Titanium Dioxide-Based Nanocomposites for Enhanced Gas-Phase Photodehydrogenation



 ,
,  ,
,  ,
,  and
and 
Abstract
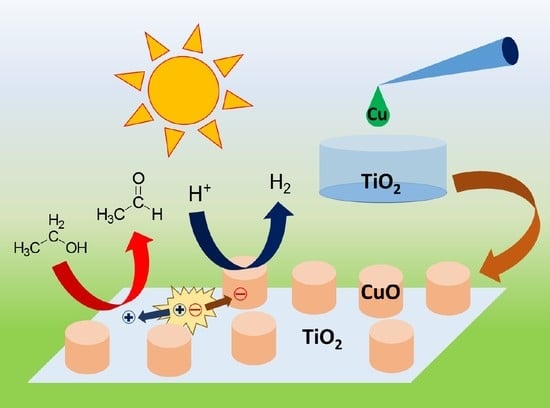
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Photocatalysts
2.2.1. Titanium Dioxide Synthesis
2.2.2. Copper Oxide Loading by Impregnation
2.2.3. Copper Oxide Loading by Complex-Precipitation
2.3. Characterization of the Nanocomposite Materials
2.4. Photocatalytic Activity Tests
3. Results and Discussion
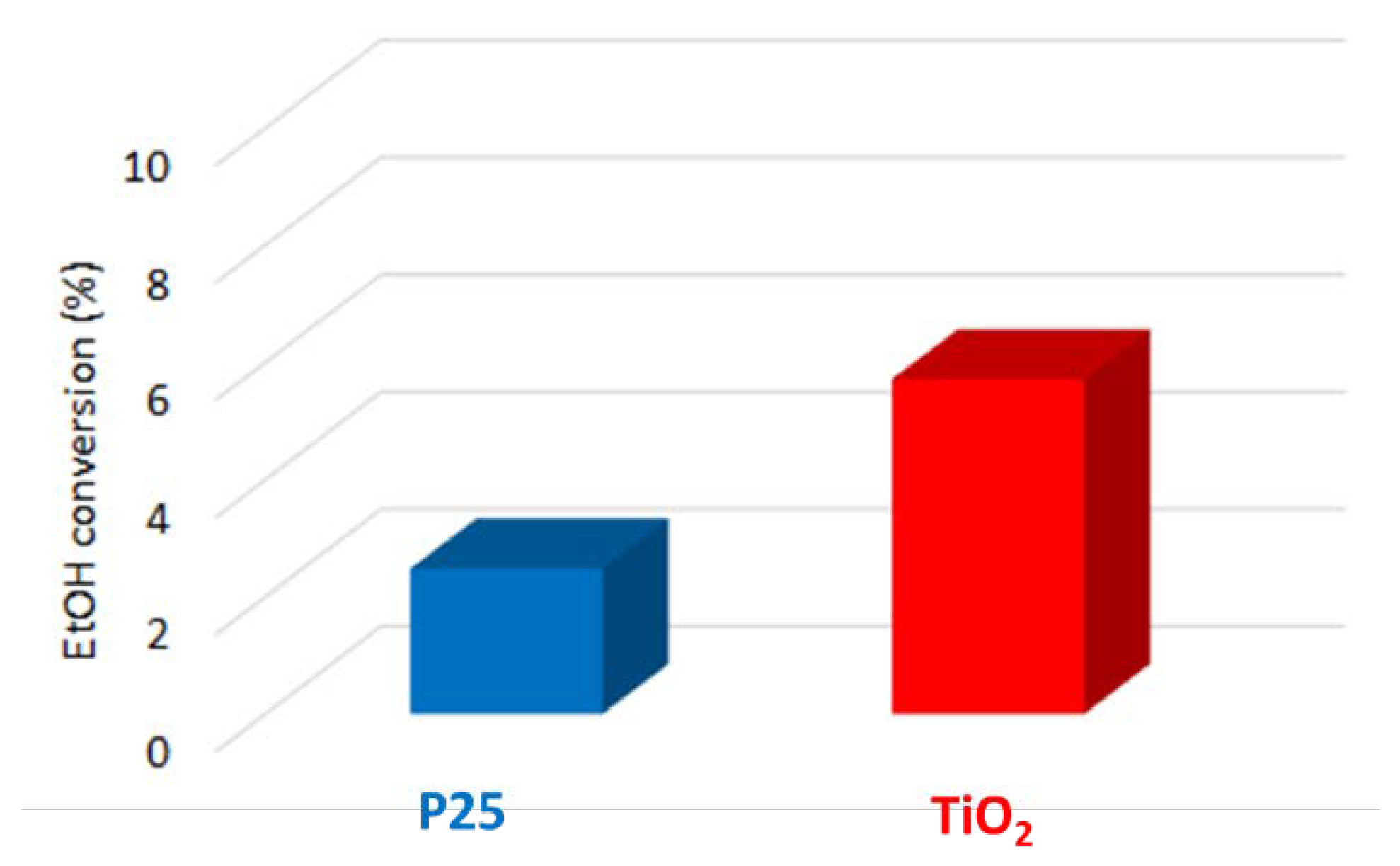
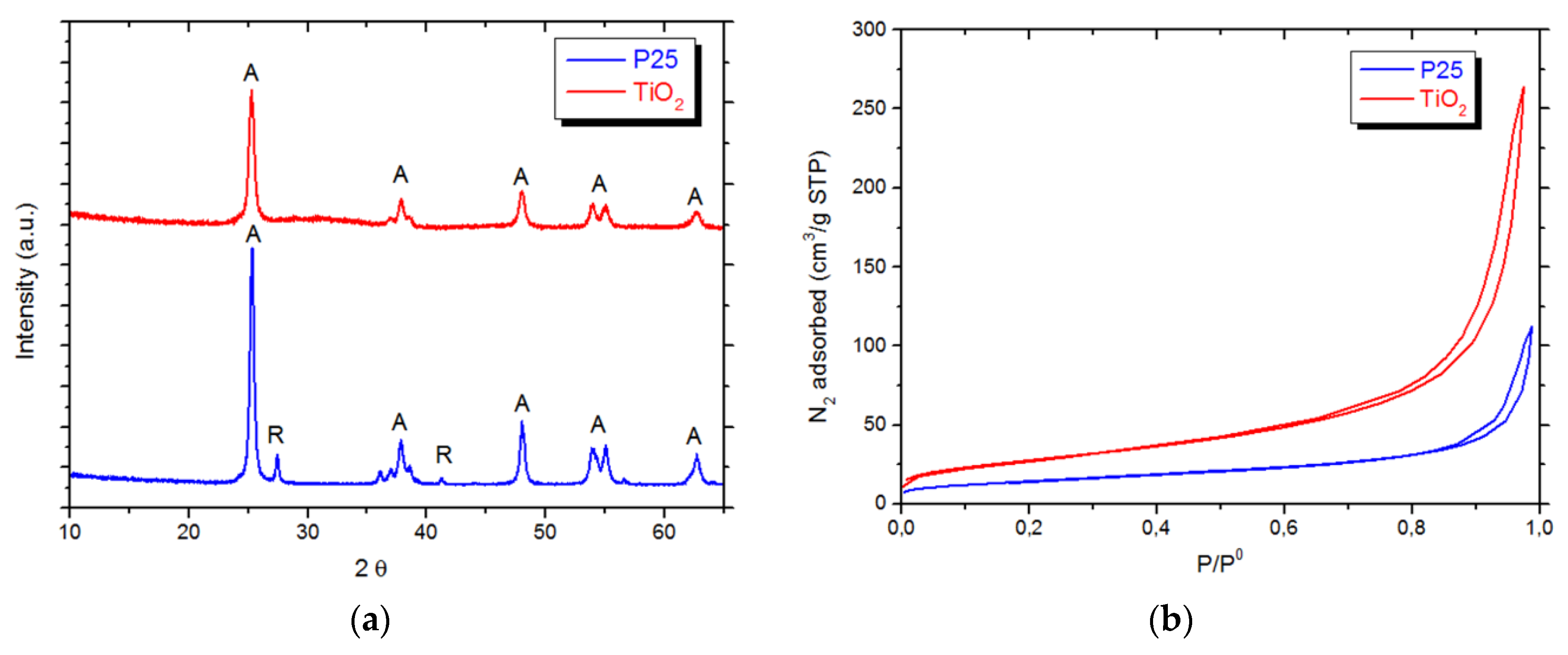
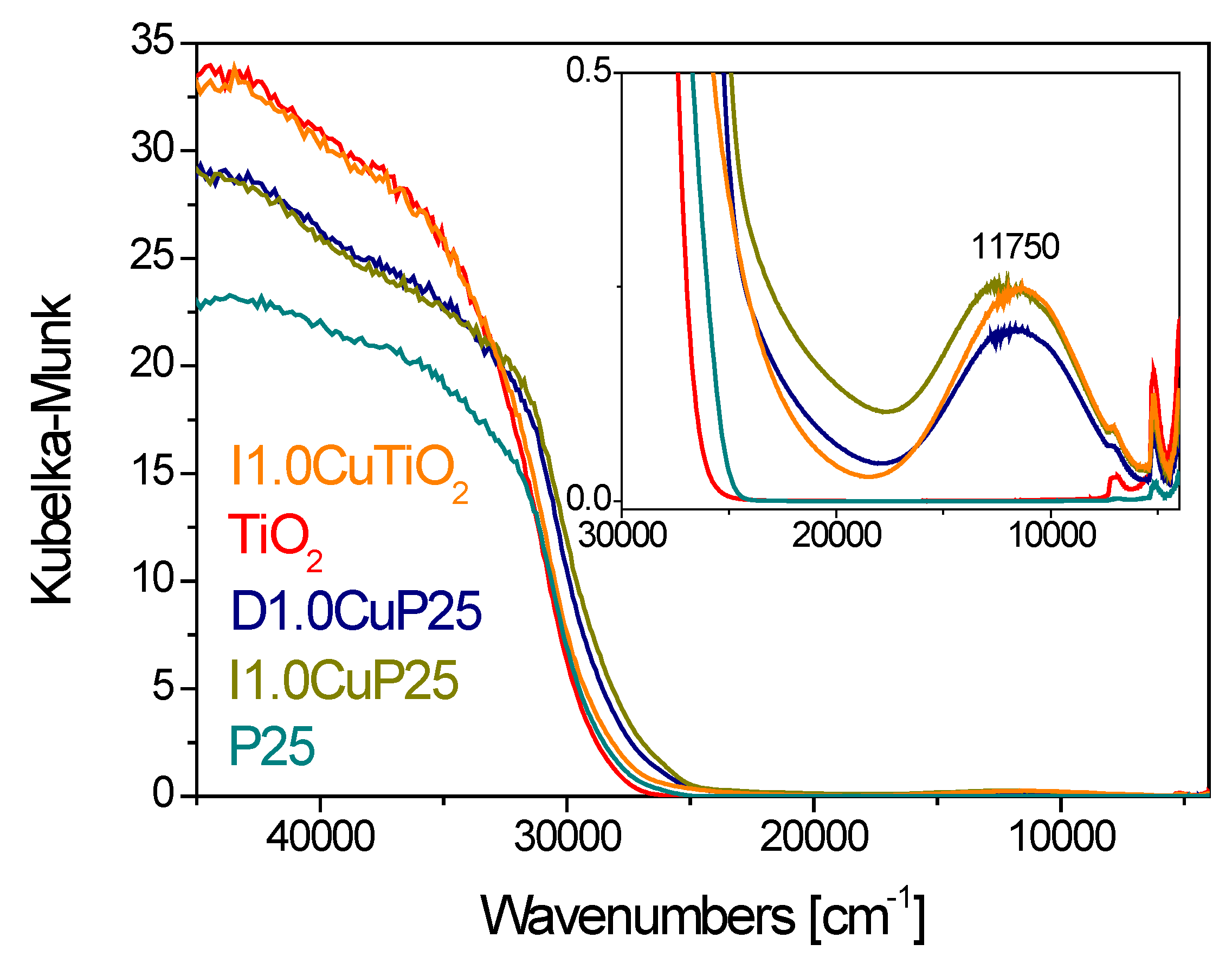
3.1. Pristine Titanium Dioxide Materials
3.2. Copper Oxide-Titania Nanocomposites
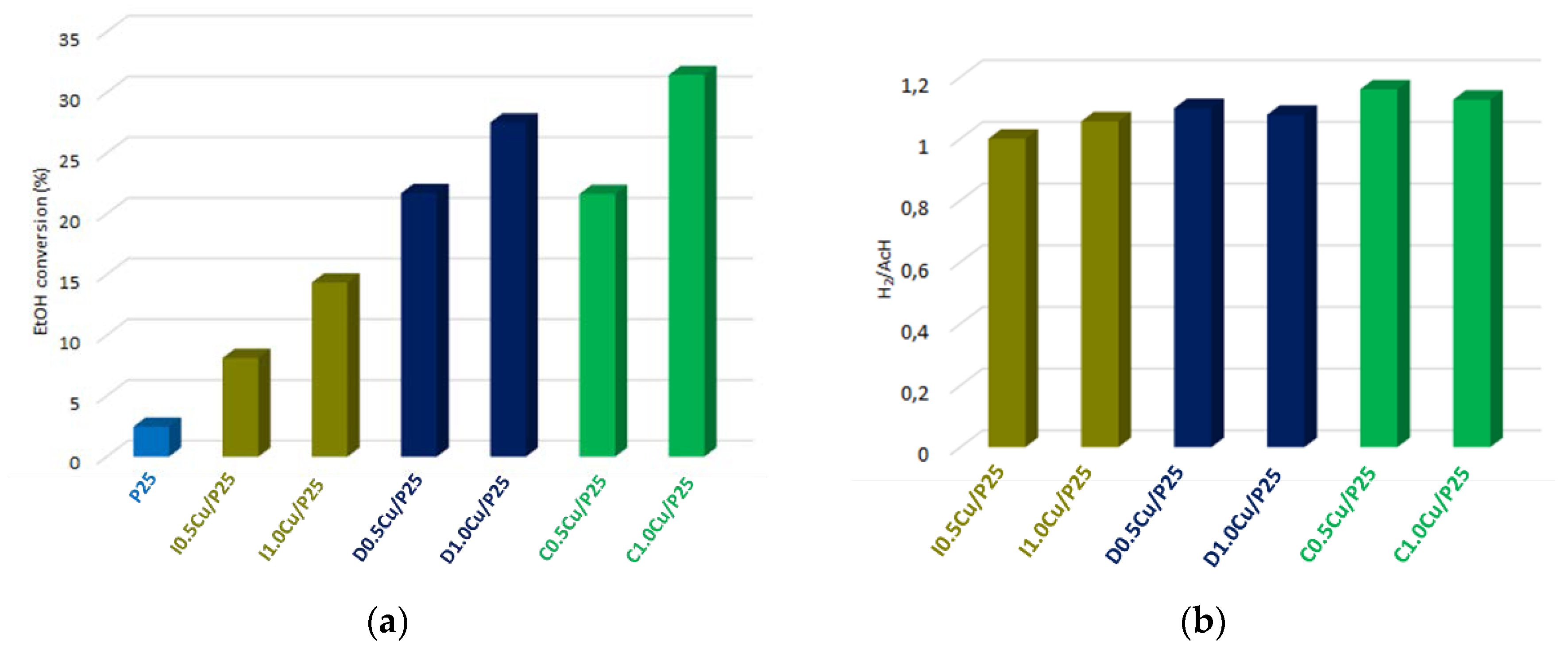
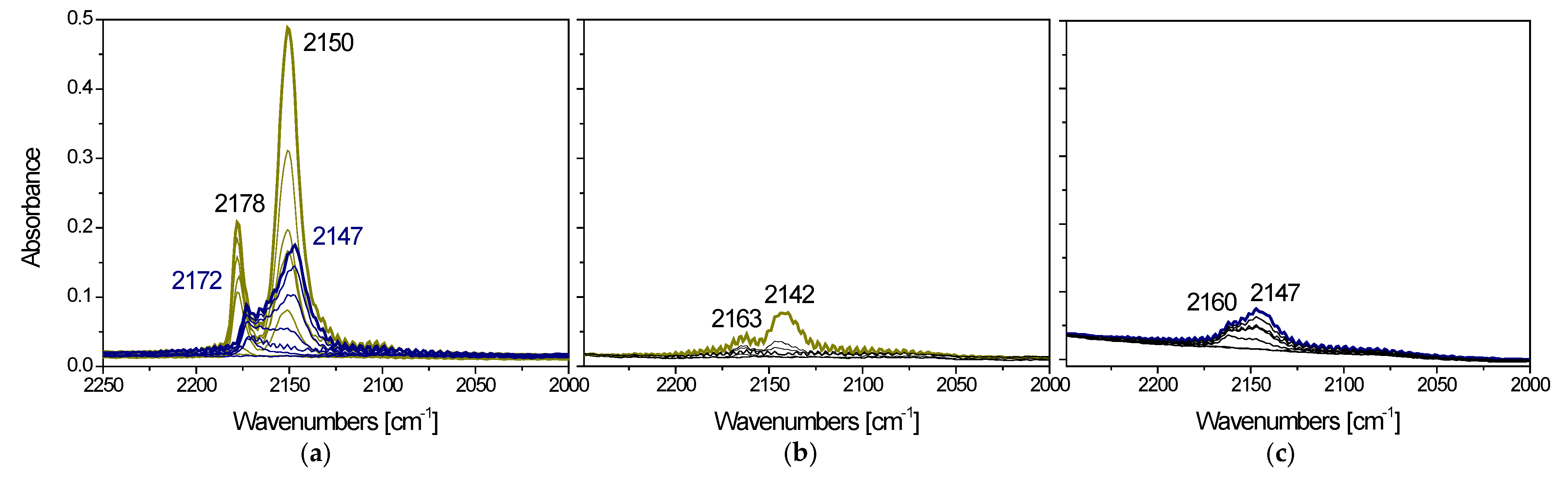
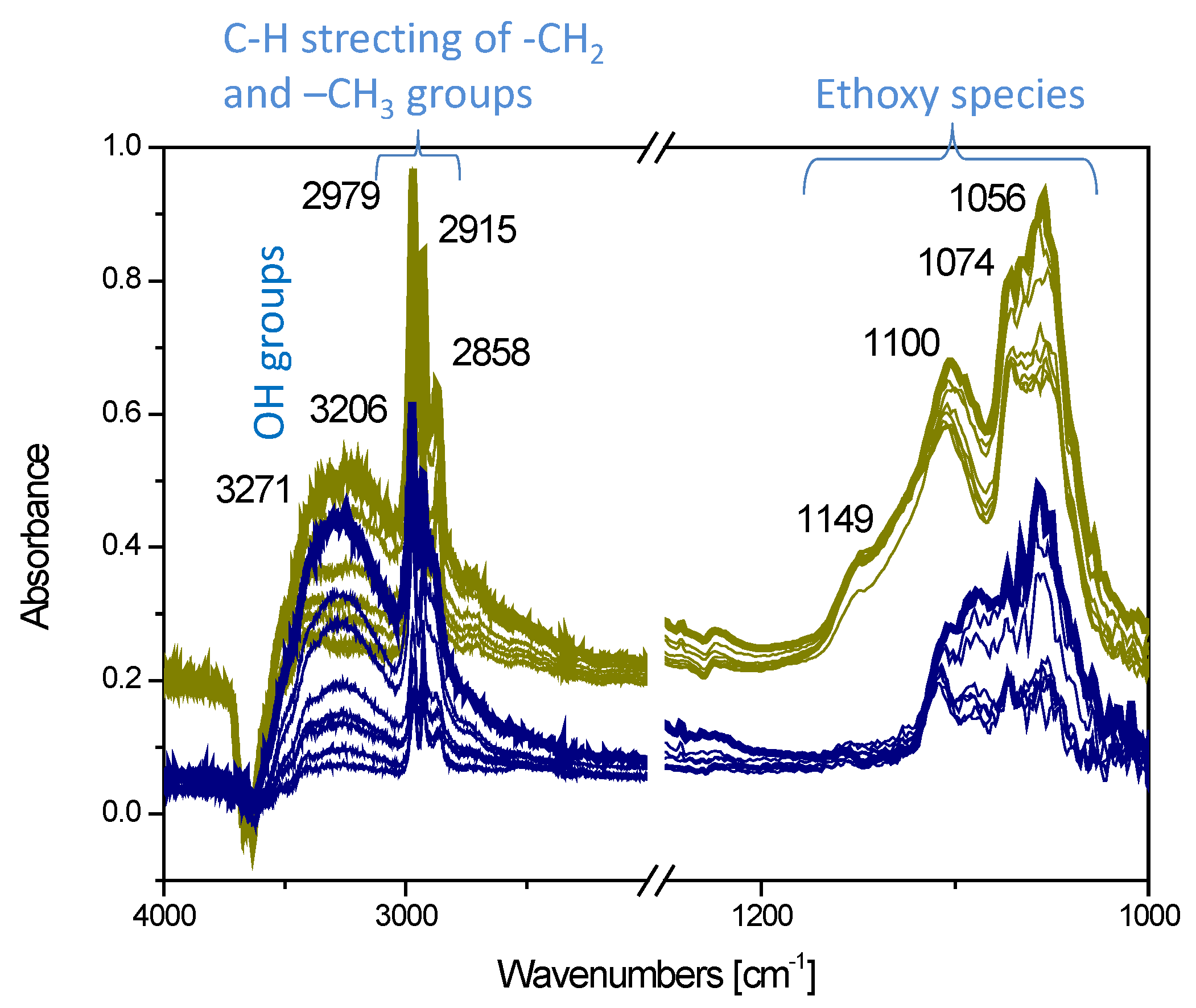
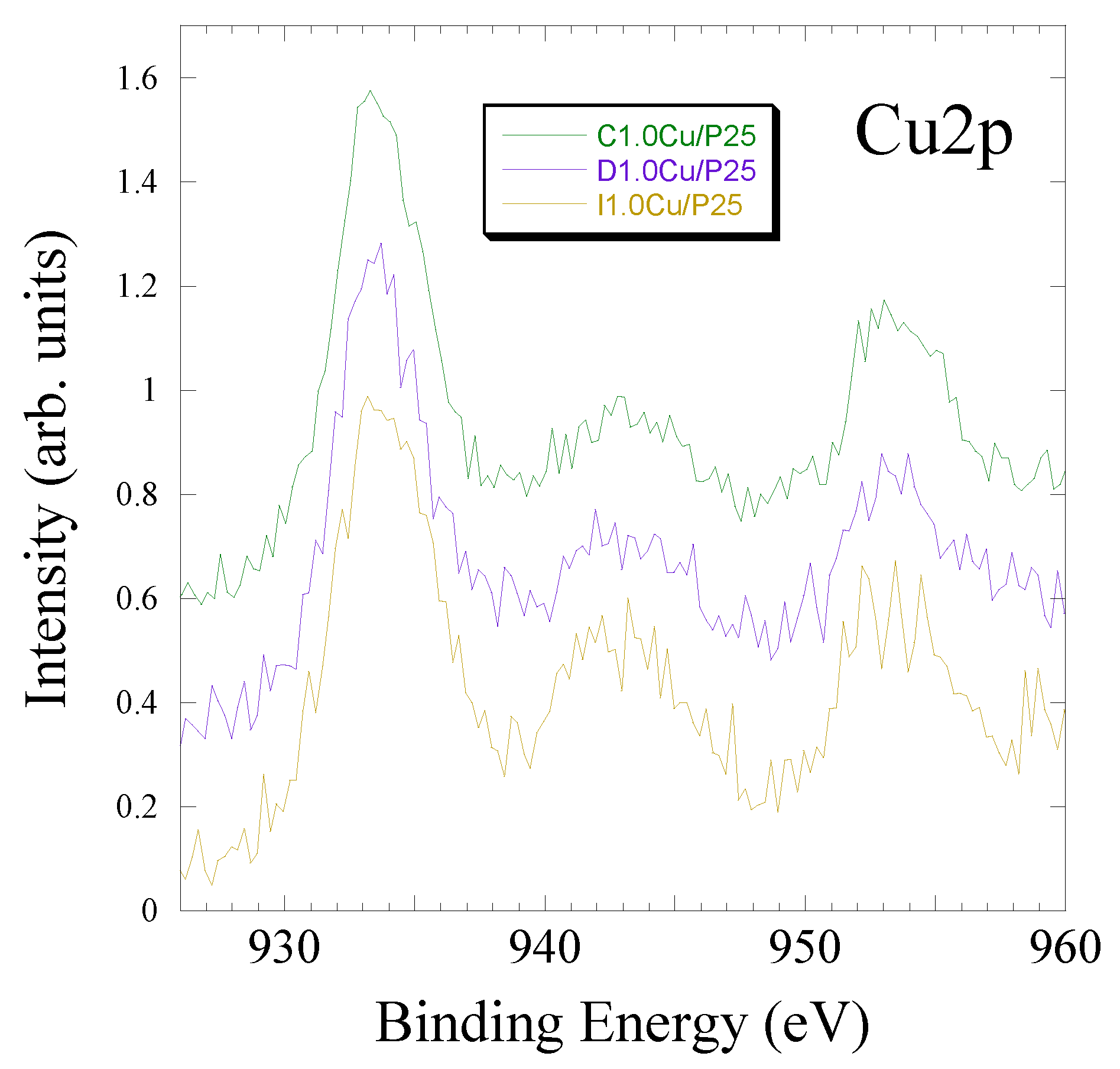
3.2.1. Copper Oxide-P25 Nanocomposites
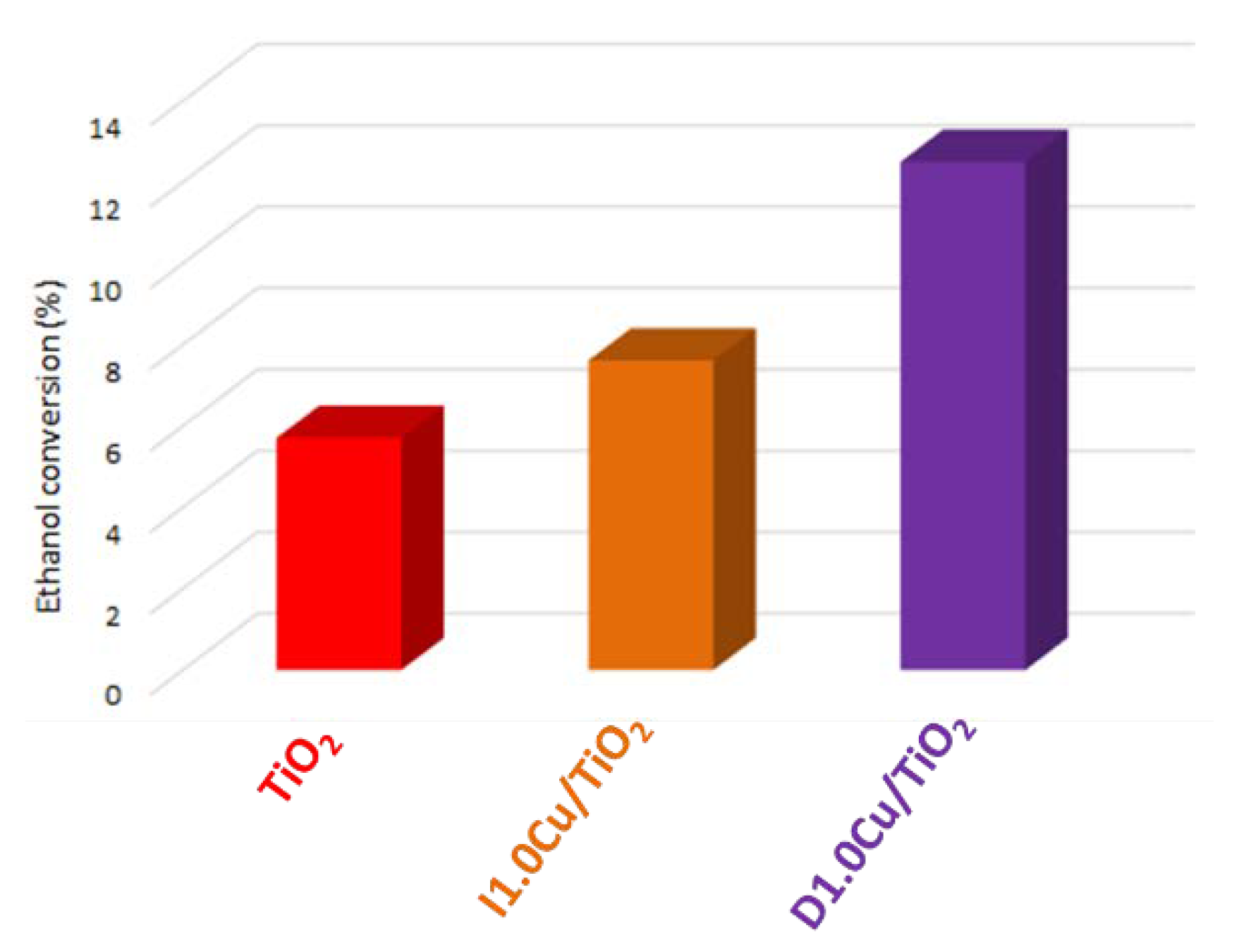
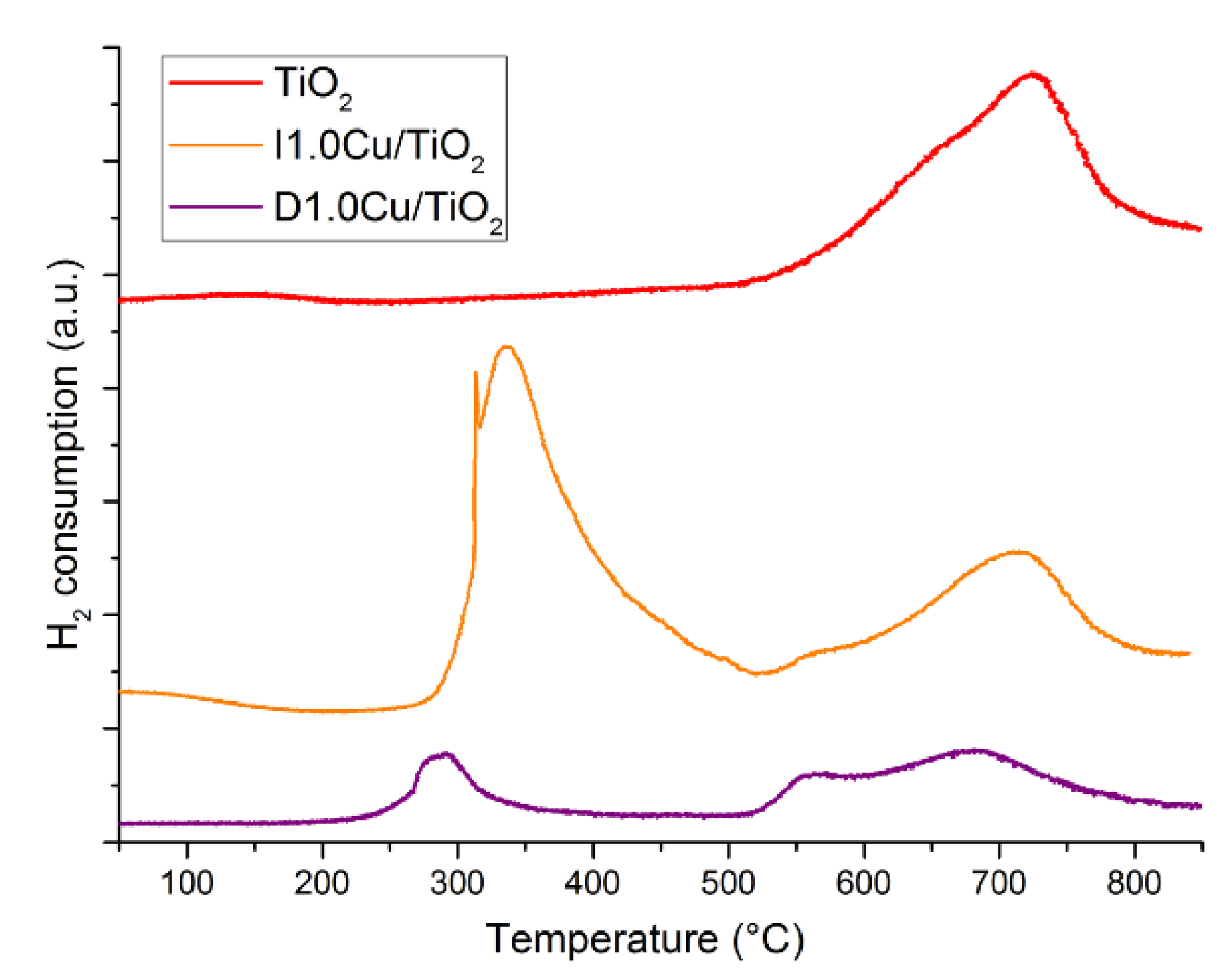
3.2.2. Copper Oxide-TiO2 Nanocomposite
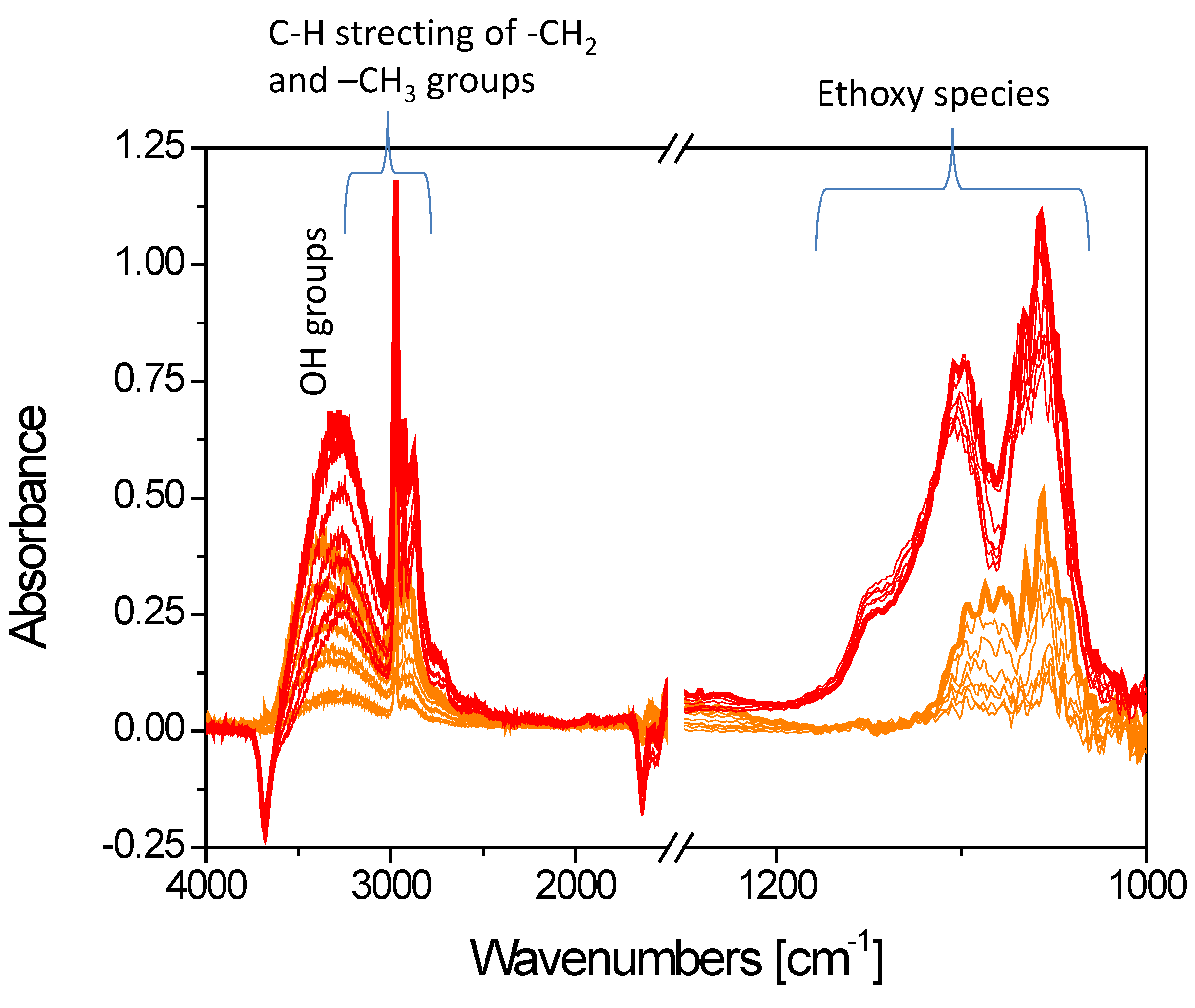
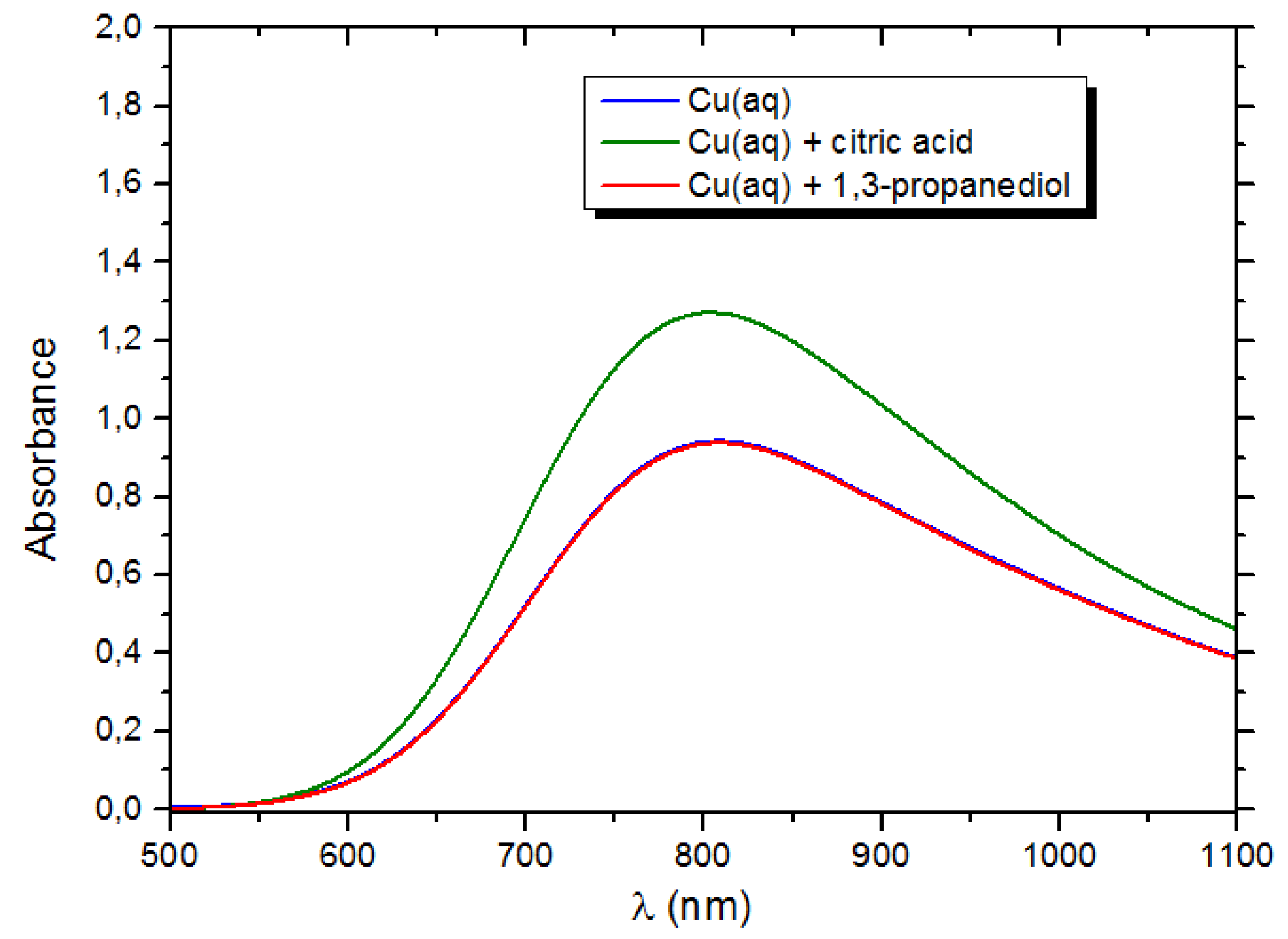
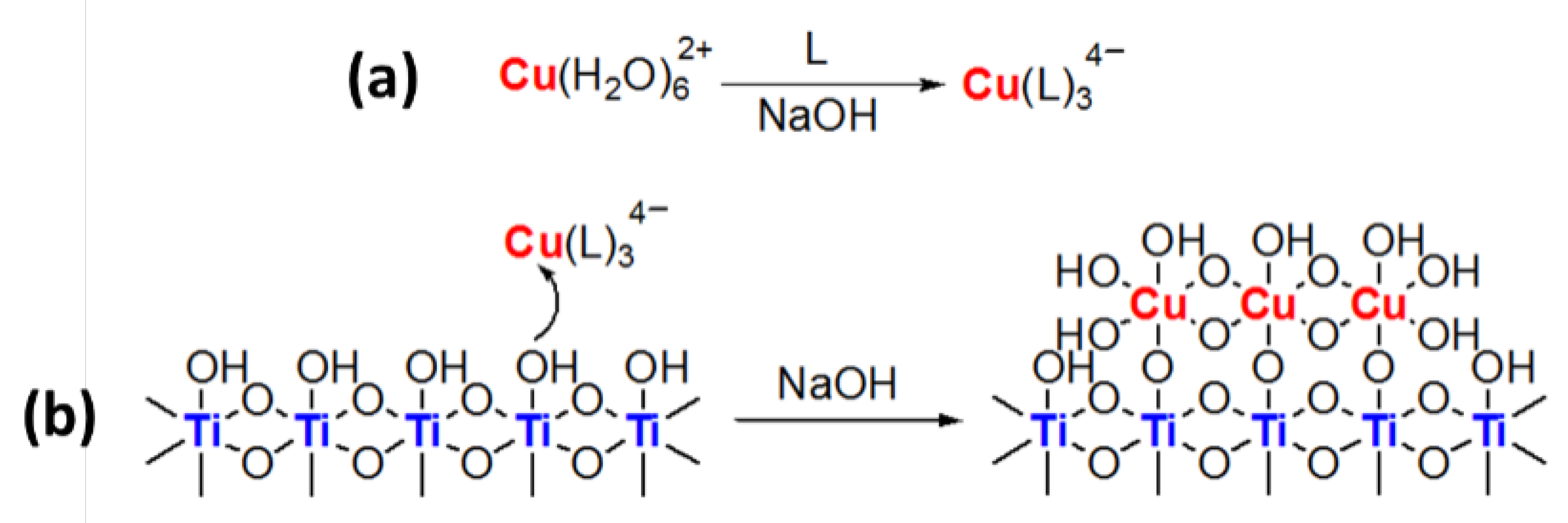
3.2.3. Mechanism of Complex-Precipitation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Szulejk, J.E.; Kumar, P.; Deep, A.; Kim, K.-H. Global warming projections to 2100 using simple CO2 greenhouse gas modeling and comments on CO2 climate sensitivity factor. Atmos. Pollut. Res. 2017, 8, 136–140. [Google Scholar] [CrossRef]
- Kannan, N.; Vakeesan, D. Solar energy for future world: - A review. Renew. Sustain. Energy Rev. 2016, 62, 1092–1105. [Google Scholar] [CrossRef]
- Olivo, A.; Zanardo, D.; Ghedini, E.; Menegazzo, F.; Signoretto, M. Solar Fuels by Heterogeneous Photocatalysis: From Understanding Chemical Bases to Process Development. ChemEngineering 2018, 2, 42. [Google Scholar] [CrossRef]
