Electronic, Mechanical and Elastic Anisotropy Properties of X-Diamondyne (X = Si, Ge)

Abstract
1. Introduction
2. Theoretical Methods
3. Results and Discussion
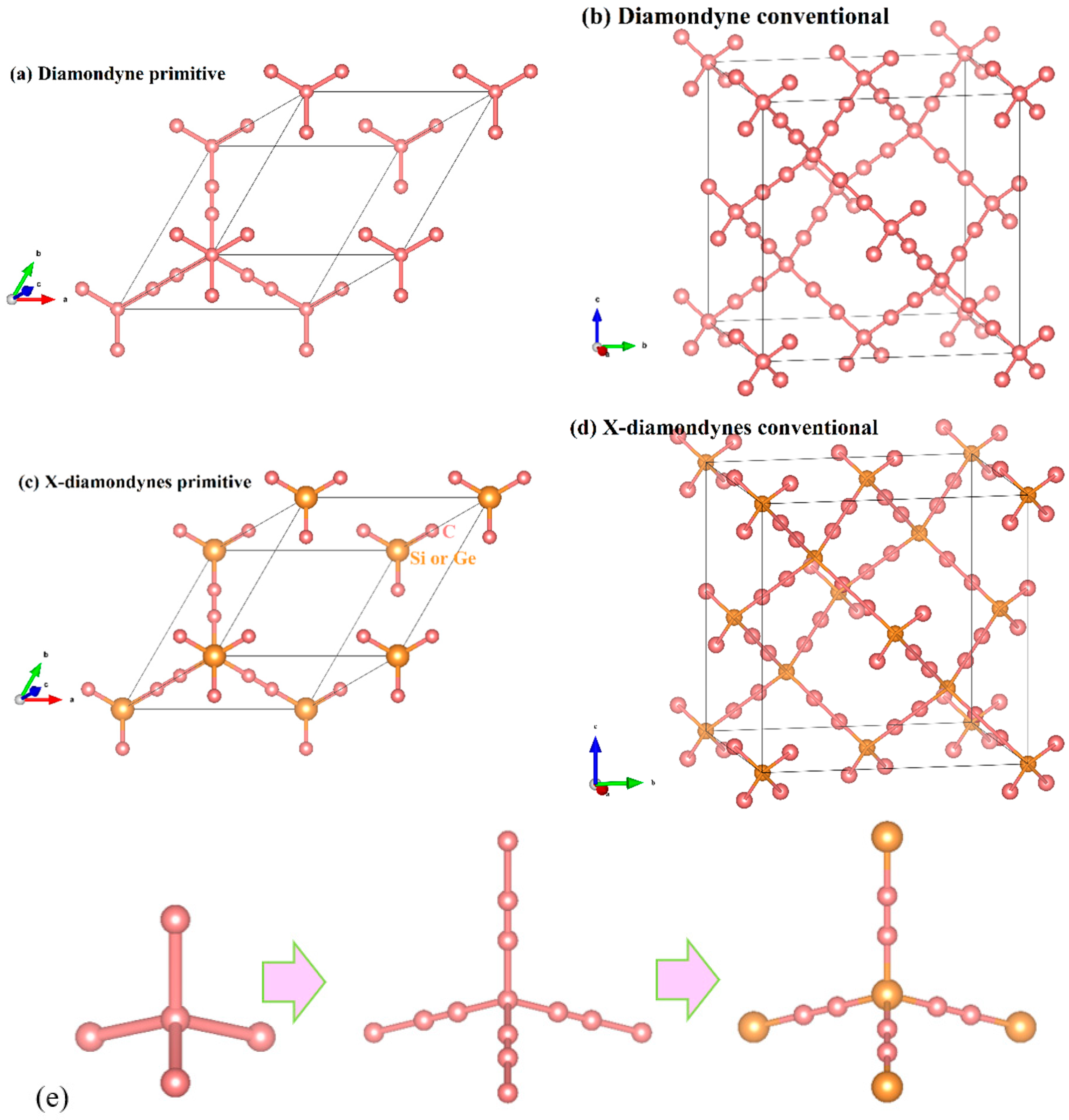
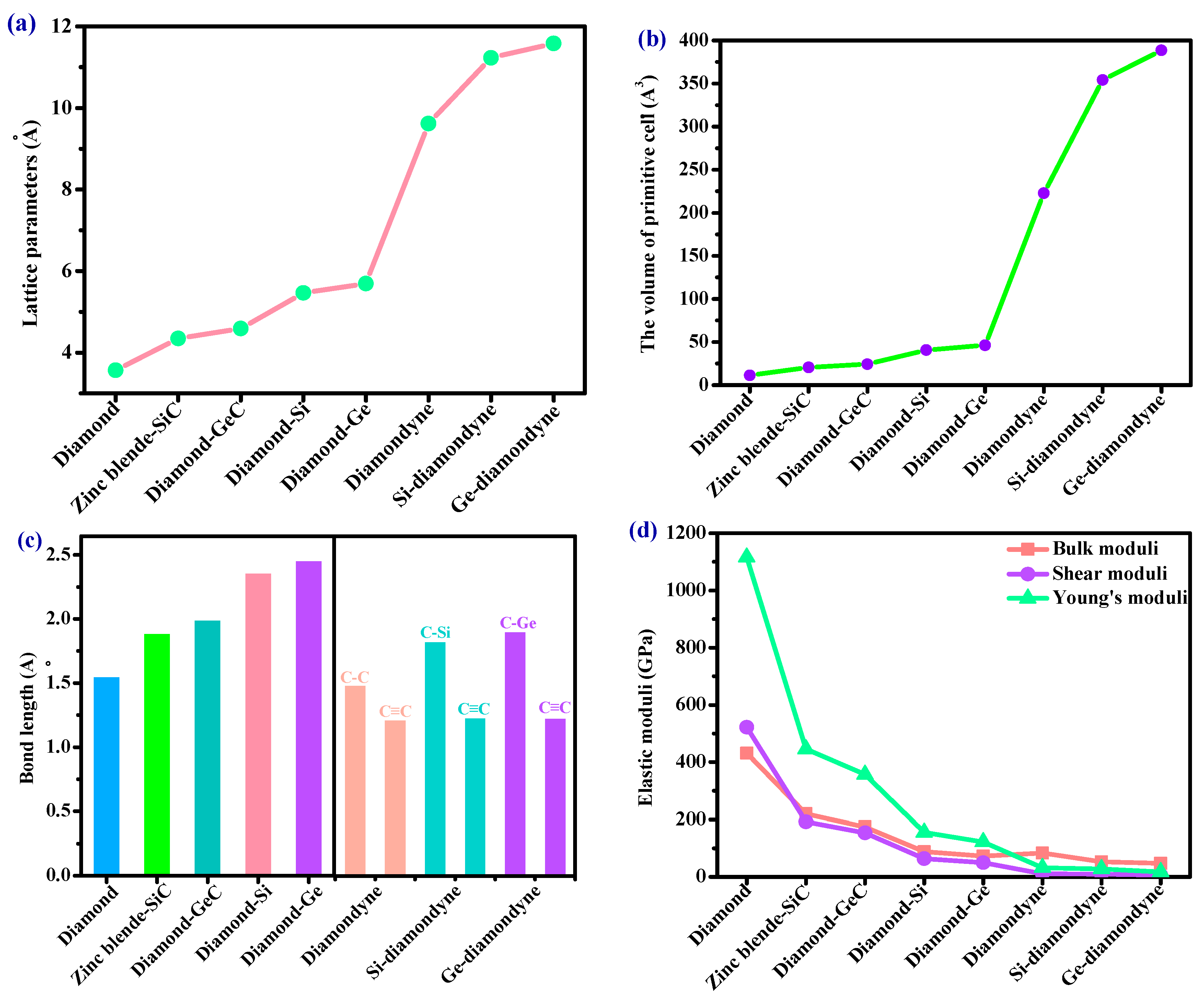
3.1. Structural Properties
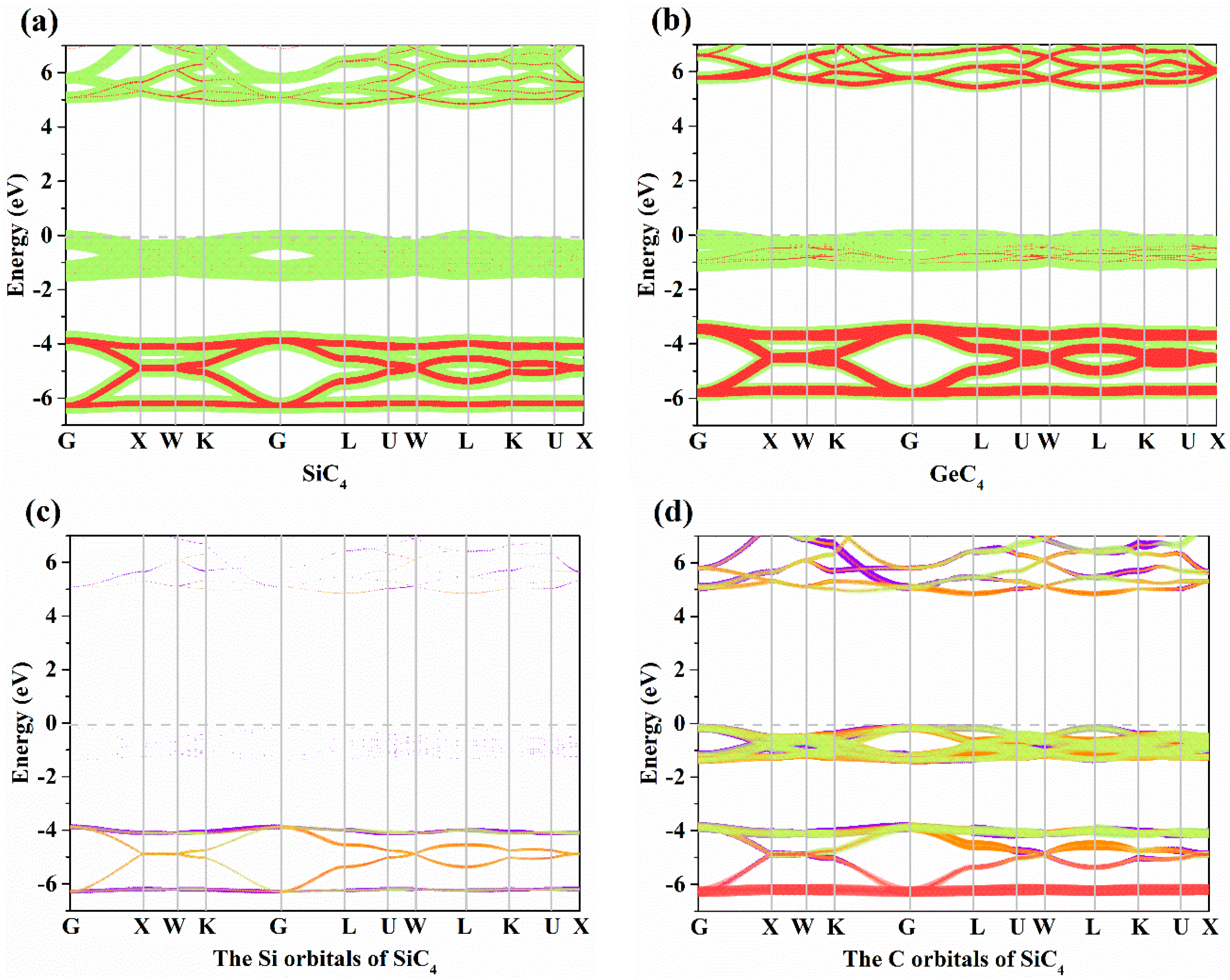
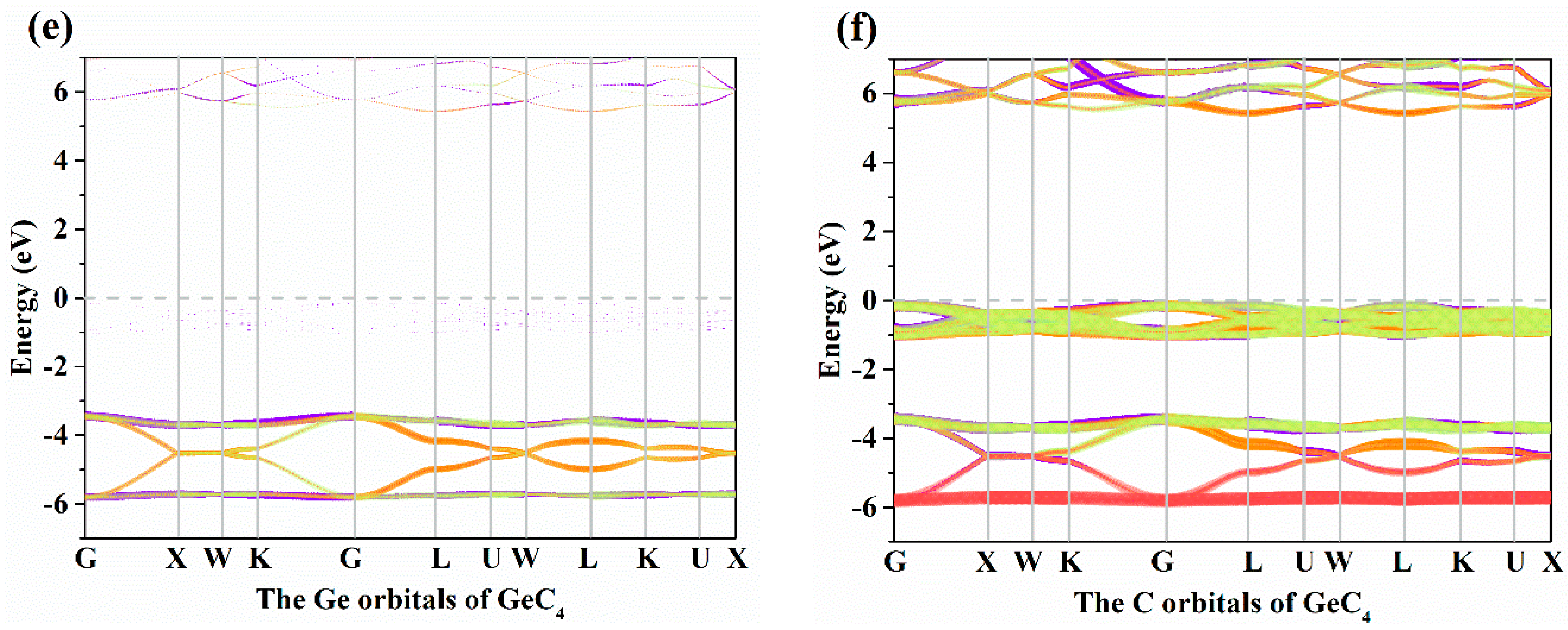
3.2. Electronic Properties
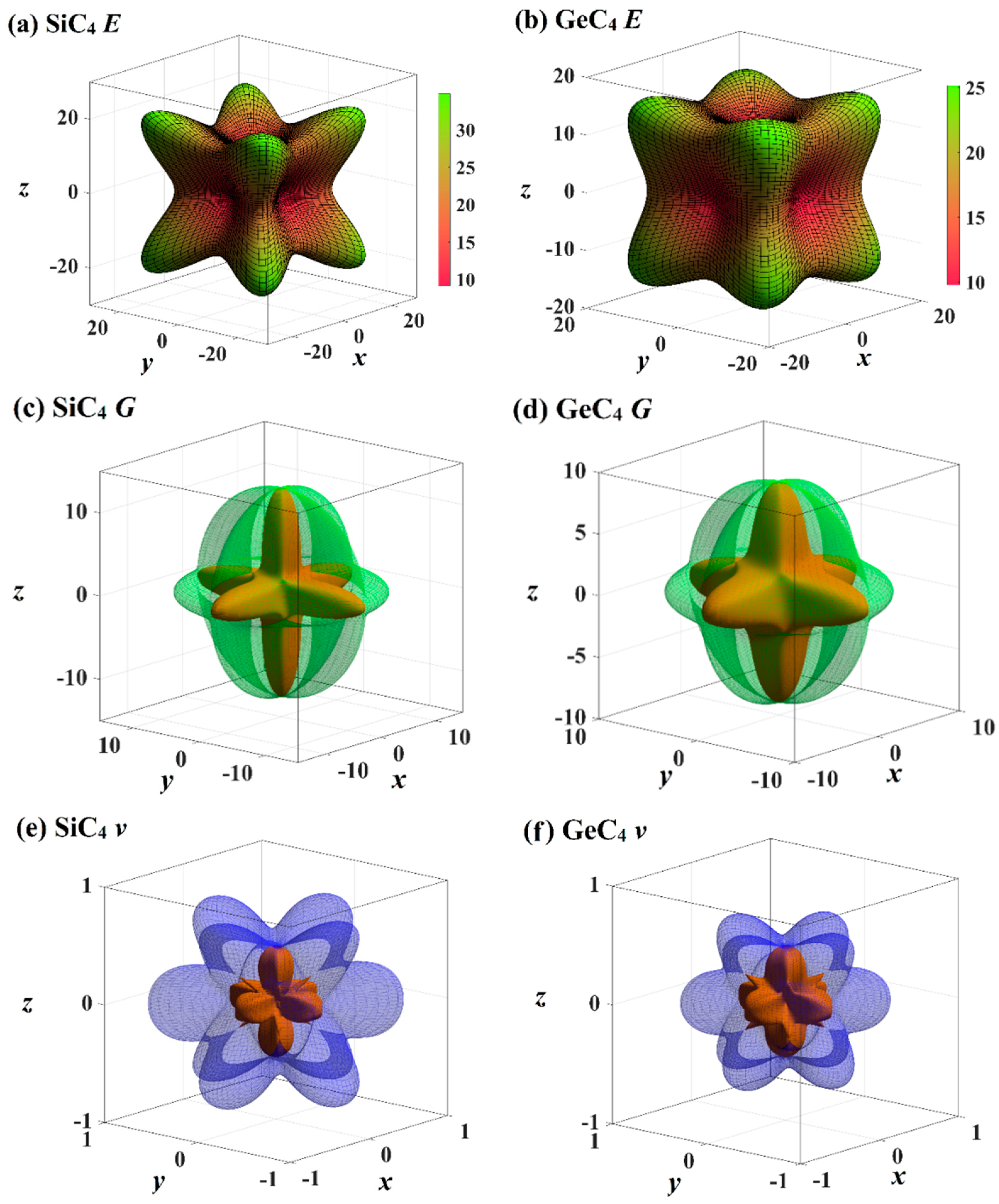
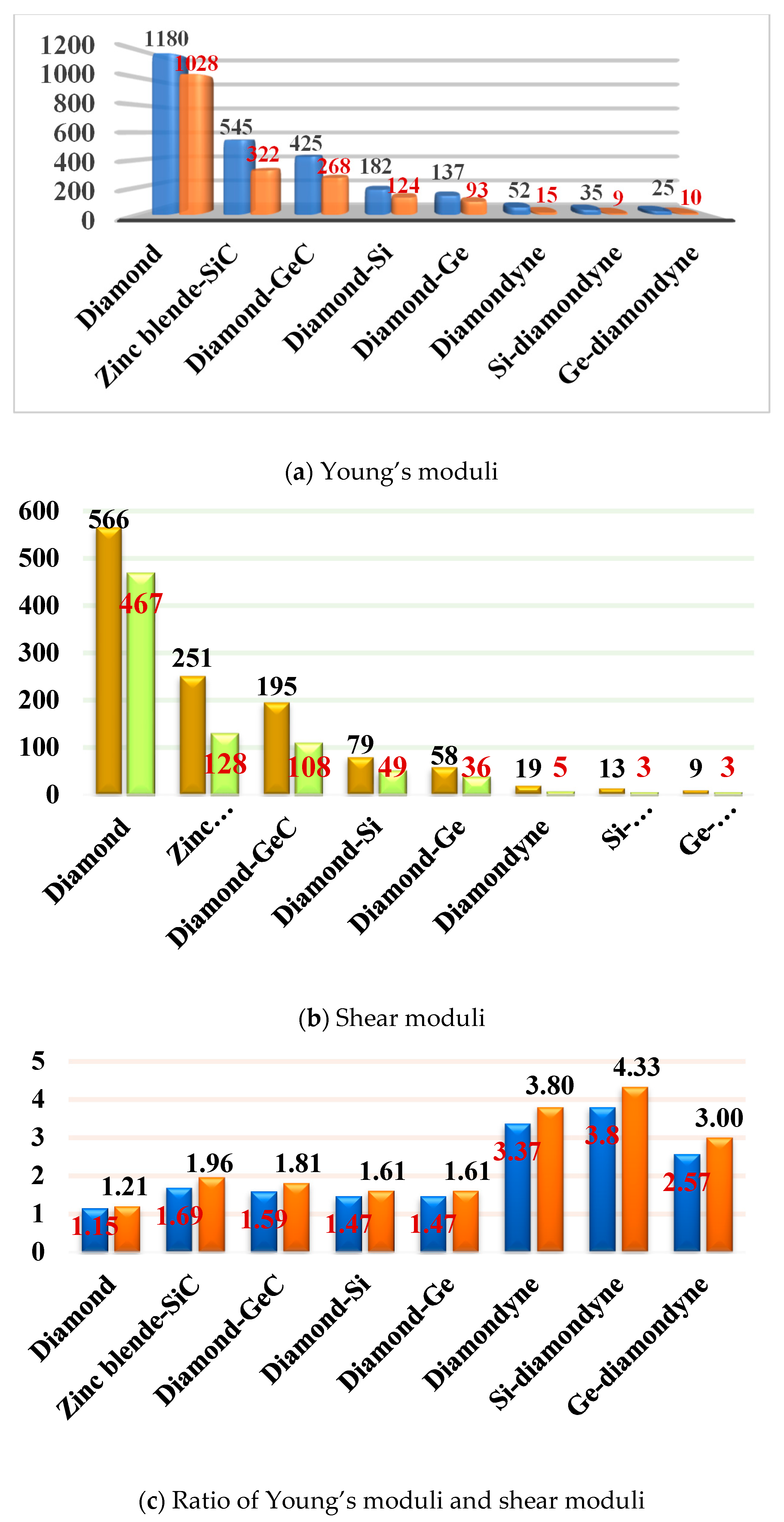
3.3. Elastic Properties and Mechanical Anisotropy Properties
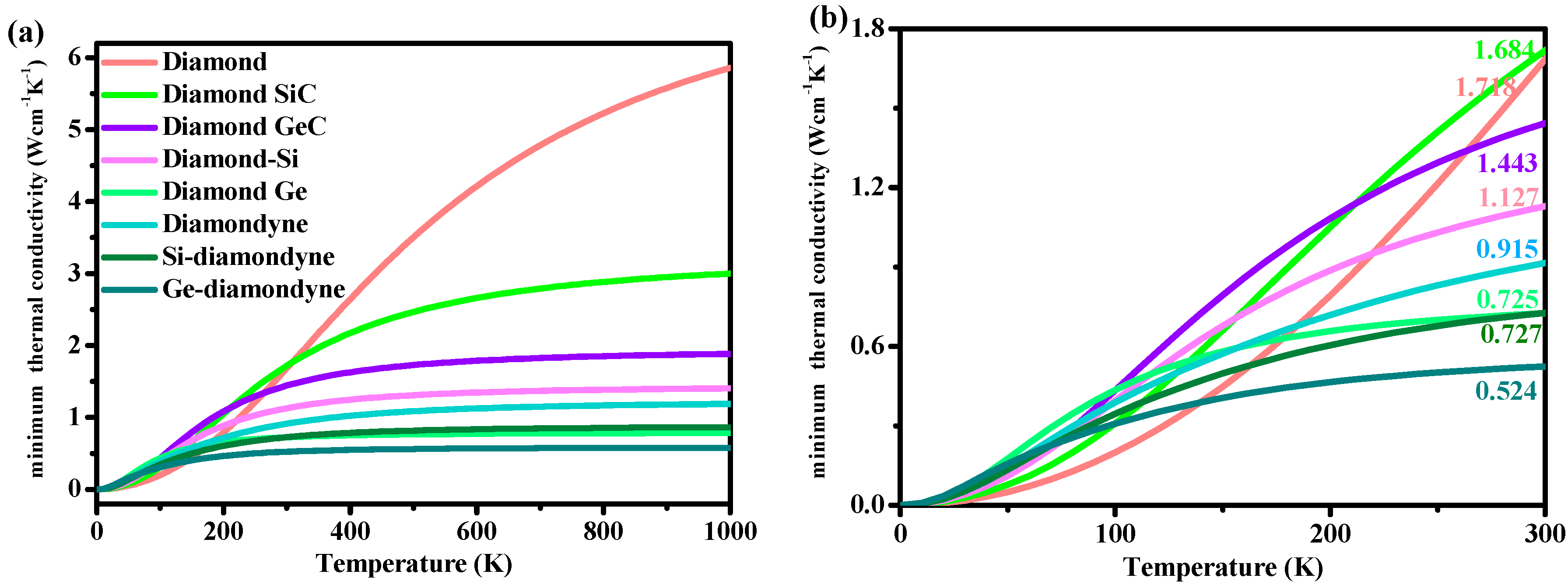
3.4. The Minimum Thermal Conductivity
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, J.T.; Chen, C.; Kawazoe, Y. Low-temperature phase transformation from graphite to sp3 orthorhombic carbon. Phys. Rev. Lett. 2011, 106, 075501. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.Y.; Wang, X.Q.; Wang, J.T. K-6 carbon: A metallic carbon allotrope in sp3 bonding networks. J. Chem. Phys. 2014, 140, 054514. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xu, B.; Zhou, X.F.; Wang, L.M.; Wen, B.; He, J.; Liu, Z.; Wang, H.T.; Tian, Y. Novel superhard carbon: C-centered orthorhombic C8. Phys. Rev. Lett. 2011, 107, 215502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y.; Lv, J.; Zhu, C.; Li, Q.; Zhang, M.; Li, Q.; Ma, Y. First-principles structural design of superhard materials. J. Chem. Phys. 2013, 138, 114101. [Google Scholar] [CrossRef]
- Li, Q.; Ma, Y.; Oganov, A.; Wang, R.H.; Wang, H.; Xu, Y.; Cui, T.; Mao, H.K.; Zou, G. Superhard monoclinic polymorph of carbon. Phys. Rev. Lett. 2009, 102, 175506. [Google Scholar] [CrossRef]
- Li, Z.; Gao, F.; Xu, Z. Strength, hardness, and lattice vibrations of Z-carbon and W-carbon: First-principles calculations. Phys. Rev. B 2012, 85, 144115. [Google Scholar] [CrossRef]
