation Catalysts. Inorg. Chem. 1996, 35, 387–396. [Google Scholar] [CrossRef]
- Zolezzi, S.; Decinti, A.; Spodine, E. Syntheses and characterization of copper(II) complexes with Schiff-base ligands derived from ethylenediamine, diphenylethylenediamine and nitro, bromo and methoxy salicylaldehyde. Polyhedron 1999, 18, 897–904. [Google Scholar] [CrossRef]
- Hirotsu, M.; Kuwamura, N.; Kinoshita, I.; Kojima, M.; Yoshikawa, Y.; Ueno, K. Steric, geometrical and solvent effects on redox potentials in salen-type copper(II) complexes. Dalton Trans. 2009, 37, 7678. [Google Scholar] [CrossRef]
- Kleij, A.W. Nonsymmetrical Salen Ligands and Their Complexes: Synthesis and Applications. Eur. J. Inorg. Chem. 2009, 2009, 193–205. [Google Scholar] [CrossRef]
- Wang, Y.; Stack, T.D.P. Galactose Oxidase Model Complexes: Catalytic Reactivities. J. Am. Chem. Soc. 1996, 118, 13097–13098. [Google Scholar] [CrossRef]
- Belokon, Y.N.; North, M.; Kublitski, V.S.; Ikonnikov, N.S.; Krasik, P.E.; Maleev, V.I. Chiral salen-metal complexes as novel catalysts for asymmetric phase transfer alkylations. Tetrahedron Lett. 1999, 40, 6105–6108. [Google Scholar] [CrossRef]
- Gao, J.; Reibenspies, J.H.; Martell, A.E. Structurally Defined Catalysts for Enantioselective Oxidative Coupling Reactions. Angew. Chem. Int. Ed. 2003, 42, 6008–6012. [Google Scholar] [CrossRef]
- Bania, K.K.; Karunakar, G.V.; Goutham, K.; Deka, R.C. Enantioselective Henry Reaction Catalyzed by “Ship in a Bottle” Complexes. Inorg. Chem. 2013, 52, 8017–8029. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, C.; Theetharappan, M.; Kowsalya, P.; Natarajan, S.; Neelakantan, M.A.; Mariappan, S.S. Biocatalysis, DNA-protein interactions, cytotoxicity and molecular docking of Cu(II), Ni(II), Zn(II) and V(IV) Schiff base complexes. Appl. Organomet. Chem. 2017, 31, e3776. [Google Scholar] [CrossRef]
- Behzad, M.; Seifikar Ghomi, L.; Damercheli, M.; Mehravi, B.; Shafiee Ardestani, M.; Samari Jahromi, H.; Abbasi, Z. Crystal structures and in vitro anticancer studies on new unsymmetrical copper(II) Schiff base complexes derived from meso-1,2-diphenyl-1,2-ethylenediamine: A comparison with related symmetrical ones. J. Coord. Chem. 2016, 69, 2469–2481. [Google Scholar] [CrossRef]
- Bian, H.-D.; Wang, J.; Wei, Y.; Tang, J.; Huang, F.-P.; Yao, D.; Yu, Q.; Liang, H. Superoxide dismutase activity studies of Mn(III)/Cu(II)/Ni(II) complexes with Schiff base ligands. Polyhedron 2015, 90, 147–153. [Google Scholar] [CrossRef]
- Korupoju, S.R.; Mangayarkarasi, N.; Ameerunisha, S.; Valente, E.J.; Zacharias, P.S. Formation of dinuclear macrocyclic and mononuclear acyclic complexes of a new trinucleating hexaaza triphenolic Schiff base macrocycle: Structure and NLO properties. J. Chem. Soc. Dalton Trans. 2000, 16, 2845–2852. [Google Scholar] [CrossRef]
- Margeat, O.; Lacroix, P.G.; Costes, J.P.; Donnadieu, B.; Lepetit, C.; Nakatani, K. Synthesis, Structures, and Physical Properties of Copper(II)−Gadolinium(III) Complexes Combining Ferromagnetic Coupling and Quadratic Nonlinear Optical Properties. Inorg. Chem. 2004, 43, 4743–4750. [Google Scholar] [CrossRef]