- Armaroli, N.; Balzani, V. Energy for a Sustainable World, 1st ed.; Wiley-VCH Verlag & Co. KGaA: Weinheim, Germany, 2011; pp. 279–299. ISBN 978-3-527-32540-5. [Google Scholar]
- Fusjishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Sakata, T. Conversion of carbohydrate into hydrogen fuel by a photocatalytic process. Nature 1980, 286, 474–476. [Google Scholar] [CrossRef]
- Rossetti, I. Hydrogen Production by Photoreforming of Renewable Substrates. ISRN Chem. Eng. 2012. [Google Scholar] [CrossRef]
- Contreras, J.L.; Salmones, J.; Colín-Luna, J.A.; Nuño, L.; Quintana, B.; Cordova, I.; Zeifert, B.; Tapia, C.; Fuentes, G.A. Catalysts for H2 production using the ethanol steam reforming (a review). Int. J. Hydrogen Energy 2014, 39, 18835–18853. [Google Scholar] [CrossRef]
- Kousi, K.; Chourdakis, N.; Matralis, H.; Kontarides, D.; Papadopouloua, C.; Verykios, X. Glycerol steam reforming over modified Ni-based catalysts. Appl. Catal. A 2016, 518, 129–141. [Google Scholar] [CrossRef]
- Daskalakis, V.M.; Kondarides, D.I. Efficient production of hydrogen by photo-induced reforming of glycerol at ambient conditions. Catal. Today 2009, 144, 75–80. [Google Scholar] [CrossRef]
- Fu, X.; Wang, X.; Leung, D.Y.C.; Xue, W.; Ding, Z.; Huang, H.; Fu, X. Photocatalytic reforming of glucose over La doped alkali tantalate photocatalysts for H2 production. Catal. Commun. 2010, 12, 184–187. [Google Scholar] [CrossRef]
- Bahruji, H.; Bowker, M.; Davies, P.R.; Pedrono, F. New insights into the mechanism of photocatalytic reforming on Pd/TiO2. Appl. Catal. B 2011, 17, 205–209. [Google Scholar] [CrossRef]
- Ampelli, C.; Genovese, C.; Passalacqua, R.; Perathoner, S.; Centi, G. A gas-phase reactor powered by solar energy and ethanol for H2 production. Appl. Therm. Eng. 2014, 70, 1270–1275. [Google Scholar] [CrossRef]