- Cheng, Y.; Melnik, R.; Kawazoe, Y.; Wen, B. Three dimensional metallic carbon from distorting sp3-bond. Cryst. Growth Des. 2016, 16, 1360. [Google Scholar] [CrossRef]
- Li, D.; Tian, F.B.; Chu, B.H.; Duan, D.; Wei, S.; Lv, Y.; Zhang, H.; Wang, L.; Lu, N.; Liu, B.; et al. Cubic C-96: A novel carbon allotrope with a porous nanocube network. J. Mater. Chem. A 2015, 3, 10448. [Google Scholar] [CrossRef]
- Xing, M.J.; Li, B.H.; Yu, Z.T.; Chen, Q. C2/m-carbon: Structural, mechanical, and electronic properties. J. Mater. Sci. 2015, 50, 7104–7114. [Google Scholar] [CrossRef]
- Wang, J.T.; Chen, C.; Mizusekid, H.; Kawazoe, Y. New carbon allotropes in sp + sp3 bonding networks consisting of C8 cubes. Phys. Chem. Chem. Phys. 2018, 20, 7962–7967. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.C.; Fan, Q.Y.; Song, Y.X. Two novel superhard carbon allotropes with honeycomb structures. J. Appl. Phys. 2019, 126, 145704. [Google Scholar] [CrossRef]
- Xing, M.J.; Li, B.H.; Yu, Z.T.; Chen, Q. Monoclinic C2/m-20 carbon: A novel superhard sp3 carbon allotrope. RSC Adv. 2016, 6, 32740–32745. [Google Scholar] [CrossRef]
- Cheng, Y.; Feng, X.; Cao, X.T.; Wen, B.; Wang, Q.; Kawazoe, Y. Body-centered tetragonal C-16: A novel topological node-line semimetallic carbon composed of tetrarings. Small 2017, 13, 1602894. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Shi, X.; Yao, M.; Liu, S.; Yang, X.; Zhu, L.; Cui, T.; Liu, B. Superhard three-dimensional carbon with metallic conductivity. Carbon 2017, 123, 311–317. [Google Scholar] [CrossRef]
- Sheng, X.L.; Yan, Q.B.; Ye, F.; Zheng, Q.R.; Su, G. T-carbon: A novel carbon allotrope. Phys. Rev. Lett. 2011, 106, 155703. [Google Scholar] [CrossRef]