- Leslie, A.G.W. Jnt CCP4/ESF-EACMB. Newslett. Protein Crystallogr. 1992, 27, 30. [Google Scholar]
- Evans, P. Jnt CCP4/ESF-EACMB. Newslett. Protein Crystallogr. 1997, 33, 22. [Google Scholar]
- Bruker, SMART, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 1997.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Johnson, L.E.; Dalton, L.R.; Robinson, B.H. Optimizing Calculations of Electronic Excitations and Relative Hyperpolarizabilities of Electrooptic Chromophores. Acc. Chem. Res. 2014, 47, 3258–3265. [Google Scholar] [CrossRef] [PubMed]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef] [PubMed]
- Renehan, M.F.; Schanz, H.-J.; McGarrigle, E.M.; Dalton, C.T.; Daly, A.M.; Gilheany, D.G. Unsymmetrical chiral salen Schiff base ligands. J. Mol. Cat. A Chem. 2005, 231, 205–220. [Google Scholar] [CrossRef]
- Costes, J.-P.; Dahan, F.; Fernandez Fernandez, M.B.; Fernandez Garcia, M.I.; Garcia Deibe, A.M.; Sanmartin, J. General synthesis of ‘salicylaldehyde half-unit complexes’: Structural determination and use as synthon for the synthesis of dimetallic or trimetallic complexes and of ‘self-assembling ligand complexes’. Inorg. Chim. Acta 1998, 274, 73–81. [Google Scholar] [CrossRef]
- Rigamonti, L.; Cinti, A.; Forni, A.; Pasini, A.; Piovesana, O. Copper(II) Complexes of Tridentate Schiff Bases of 5-Substituted Salicylaldehydes and Diamines-The Role of the Substituent and the Diamine in the Formation of Mono-, Di- and Trinuclear Species-Crystal Structures and Magnetic Properties. Eur. J. Inorg. Chem. 2008, 2008, 3633–3647. [Google Scholar] [CrossRef]
- Rigamonti, L.; Forni, A.; Pievo, R.; Reedijk, J.; Pasini, A. Copper(II) compounds with NNO tridentate Schiff base ligands: Effect of subtle variations in ligands on complex formation, structures and magnetic properties. Inorg. Chim. Acta 2012, 387, 373–382. [Google Scholar] [CrossRef]
- Fernandez Garcia, M.I.; Fondo, M.; Garcia Deibe, A.M.; Fernandez Fernandez, M.B.; gonzalez, A.M. Copper(II) Complexes with Asymmetrical Schiff Base Ligands Derived from 2-Acetylpyrazine. Z. Anorg. Allg. Chem. 2000, 626, 1985–1991. [Google Scholar] [CrossRef]
- Waters, T.N.; Wright, P.E. Electronic absorption band assignments for copper(II) salicylaldimine complexes. J. Inorg. Nucl. Chem. 1971, 33, 359–363. [Google Scholar] [CrossRef]
- Rigamonti, L.; Forni, A.; Pievo, R.; Reedijk, J.; Pasini, A. Synthesis, crystal structures and magnetic properties of dinuclear copper(II) compounds with NNO tridentate Schiff base ligands and bridging aliphatic diamine and aromatic diimine linkers. Dalton Trans. 2011, 40, 3381–3393. [Google Scholar] [CrossRef] [PubMed]
- Kanis, D.R.; Ratner, M.A.; Marks, T.J. Design and construction of molecular assemblies with large second-order optical nonlinearities. Quantum chemical aspects. Chem. Rev. 1994, 94, 195–242. [Google Scholar] [CrossRef]
- Willetts, A.; Rice, J.E.; Burland, D.M.; Shelton, D.P. Problems in the comparison of theoretical and experimental hyperpolarizabilities. J. Chem. Phys. 1992, 97, 7590–7599. [Google Scholar] [CrossRef]