- Shimura, K.; Kato, S.; Yoshida, T.; Itoh, H.; Hattori, T.; Yoshida, H. Photocatalytic Steam Reforming of Methane over Sodium Tantalate. J. Phys. Chem. C 2010, 114, 3493–3503. [Google Scholar] [CrossRef]
- Chiarello, G.L.; Aguirre, M.H.; Selli, E. Hydrogen production by photocatalytic steam reforming of methanol on noble metal-modified TiO2. J. Catal. 2010, 273, 182–190. [Google Scholar] [CrossRef]
- Taboada, E.; Angurell, I.; Llorca, J. Dynamic photocatalytic hydrogen production from ethanol–water mixtures in an optical fiber honeycomb reactor loaded with Au/TiO2. J. Catal. 2014, 309, 460–467. [Google Scholar] [CrossRef]
- Zabed, H.; Sahu, J.N.; Boyce, A.N.; Faruq, G. Fuel ethanol production from lignocellulosic biomass: An overview on feedstocks and technological approaches. Renew. Sustain. Energy Rev. 2016, 66, 751–774. [Google Scholar] [CrossRef]
- Puga, A.V.; Forneli, A.; García, H.; Corma, A. Production of H2 by Ethanol Photoreforming on Au/TiO2. Adv. Funct. Mater. 2014, 24, 241–248. [Google Scholar] [CrossRef]
- Woutersen, R.A.; Appleman, L.M.; Van Garderen-Hoetmer, A.; Feron, J.V. Inhalation toxicity of acetaldehyde in rats. III. Carcinogenicity study. Toxicology 1986, 41, 213–231. [Google Scholar] [CrossRef]
- Cherubini, F. The biorefinery concept: Using biomass instead of oil for producing energy and chemicals. Energy Convers. Manag. 2010, 15, 1412–1421. [Google Scholar] [CrossRef]
- Weissermel, K.; Arpe, H.-J. Industrial Organic Chemistry, 3rd ed.; Wiley-VCH Verlagsgesellschaft mbH: Weinheim, Germany, 1997; ISBN 3-527-28838-4. [Google Scholar]
- Chae, H.-J.; Kim, T.-W.; Moon, Y.-K.; Kim, H.-K.; Jeong, K.-E.; Kim, C.-U.; Jeong, S.-Y. Butadiene production from bioethanol and acetaldehyde over tantalum oxide-supported ordered mesoporous silica catalysts. Appl. Catal. B 2014, 150–151, 596–604. [Google Scholar] [CrossRef]