- Xing, M.; Li, B.; Yu, Z.; Chen, Q. A Reinvestigation of a superhard tetragonal sp3 carbon allotrope. Materials 2016, 9, 484. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, M. Porous CY carbon: A new semiconducting phase with an sp1-sp2-sp3 bonding network. RSC Adv. 2016, 6, 112035–112039. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Chen, S.; Xiang, H.; Gong, X.G. Hybrid crystalline sp2-sp3 carbon as a high-efficiency solar cell absorber. Carbon 2016, 109, 246–252. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, H.; Xu, S.; Hu, Y.; Liu, N.; Xu, X. Multiporous carbon allotropes transformed from symmetry-matched carbon nanotubes. AIP Adv. 2016, 6, 065225. [Google Scholar] [CrossRef]
- He, C.; Sun, L.; Zhang, C.; Zhong, J. Two viable three-dimensional carbon semiconductors with an entirely sp2 configuration. Phys. Chem. Chem. Phys. 2013, 15, 680–684. [Google Scholar] [CrossRef]
- Yang, B.; Zhou, H.; Zhang, X.; Liu, X.; Zhao, M. Dirac cones and highly anisotropic electronic structure of super-graphyne. Carbon 2017, 113, 40–45. [Google Scholar] [CrossRef]
- Li, Z.Z.; Chen, J.; Nie, S.; Xu, L.; Mizuseki, H.; Weng, H.; Wang, J.T. Orthorhombic carbon oC24: A novel topological nodal line semimetal. Carbon 2018, 133, 39–43. [Google Scholar] [CrossRef]
- Cheng, Y.; Du, J.; Melnik, R.; Kawazoe, Y.; Wen, B. Novel three dimensional topological nodal line semimetallic carbon. Carbon 2016, 98, 468–473. [Google Scholar] [CrossRef]
- Wang, J.T.; Weng, H.M.; Nie, S.; Fang, Z.; Kawazoe, Y.S.; Chen, C.F. Body-centered orthorhombic C16: A novel topological node-line semimetal. Phys. Rev. Lett. 2016, 116, 195501. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Liang, Y.; Xu, Q.; Yu, R.; Fang, Z.; Dai, X. Topological node-line semimetal in three-dimensional graphene networks. Phys. Rev. B 2015, 92, 045108. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, Y.; Yang, S.Y.A.; Pan, H.; Zhang, F.; Cohen, M.L. Nanostructured carbon allotropes with Weyl-like loops and points. Nano Lett. 2015, 15, 6974–6978. [Google Scholar] [CrossRef]