- Zhu, J.; Song, H.; Sun, J.; Yan, P.; Hou, G.; Li, G. Luminescence and nonlinear optics of 1D N,N′-bis(salicylidene)-1,2-cyclohexanediamine lanthanide coordination polymers. Synth. Met. 2014, 192, 29–36. [Google Scholar] [CrossRef]
- Buda, A.B.; der Heyde, T.A.; Mislow, K. On Quantifying Chirality. Angew. Chem. Int. Ed. Engl. 1992, 31, 989–1007. [Google Scholar] [CrossRef]
- Zayit, A.; Pinsky, M.; Elgavi, H.; Dryzun, C.; Avnir, D. A web site for calculating the degree of chirality. Chirality 2011, 23, 17–23. [Google Scholar] [CrossRef]
- Cariati, E.; Roberto, D.; Ugo, R.; Ford, P.C.; Galli, S.; Sironi, A. New Structural Motifs, Unusual Quenching of the Emission, and Second Harmonic Generation of Copper(I) Iodide Polymeric or Oligomeric Adducts with Para-Substituted Pyridines or trans-Stilbazoles. Inorg. Chem. 2005, 44, 4077–4085. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1a∙0.5H2O | 1b | 1c | |
|---|---|---|---|
| Crystal Data | |||
| Moiety formula | [C26H23CuN4O3](ClO4)∙0.5H2O | [C26H23CuN4O3](ClO4) | [C18H21CuN4O3](ClO4) |
| Sum formula | C26H24ClCuN4O7.5 | C26H23ClCuN4O7 | C18H21ClCuN4O7 |
| M | 611.47 | 602.47 | 504.38 |
| Crystal system | monoclinic | orthorhombic | tetragonal |
| Space group | P21/n (n. 14) | P212121 (n. 19) | P43 (n. 78) |
| a/Å | 13.232(3) | 7.1810(7) | 11.6613(6) |
| b/Å | 13.329(3) | 21.970(2) | 11.6613(6) |
| c/Å | 15.383(3) | 35.121(3) | 15.3217(8) |
| α/° | 90 | 90 | 90 |
| β/° | 92.40(3) | 90 | 90 |
| γ/° | 90 | 90 | 90 |
| V/Å3, Z | 293(2), 4 | 5541.0(9), 8 | 2083.5(2), 4 |
| Reflns for cell det | 130 | 6497 | 9386 |
| 2θ/° for cell det | 16.0–52.0 | 4.64–40.24 | 4.39–60.26 |
| Dx/Mg m−3 | 1.515 | 1.444 | 1.608 |
| μ/mm−1 | 1.318 | 0.935 | 1.226 |
| Colour, habit | green, needle | violet, needle | red, block |
| Dimensions/mm | 0.20 × 0.01 × 0.01 | 0.58 × 0.14 × 0.08 | 0.36 × 0.34 × 0.30 |
| Data Collection | |||
| Temperature/K | 293(2) | 293(2) | 294(2) |
| radiation λ/Å | synchrotron, 0.800 | Mo–Kα, 0.71073 | Mo–Kα, 0.71073 |
| Scan type | φ | φ and ω | φ and ω |
| 2θ max/° | 57.4 | 49.7 | 64.5 |
| h range | −16 → 16 | −8 → 8 | −17 → 17 |
| k range | −17 → 17 | −25 → 25 | −17 → 17 |
| l range | −17 → 17 | −41 → 41 | −22 → 22 |
| Intensity decay | None | None | None |
| Measured reflns | 31,811 | 66,969 | 44,359 |
| Independent reflns | 4628 | 9600 | 7153 |
| Reflns with I > 2σ(I) | 4001 | 7633 | 6226 |
| Rint | 0.0390 | 0.0415 | 0.0214 |
| Refinement on F2 | |||
| R1, wR2 [F2 > 2σ(F2)] | 0.0429,0.1165 | 0.0430, 0.1073 | 0.0321, 0.0901 |
| R1, wR2 [all data] | 0.0499, 0.1231 | 0.0604, 0.1176 | 0.0394, 0.0949 |
| S | 1.101 | 1.016 | 1.029 |
| Flack parameter | 0.007(5) | 0.014(3) | |
| Params, restraints | 455, 159 | 703, 0 | 280, 1 |
| (Δ/σ)max | 0.001 | 0.001 | 0.001 |
| Δρmax, Δρmin/e Å−3 | 0.293, −0.343 | 0.375, −0.288 | 0.396, −0.345 |
| 1a∙0.5H2O | 1b | 1c | |
|---|---|---|---|
| Cu1–N1/Cu2–N5 1 | 2.011(5) | 1.998(4)/2.002(5) 1 | 2.007(2) |
| 2.066 | 2.054 | 2.060 | |
| Cu1–N2/Cu2–N6 1 | 1.949(4) | 1.958(4)/1.951(4) 1 | 1.951(2) |
| 1.940 | 1.948 | 1.952 | |
| Cu1–O1/Cu2–O4 1 | 1.903(4) | 1.901(4)/1.873(4) 1 | 1.902(2) |