- Zhu, Q.; Wang, B.; Tan, T. Conversion of Ethanol and Acetaldehyde to Butadiene over MgO–SiO2Catalysts: Effect of Reaction Parameters and Interaction between MgO and SiO2 on Catalytic Performance. ACS Sustainable Chem. Eng. 2017, 5, 722–733. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, X.; Jia, Y.; Chen, X.; Han, H.; Li, C. Titanium Dioxide-Based Nanomaterials for Photocatalytic Fuel Generations. Chem. Rev. 2014, 114, 9987–10043. [Google Scholar] [CrossRef] [PubMed]
- Pulido Melián, E.; González Díaz, O.; Ortega Méndez, A.; López, C.R.; Nereida Suárez, M.; Doña Rodríguez, J.M.; Navío, J.A.; Fernández Hevia, D.; Pérez Peña, J. Efficient and affordable hydrogen production by water photo-splitting using TiO2-based photocatalysts. Int. J. Hydrogen Energy 2013, 38, 2144–2155. [Google Scholar] [CrossRef]
- Sreethawong, T.; Suzuki, Y.; Yoshikawa, S. Synthesis, characterization, and photocatalytic activity for hydrogen evolution of nanocrystalline mesoporous titania prepared by surfactant-assisted templating sol-gel process. J. Solid State Chem. 2005, 178, 329–338. [Google Scholar] [CrossRef]
- Romero Ocaña, I.; Beltram, A.; Delgado Jaén, J.J.; Adami, G.; Montini, T.; Fornasiero, P. Photocatalytic H2 production by ethanol photodehydrogenation: Effect of anatase/brookite nanocomposites composition. Inorganica Chim. Acta 2015, 431, 197–205. [Google Scholar] [CrossRef]
- Dozzi, M.V.; Chiarello, G.L.; Pedroni, M.; Livraghi, S.; Giamello, E.; Selli, E. High photocatalytic hydrogen production on Cu(II) pre-grafted Pt/TiO2. Appl. Catal. B 2017, 209, 417–428. [Google Scholar] [CrossRef]
- Kondarides, D.I.; Daskalaki, V.M.; Patsoura, A.; Verykios, X.E. Hydrogen Production by Photo-Induced Reforming of Biomass Components and Derivatives at Ambient Conditions. Catal. Lett. 2008, 122, 26–32. [Google Scholar] [CrossRef]