- Jo, J.Y.; Kim, B.G. Carbon allotropes with triple bond predicted by first-principle calculation: Triple bond modified diamond and T-carbon. Phys. Rev. B 2012, 86, 075151. [Google Scholar] [CrossRef]
- Costa, D.G.; Henrique, F.J.F.S.; Oliveira, F.L. n- Diamondynes: Expanding the family of carbon allotropes. Carbon 2018, 136, 337–344. [Google Scholar] [CrossRef]
- Sun, M.J.; Cao, X.; Cao, Z. Si(C≡C)4-based single-crystalline semiconductor: Diamond-like superlight and superflexible wide-bandgap material for the UV photoconductive device. ACS Appl. Mater. Interfaces 2016, 8, 16551–16554. [Google Scholar] [CrossRef]
- Cao, X.; Li, X.F.; Zhu, Z.Z. Superlight and superflexible three-dimensional semiconductor frameworks A(X≡Y)4 (A = Si, Ge; X/Y = C, B, N) with tunable optoelectronic. Chem. Asian J. 2017, 12, 804–810. [Google Scholar] [CrossRef]
- Fang, L.; Sun, M.; Cao, X.; Cao, Z. Mechanical and optical properties of a novel diamond-like Si(C equivalent to C-C6H4-C equivalent to C)(4) single-crystalline semiconductor: A first-principles study. Acta Phys. Chim. Sin. 2018, 34, 296–302. [Google Scholar]
- Hohenberg, P.; Kohn, W. SemiclasSiCal origin of density functionals. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Yang, L.; Liu, T.; Zhang, C.; Dong, H. Climatic and seasonal suitability of phase change materials coupled with night ventilation for office buildings in Western China. Renew. Energy 2020, 147, 356–373. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M.J. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Petrescu, M.I. Boron nitride theoretical hardness compared to carbon polymorphs. Diam. Relat. Mater. 2004, 13, 1848–1853. [Google Scholar] [CrossRef]
- Grimsditch, M.; Zouboulis, E.S.; Polian, A. Elastic constants of boron nitride. J. Appl. Phys. 1994, 76, 832–834. [Google Scholar] [CrossRef]