| 1.899 | 1.896 | 1.894 | |
| Cu1–N3/Cu2–N7 1 | 1.994(5) | 2.011(5)/2.032(5) 1 | 2.015(2) |
| 2.014 | 2.030 | 2.028 | |
| Cu1∙∙∙O1p, Cu1∙∙∙O2p 2 | 2.59(1), absent | 2.418(5), 2.557(5) 2 | 2.654(5), 2.857(5) |
| N1–Cu1–N2/N5–Cu2–N6 1 | 83.38(18) | 83.43(18)/83.27(18) 1 | 84.36(9) |
| 83.44 | 83.35 | 83.49 | |
| N2–Cu1–O1/N6–Cu2–O4 1 | 92.65(17) | 93.60(18)/92.27(18) 1 | 93.58(9) |
| 93.31 | 93.52 | 93.84 | |
| O1–Cu1–N3/O4–Cu2–N7 1 | 87.86(18) | 89.25(18)/88.9(2) 1 | 91.02(9) |
| 89.84 | 88.45 | 88.76 | |
| N3–Cu1–N1/N7–Cu2–N5 1 | 96.19(19) | 93.46(19)/96.2(2) 1 | 91.02(10) |
| 95.29 | 94.69 | 94.44 | |
| N1–Cu1–O1/N5–Cu2–O4 1 | 173.86(18) | 175.0(2)/172.8(2) 1 | 177.24(10) |
| 168.50 | 175.78 | 171.61 | |
| N2–Cu1–N3/N6–Cu2–N7 1 | 178.8(2) | 175.3(2)/174.0(2) 1 | 175.36(9) |
| 169.69 | 178.02 | 175.51 | |
| Cu1∙∙∙N3O/Cu2∙∙∙N3O (Å) 1 | 0.030 | 0.066/0.006 1 | 0.020 |
| 0.005 | 0.022 | 0.034 | |
| N3O ∠ py (°) | 63.0(1) | 54.2(3)/14.1(1) 1 | 58.5(1) |
| 41.27 | 36.54 | 38.86 |
| 2a (NO2, H) | 2b (NO2, OMe) | 2c (NO2, OMe) | |
|---|---|---|---|
| Cu–N1 1 | 1.952 | 1.954 | 1.957 |
| Cu–N2 1 | 1.966 | 1.967 | 1.969 |
| Cu–O1 1 | 1.928 | 1.913 | 1.931 |
| Cu–O2 1 | 1.903 | 1.897 | 1.895 |
| O1–Cu–O2 | 90.51 | 90.62 | 91.23 |
| N1–Cu–N2 | 84.34 | 84.30 | 84.02 |
| A–sal ∠ D–sal 2 | 10.20 | 10.28 | 12.33 |
| 1a | 1b | 1c | 2a | 2b | 2c | |
|---|---|---|---|---|---|---|
| μx | 12.66 | 12.34 | 12.66 | 7.86 | 8.47 | 8.48 |
| μy | 4.69 | 5.42 | 4.71 | 10.35 | 8.81 | 8.96 |
| μz | −2.12 | −0.56 | 0.71 | 0.88 | 0.95 | 0.12 |
| μg | 13.66 | 13.49 | 13.53 | 13.02 | 12.26 | 12.34 |
| βx | 23.72 | 22.79 | 23.61 | 37.70 | 47.02 | 48.75 |
| βy | −4.11 | −2.78 | −3.01 | −12.44 | −14.50 | −8.98 |
| βz | 0.64 | −1.06 | 0.56 | −0.65 | −0.77 | −1.93 |
| βtot | 24.08 | 22.98 | 23.81 | 39.71 | 49.22 | 49.60 |
| βvec | 20.46 | 19.78 | 21.08 | 12.83 | 22.00 | 26.98 |
| θ | 31.8 | 30.6 | 27.7 | 71.2 | 63.5 | 57.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rigamonti, L.; Forni, A.; Cariati, E.; Malavasi, G.; Pasini, A. Solid-State Nonlinear Optical Properties of Mononuclear Copper(II) Complexes with Chiral Tridentate and Tetradentate Schiff Base Ligands. Materials 2019, 12, 3595. https://doi.org/10.3390/ma12213595
Rigamonti L, Forni A, Cariati E, Malavasi G, Pasini A. Solid-State Nonlinear Optical Properties of Mononuclear Copper(II) Complexes with Chiral Tridentate and Tetradentate Schiff Base Ligands. Materials. 2019; 12(21):3595. https://doi.org/10.3390/ma12213595
Chicago/Turabian StyleRigamonti, Luca, Alessandra Forni, Elena Cariati, Gianluca Malavasi, and Alessandro Pasini. 2019. "Solid-State Nonlinear Optical Properties of Mononuclear Copper(II) Complexes with Chiral Tridentate and Tetradentate Schiff Base Ligands" Materials 12, no. 21: 3595. https://doi.org/10.3390/ma12213595
APA StyleRigamonti, L., Forni, A., Cariati, E., Malavasi, G., & Pasini, A. (2019). Solid-State Nonlinear Optical Properties of Mononuclear Copper(II) Complexes with Chiral Tridentate and Tetradentate Schiff Base Ligands. Materials, 12(21), 3595. https://doi.org/10.3390/ma12213595