- Wu, G.; Chen, T.; Su, W.; Zhou, G.; Zong, X.; Lei, Z.; Li, C. H2 production with ultra-low CO selectivity via photocatalytic reforming of methanol on Au/TiO2 catalyst. Int. J. Hydrogen Energy 2008, 33, 1243–1251. [Google Scholar] [CrossRef]
- Puga, A.V. Photocatalytic production of hydrogen from biomass-derived feedstocks. Coord. Chem. Rev. 2016, 315, 1–66. [Google Scholar] [CrossRef]
- Menegazzo, F.; Pizzolitto, C.; Ghedini, E.; Di Michele, A.; Cruciani, G.; Signoretto, M. Development of La Doped Ni/CeO2 for CH4/CO2 Reforming. J. Carbon Res. 2018, 4, 60. [Google Scholar] [CrossRef]
- Nagpure, A.S.; Venugopal, A.K.; Lucas, N.; Manikandan, M.; Thirumalaiswamy, R.; Chilukuri, S. Renewable fuels from biomass-derived compounds: Ru-containing hydrotalcites as catalysts for conversion of HMF to 2,5-dimethylfuran. Catal. Sci. Technol. 2015, 5, 1463–1472. [Google Scholar] [CrossRef]
- Sakthivel, S.; Hidalgo, M.C.; Bahnemann, D.W.; Geissen, S.-U.; Murugesan, V.; Vogelpohl, A. A fine route to tune the photocatalytic activity of TiO2. Appl. Catal. B 2006, 63, 31–40. [Google Scholar] [CrossRef]
- Olivo, A.; Trevisan, V.; Ghedini, E.; Pinna, F.; Bianchi, C.L.; Naldoni, A.; Cruciani, G.; Signoretto, M. CO2 photoreduction with water: Catalyst and process investigation. J. CO2 Utiliz. 2015, 12, 86–94. [Google Scholar] [CrossRef]
- Yang, J.; Wang, D.; Han, H.; Li, C. Roles of Cocatalysts in Photocatalysis and Photoelectrocatalysis. Acc. Chem. Res. 2013, 46, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.; Wolf, E.E.; Kamat, P.V. Catalysis with TiO2/Gold Nanocomposites. Effect of Metal Particle Size on the Fermi Level Equilibration. J. Am. Chem. Soc. 2004, 126, 4943–4950. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Hashimoto, K.; Kominami, H. Visible-Light-Induced Hydrogen and Oxygen Formation over Pt/Au/ WO3 Photocatalyst Utilizing Two Types of Photoabsorption Due to Surface Plasmon Resonance and Band-Gap Excitation. J. Am. Chem. Soc. 2014, 136, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Chena, W.