- Van Camp, P.E.; Doren, V.E.; Devreese, J.T. First-principles calculation of the pressure coefficient of the indirect band gap and of the charge density of C and Si. Phys. Rev. B 1986, 34, 1314. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, W.R.L.; Segall, B.; Methfessel, M.; van Schilfgaarde, M. Calculated elastic constants and deformation potentials of cubic SiC. Phys. Rev. B 1991, 44, 3685. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.P.; Wei, H.D.; Zhu, J.; Yang, X.D. First-principles study of pressure-induced phase transition in silicon carbide. Phys. B 2008, 403, 3543–3546. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Xu, J.; Zhang, W.Z.; Song, Y.X.; Yun, S.N. Physical properties of group 14 semiconductor alloys in orthorhombic phase. J. Appl. Phys. 2019, 126, 045709. [Google Scholar] [CrossRef]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 73rd ed.; Chemical Rubber: Boca Raton, FL, USA, 1994. [Google Scholar]
- Zhang, X.; Quan, S.; Ying, C.; Li, Z. Theoretical investigations on the structural, lattice dynamical and thermodynamic properties of XC (X = Si, Ge, Sn). Solid State Commun. 2011, 151, 1545–1549. [Google Scholar] [CrossRef]
- Hao, A.M.; Yang, X.C.; Wang, X.M.; Zhu, Y.; Liu, X.; Liu, R.P. First-principles investigations on electronic, elastic and optical properties of XC (X = Si, Ge, and Sn) under high pressure. J. Appl. Phys. 2010, 108, 063531. [Google Scholar] [CrossRef]
- Anderson, O.L. A simplified method for calculating the debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Panda, K.B.; Ravi, K.S. Determination of elastic constants of titanium diboride (TiB2) from first principles using FLAPW implementation of the density functional theory. Comput. Mater. Sci. 2006, 35, 134–150. [Google Scholar] [CrossRef]
- Zywietz, A.; Karch, K.; Bechstedt, F. Influence of polytypism on thermal properties of silicon carbide. Phys. Rev. B 1996, 54, 1791. [Google Scholar] [CrossRef]
- Hu, W.C.; Liu, Y.; Li, D.J.; Zeng, X.Q.; Xu, C.S. First-principles study of structural and electronic properties of C14-type Laves phase Al2Zr and Al2Hf. Comput. Mater. Sci. 2014, 83, 27–34. [Google Scholar] [CrossRef]
- Fan, Q.; Chai, C.; Wei, Q.; Zhou, P.; Yang, Y. Two novel Ge phases and their Si-Ge alloys with excellent electronic and optical properties. Mater. Des. 2017, 132, 539–551. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.; Song, Y.; Fan, Q.; Yang, Y. Structural, mechanical, anisotropic, and thermal properties of AlAs in oC12 and hP6 phases under pressure. Materials 2018, 11, 740. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.G.; Watson, S.K.; Pohl, R.O. Lower limit to the thermal conductivity of disordered crystals. Phys. Rev. B. 1992, 46, 6131. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.Y.; Yang, R.L.; Zhang, W.; Yun, S.N. Elastic anisotropy and thermal conductivity of silicon allotropes. Results Phys. 2019, 15, 102580. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Wang, P.; Yan, F.; Shi, C.L.; Tian, Y. Physical properties of B4N4-I and B4N4-II: First-principles study. Chin. Phys. B 2019, 28, 036101. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Wang, H.Q.; Zhang, W.Z.; Wei, M.F.; Song, Y.X.; Zhang, W.; Yun, S.N. Si–Ge alloys in C2/c phase with tunable direct band gaps: A comprehensive study. Curr. Appl. Phys. 2019, 19, 1325–1333. [Google Scholar] [CrossRef]
- Fan, Q.; Niu, R.; Zhang, W.; Zhang, W.; Ding, Y.; Yun, S. t-Si64: A Novel Silicon Allotrope. ChemPhysChem 2019, 20, 128–133. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | ρ | V | C11 | C12 | C44 | B | G | E | v | |
|---|---|---|---|---|---|---|---|---|---|---|
| Diamond | 3.566 | 3.518 | 11.340 | 1053 | 120 | 563 | 431 | 522 | 1116 | 0.07 |
| 3.567 a | 1076 b | 125 | 577 | 442 | ||||||