-T.; Chana, A.; Sun-Waterhouse, D.; Moriga, T.; Idriss, H.; Waterhouse, G.I.N. Ni/TiO2: A promising low-cost photocatalytic system for solar H2 production from ethanol–water mixtures. J. Catal. 2015, 316, 43–53. [Google Scholar] [CrossRef]
- Sreethawong, T.; Yoshikawa, S. Comparative investigation on photocatalytic hydrogen evolution over Cu-, Pd-, and Au-loaded mesoporous TiO2 photocatalysts. Catal. Commun. 2005, 6, 661–668. [Google Scholar] [CrossRef]
- Zhou, W.; Yin, Z.; Du, Y.; Huang, X.; Zeng, Z.; Fan, Z.; Liu, H.; Wang, J.; Zhang, H. Synthesis of Few-Layer MoS2 Nanosheet-Coated TiO2 Nanobelt Heterostructures for Enhanced Photocatalytic Activities. Small 2013, 9, 140–147. [Google Scholar] [CrossRef]
- Caravaca, A.; Daly, H.; Smith, M.; Mills, A.; Chansai, S.; Hardacre, C. Continuous flow gas phase photoreforming of methanol at elevated reaction temperatures sensitised by Pt/TiO2. React. Chem. Eng. 2016, 1, 649–657. [Google Scholar] [CrossRef]
- Clarizia, L.; Spasiano, D.; Di Somma, I.; Marotta, R.; Andreozzi, R.; Dionysiou, D.D. Copper modified-TiO2 catalysts for hydrogen generation through photoreforming of organics. A short review. Int. J. Hydrogen Energy 2014, 39, 16812–16831. [Google Scholar] [CrossRef]
- Xu, S.; Sun, D. Significant improvement of photocatalytic hydrogen generation rate over TiO2 with deposited CuO. Int. J. Hydrogen Energy 2009, 34, 6096–6104. [Google Scholar] [CrossRef]
- Montini, T.; Gombac, V.; Sordelli, L.; José Delgado, J.; Chen, X.; Adami, G.; Fornasiero, P. Nanostructured Cu/TiO2 Photocatalysts for H2 Production from Ethanol and Glycerol Aqueous Solutions. ChemCatChem 2011, 3, 574–577. [Google Scholar] [CrossRef]