| Zinc blende-SiC | 4.348 | 3.240 | 20.550 | 390 | 134 | 251 | 220 | 192 | 446 | 0.16 |
| 4.360 c | 390 d | 142 | 256 | 227 e | ||||||
| Diamond-Si | 5.464, 5.465 f | 2.288 | 40.773 | 154 | 56 | 79 | 88 | 64 | 155 | 0.21 |
| 5.430 g | 165 | 64 | 87 | |||||||
| Diamond-GeC | 4.590 | 5.811 | 24.175 | 318 | 102 | 195 | 174 | 154 | 357 | 0.16 |
| 4.590 h | 175 i | |||||||||
| Diamond-Ge | 5.694 | 5.224 | 46.152 | 121 | 49 | 62 | 73 | 50 | 122 | 0.22 |
| 5.660 g | 5.318 | 45.330 | 129 | 48 | 67 | 77 | ||||
| Diamondyne | 9.621 | 0.896 | 222.65 | 90 | 79 | 19 | 83 | 11 | 32 | 0.44 |
| 9.628 j | 0.894 | 223.10 | 95 | |||||||
| 9.636 k | 0.892 | 223.68 | 83 | |||||||
| Si-diamondyne | 11.233 | 0.713 | 354.36 | 58 | 51 | 13 | 53 | 10 | 28 | 0.41 |
| 11.220 l | 0.740 | 353.12 | 59 | 54 | 14 | 55 | 7 | 20 | 0.48 | |
| Ge-diamondyne | 11.584 | 1.031 | 388.64 | 51 | 45 | 9 | 47 | 6 | 17 | 0.44 |
| 11.590 m | 1.030 | 389.22 | 55 | 48 | 7 | 50 | 5 | 15 | 0.48 |
| Diamond | Zinc Blende-SiC | Diamond-GeC | Diamond-Si | Diamond-Ge | Diamondyne | Si-Diamondyne | Ge-Diamondyne | |
|---|---|---|---|---|---|---|---|---|
| vp | 17,898 | 12,121 | 8080 | 8704 | 5171 | 10,440 | 9645 | 7304 |
| vs | 12,181 | 7698 | 5148 | 5289 | 3094 | 3504 | 3745 | 2412 |
| vm | 13,280 | 8465 | 5659 | 5843 | 3423 | 3986 | 4246 | 3745 |
| ΘD | 2220 | 1161, 1232 a | 734 | 638 | 358 | 422 | 385 | 242 |
| κmin | 1.684 | 1.718 | 1.443 | 1.127 | 0.725 | 0.915 | 0.727 | 0.524 |
| (001) (010) (100) | (011) (101) (110) | (111) | All | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Emax | Emin | Ratio | Emax | Emin | Ratio | Emax | Emin | Ratio | Emax | Emin | Ratio | |
| Diamond | 1138 | 1028 | 1.11 | 1180 | 1028 | 1.15 | 1138 | 1138 | 1.00 | 1180 | 1028 | 1.15 |
| Zinc blende-SiC | 465 | 322 | 1.44 | 545 | 322 | 1.69 | 465 | 465 | 1.00 | 545 | 322 | 1.69 |
| Diamond-GeC | 371 | 268 | 1.38 | 425 | 268 | 1.59 | 371 | 371 | 1.00 | 425 | 268 | 1.59 |
| Diamond-Si | 163 | 124 | 1.31 | 182 | 124 | 1.47 | 163 | 163 | 1.00 | 182 | 124 | 1.47 |
| Diamond-Ge | 123 | 93 | 1.32 | 137 | 93 | 1.47 | 123 | 123 | 1.00 | 137 | 93 | 1.47 |
| Diamondyne | 33 | 15 | 2.20 | 52 | 15 | 3.47 | 33 | 33 | 1.00 | 52 | 15 | 3.47 |
| Si-diamondyne | 21 | 9 | 2.33 | 35 | 9 | 3.89 | 21 | 21 | 1.00 | 35 | 9 | 3.89 |
| Ge-diamondyne | 18 | 10 | 1.80 | 25 | 10 | 2.50 | 18 | 18 | 1.00 | 25 | 10 | 2.50 |
| V | G | ||||
|---|---|---|---|---|---|
| vmax | vmin | Gmax | Gmin | Gmax/Gmin | |
| Diamond | 0.11 | 0.01 | 566 | 467 | 1.21 |
| Zinc blende-SiC | 0.37 | 0.00 | 251 | 128 | 1.96 |
| Diamond-GeC | 0.34 | 0.00 | 195 | 108 | 1.81 |
| Diamond-Si | 0.35 | 0.04 | 79 | 49 | 1.61 |
| Diamond-Ge | 0.38 | 0.06 | 58 | 36 | 1.61 |
| Diamondyne | 0.99 | 0.00 | 19 | 5 | 3.80 |
| Si-diamondyne | 1.06 | 0.00 | 13 | 3 | 4.33 |
| Ge-diamondyne | 0.86 | 0.01 | 9 | 3 | 3.00 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Q.; Duan, Z.; Song, Y.; Zhang, W.; Zhang, Q.; Yun, S. Electronic, Mechanical and Elastic Anisotropy Properties of X-Diamondyne (X = Si, Ge). Materials 2019, 12, 3589. https://doi.org/10.3390/ma12213589
Fan Q, Duan Z, Song Y, Zhang W, Zhang Q, Yun S. Electronic, Mechanical and Elastic Anisotropy Properties of X-Diamondyne (X = Si, Ge). Materials. 2019; 12(21):3589. https://doi.org/10.3390/ma12213589
Chicago/Turabian StyleFan, Qingyang, Zhongxing Duan, Yanxing Song, Wei Zhang, Qidong Zhang, and Sining Yun. 2019. "Electronic, Mechanical and Elastic Anisotropy Properties of X-Diamondyne (X = Si, Ge)" Materials 12, no. 21: 3589. https://doi.org/10.3390/ma12213589
APA StyleFan, Q., Duan, Z., Song, Y., Zhang, W., Zhang, Q., & Yun, S. (2019). Electronic, Mechanical and Elastic Anisotropy Properties of X-Diamondyne (X = Si, Ge). Materials, 12(21), 3589. https://doi.org/10.3390/ma12213589