- Yu, J.; Hai, Y.; Jaroniec, M. Photocatalytic hydrogen production over CuO-modified titania. J. Colloid Interface Sci. 2011, 357, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Yoong, L.S.; Chong, F.K.; Dutta, B.K. Development of copper-doped TiO2 photocatalyst for hydrogen production under visible light. Energy 2009, 34, 1652–1661. [Google Scholar] [CrossRef]
- Chen, W.-T.; Jovic, V.; Sun-Waterhouse, D.; Idriss, H.; Waterhouse, G.I.N. The role of CuO in promoting photocatalytic hydrogen production over TiO2. Int. J. Hydrogen Energy 2013, 38, 15036–15048. [Google Scholar] [CrossRef]
- Bandara, J.; Udawatta, C.P.K.; Rajapakse, C.S.K. Highly stable CuO incorporated TiO2 catalyst for photocatalytic hydrogen production from H2O. Photochem. Photobiol. Sci. 2005, 4, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Lalitha, K.; Sadanandam, G.; Kumari, V.D.; Subrahmanyam, M.; Sreedhar, B.; Hebalkar, N.Y. Highly Stabilized and Finely Dispersed Cu2O/TiO2: A Promising Visible Sensitive Photocatalyst for Continuous Production of Hydrogen from Glycerol: Water Mixtures. J. Phys. Chem. C 2010, 114, 22181–22189. [Google Scholar] [CrossRef]
- Olivo, A.; Ghedini, E.; Pascalicchio, P.; Manzoli, M.; Cruciani, G.; Signoretto, M. Sustainable carbon dioxide photoreduction by a cooperative effect of reactor design and titania metal promotion. Catalysts 2018, 8, 41. [Google Scholar] [CrossRef]
- Wu, N.-L.; Lee, M.-S. Enhanced TiO2 photocatalysis by Cu in hydrogen production from aqueous methanol solution. Int. J. Hydrogen Energy 2004, 29, 1601–1605. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Kubelka, P.; Munk, F. Ein beitrag zur optik der farbanstriche. Zeitschrift Technische Physik 1931, 12, 593–601. [Google Scholar]
- NIST X-ray Photoelectron Spectroscopy Database—NIST Standard ReferenceDatabase 20, Version 4.1. Available online: http://srdata.nist.gov/xps/Default.aspx (accessed on 31 August 2019).
- Comas, J.; Mariño, F.; Laborde, M.; Amadeo, N. Bio-ethanol steam reforming on Ni/Al2O3 catalyst. Chem. Eng. J. 2004, 98, 61–68. [Google Scholar] [CrossRef]
- Braslavsky, S.E.; Braun, A.M.; Cassano, A.E.; Emeline, A.V.; Litter, M.I.; Palmisano, L.; Parmon, V.N.; Serpone, N. Glossary of terms used in photocatalysis and radiation catalysis (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 931–1014. [Google Scholar] [CrossRef]
- Kudo, A.; Miskei, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, V.; Olivo, A.; Pinna, F.; Signoretto, M.; Vindigni, F.; Cerrato, G.; Bianchi, C.L. C-N/TiO2 photocatalysts: Effect of co-doping on the catalytic performance under visible light. Appl. Catal. B 2014, 160–161, 152–160. [Google Scholar] [CrossRef]
- Ilda, Y.; Ozaki, S. Grain Growth and Phase Transformation of Titanium Oxide During Calcination. J. Am. Chem. Soc. 1961, 44, 120–127. [Google Scholar]
- Hanaor, D.A.H.; Sorrell, C.C. Review of the anatase to rutile phase transformation. J. Mater. Sci. 2011, 46, 855–874. [Google Scholar] [CrossRef]
- Boccuzzi, F.; Chiorino, A.; Martra, G.; Gargano, M.; Ravasio, N.; Carrozzini, B. Preparation, Characterization, and Activity of Cu/TiO2 Catalysts I. Influence of the Preparation Method on the Dispersion of Copper in Cu/TiO2. J. Catal. 1997, 165, 129–139. [Google Scholar] [CrossRef]
- Yashnik, S.; Ismagilov, Z.; Anufrienko, V. Catalytic properties and electronic structure of copper ions in Cu-ZSM-5. Catal. Today 2005, 110, 310–322. [Google Scholar] [CrossRef]
- Bravo-Suárez, J.J.; Subramaniam, B.; Chaudhari, R.V. Ultraviolet−Visible Spectroscopy and Temperature-Programmed Techniques as Tools for Structural Characterization of Cu in CuMgAlOx Mixed Metal Oxides. J. Phys. Chem. C 2012, 116, 18207–18221. [Google Scholar] [CrossRef]
- Hadjiivanov, K.I.; Vayssilov, G.N. Characterization of Oxide Surfaces and Zeolites by Carbon Monoxide as an IR Probe Molecule. Adv. Catal. 2002, 47, 307–511. [Google Scholar]
- Martra, G. Lewis acid and base sites at the surface of microcrystalline TiO2 anatase: relationships between surface morphologyand chemical behaviour. Appl. Catal. A 2000, 200, 275–285. [Google Scholar] [CrossRef]
- Sola, A.C.; Garzón Sousa, D.; Araña, J.; González Díaz, O.; Doña Rodríguez, J.M.; Ramírez de la Piscina, P.; Homs, N. Differences in the vapour phase photocatalytic degradation of ammonia and ethanol in the presence of water as a function of TiO2 characteristics and the presence of O2. Catal. Today 2016, 266, 53–61. [Google Scholar] [CrossRef]
- Natal-Santiago, M.A.; Dumesic, J.A. Microcalorimetric, FTIR, and DFT Studies of the Adsorption of Methanol, Ethanol, and 2,2,2-Trifluoroethanol on Silica. J. Catal. 1998, 175, 252–268. [Google Scholar] [CrossRef]
- Mazzoldi, P.; Cavaccale, F.; Cattaruzza, E.; Chakraborty, P.; Tramontin, L.; Boscolo-Boscoletto, A.; Bertoncello, R.; Trivillin, F.; Battaglin, G.; Arnold, G.W. Colloid formation in copper-implanted fused silica and silicate glasses. Nucl. Instrum. Methods Phys. Res. B 1994, 91, 505–509. [Google Scholar] [CrossRef]
- Iijima, Y.; Niimura, N.; Hiraoka, K. Prevention of the Reduction of CuO during X-ray Photoelectron Spectroscopy Analysis. Surf. Interface Anal. 1996, 24, 193–197. [Google Scholar] [CrossRef]
- Bowker, M.; James, D.; Stone, P.; Bennett, R.; Perkins, N.; Millard, L.; Greaves, J.; Dickinson, A. Catalysis at the metal-support interface: exemplified by the photocatalytic reforming of methanol on Pd/TiO2. J. Catal. 2003, 217, 427–433. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Titania | Copper-Loading (%) and Introduction Technique | Label |
|---|---|---|
| Benchmark (P25) | / | P25 |
| Lab-made TiO2 | / | TiO2 |
| Benchmark (P25) | 0.5%‒wetness impregnation | I0.5Cu/P25 |
| Benchmark (P25) | 1.0%‒wetness impregnation | I1.0Cu/P25 |
| Benchmark (P25) | 0.5%‒complex precipitation with 1,3-propanediol | D0.5Cu/P25 |
| Benchmark (P25) | 1.0%‒complex precipitation with 1,3-propanediol | D1.0Cu/P25 |
| Benchmark (P25) | 0.5%‒complex precipitation with citric acid | C0.5Cu/P25 |
| Benchmark (P25) | 1.0%‒complex precipitation with citric acid | C1.0Cu/P25 |
| Lab-made TiO2 | 1.0%‒wetness impregnation | I1.0Cu/TiO2 |
| Lab-made TiO2 | 1.0%‒complex precipitation with 1,3-propanediol | D1.0Cu/TiO2 |
| Sample | AQY (%) | TOF (mmol∙g−1∙h−1) |
|---|---|---|
| I0.5Cu/P25 | 13 ± 1 | 4.4 ± 0.5 |
| I1.0Cu/P25 | 17 ± 2 | 5.5 ± 0.6 |
| D0.5Cu/P25 | 19.0 ± 0.3 | 6.2 ± 0.1 |
| D1.0Cu/P25 | 21 ± 1 | 7.1 ± 0.2 |
| C0.5Cu/P25 | 21 ± 1 | 6.8 ± 0.4 |
| C1.0Cu/P25 | 23 ± 2 | 7.5 ± 0.6 |
| Sample | Cu (%) | SBET (m2/g) |
|---|---|---|
| P25 | - | 40 |
| I0.5Cu/P25 | 0.41 | 41 |
| I1.0Cu/P25 | 0.43 | 42 |
| D0.5Cu/P25 | 0.43 | 44 |
| D1.0Cu/P25 | 0.93 | 42 |
| C0.5Cu/P25 | 0.99 | 45 |
| C1.0Cu/P25 | 0.98 | 43 |
| I1.0Cu/P25 | D1.0Cu/P25 | C1.0Cu/P25 | |
|---|---|---|---|
| Cu | 2.3 | 2.4 | 2.8 |
| Ti | 27 | 23 | 24 |
| O | 63 | 57 | 64 |
| C | 7 | 18 | 9 |
| Sample | Cu (%) | SBET (m2/g) |
|---|---|---|
| TiO2 | - | 101 |
| I1.0Cu/TiO2 | 0.89 | 71 |
| D1.0Cu/TiO2 | 0.94 | 101 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanardo, D.; Ghedini, E.; Menegazzo, F.; Cattaruzza, E.; Manzoli, M.; Cruciani, G.; Signoretto, M. Titanium Dioxide-Based Nanocomposites for Enhanced Gas-Phase Photodehydrogenation. Materials 2019, 12, 3093. https://doi.org/10.3390/ma12193093
Zanardo D, Ghedini E, Menegazzo F, Cattaruzza E, Manzoli M, Cruciani G, Signoretto M. Titanium Dioxide-Based Nanocomposites for Enhanced Gas-Phase Photodehydrogenation. Materials. 2019; 12(19):3093. https://doi.org/10.3390/ma12193093
Chicago/Turabian StyleZanardo, Danny, Elena Ghedini, Federica Menegazzo, Elti Cattaruzza, Maela Manzoli, Giuseppe Cruciani, and Michela Signoretto. 2019. "Titanium Dioxide-Based Nanocomposites for Enhanced Gas-Phase Photodehydrogenation" Materials 12, no. 19: 3093. https://doi.org/10.3390/ma12193093
APA StyleZanardo, D., Ghedini, E., Menegazzo, F., Cattaruzza, E., Manzoli, M., Cruciani, G., & Signoretto, M. (2019). Titanium Dioxide-Based Nanocomposites for Enhanced Gas-Phase Photodehydrogenation. Materials, 12(19), 3093. https://doi.org/10.3390/ma12193093