Synthesis and Characterization of Chitosan/Reduced Graphene Oxide Hybrid Composites



Abstract
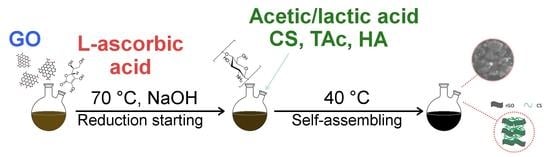
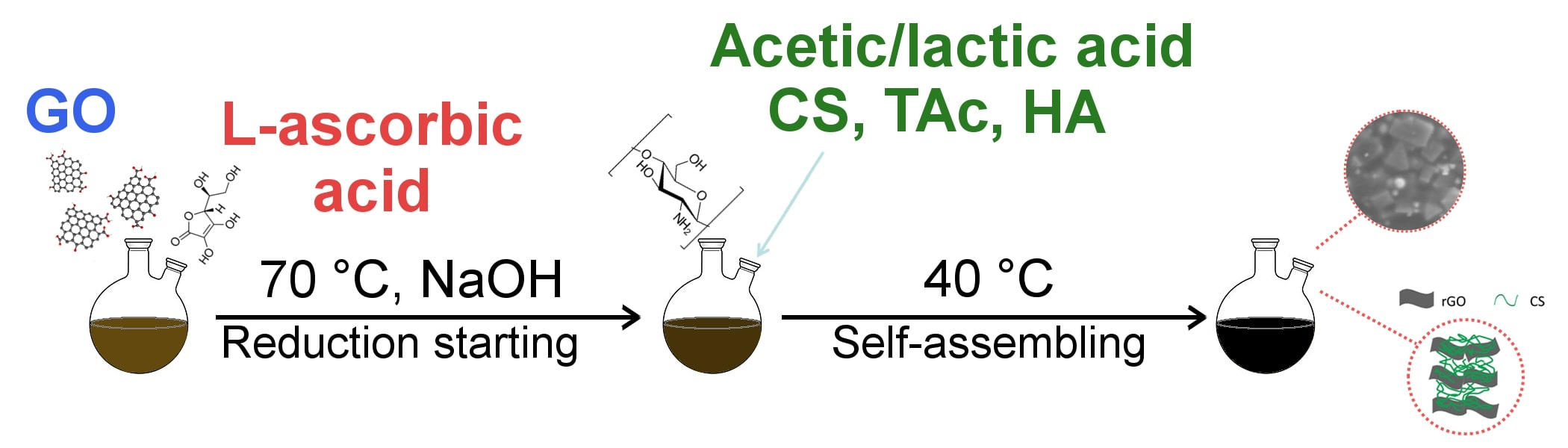{kind=link}
{kind=link}
{kind=link}
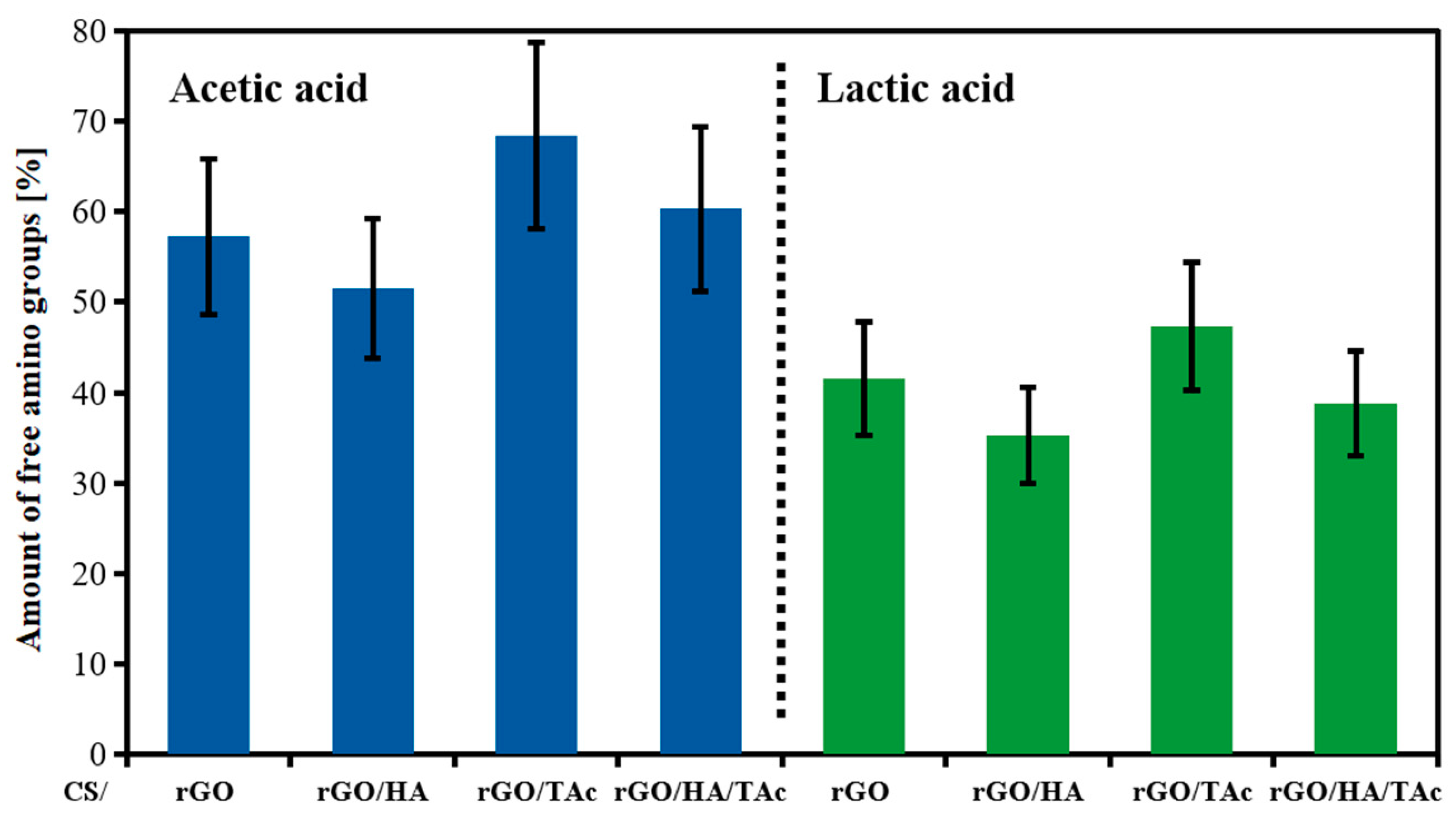{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Graphene Oxide (GO) and Reduced Graphene Oxide (rGO)
2.3. Fabrication of Chitosan/rGO Composites
2.4. Characterization
2.4.1. X-ray Photoelectron Spectroscopy (XPS)
2.4.2. X-ray Diffractometry (XRD)
2.4.3. Attenuated Total Reflection Fourier-Transform Infrared Spectroscopy (ATR-FTIR)
2.4.4. Mechanical Properties
2.4.5. In Vitro Degradation
2.4.6. Scanning Electron Microscopy (SEM)
2.4.7. In Vitro Bioactivity Test
2.4.8. Wettability
2.4.9. Determination of the Number of Free Amino Groups
2.4.10. Cytocompatibility
3. Results
3.1. Chemical Reduction of GO
3.2. Characterization of Nanocomposite Hydrogels
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.R.; Li, Y.-C.; Jang, H.L.; Khoshakhlagh, P.; Akbari, M.; Nasajpour, A.; Zhang, Y.S.; Tamayol, A.; Khademhosseini, A. Graphene-based materials for tissue engineering. Adv. Drug Deliv. Rev. 2016, 105, 255–274. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Shin, D.; Ryoo, S.-R.; Min, D.-H.; Kim, Y.-K.; Hong, B.H. Biomedical Applications of Graphene and Graphene Oxide. Accounts Chem. Res. 2013, 46, 2211–2224. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Kim, J.; Huang, J. Material Processing of Chemically Modified Graphene: Some Challenges and Solutions. Accounts Chem. Res. 2013, 46, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Feng, L.; Hong, H.; Cai, W.; Liu, Z. Preparation and functionalization of graphene nanocomposites for biomedical applications. Nat. Protoc. 2013, 8, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Cheng, H.-M.; Enoki, T.; Gogotsi, Y.; Hurt, R.H.; Koratkar, N.; Kyotani, T.; Monthioux, M.; Park, C.R.; Tascón, J.M.; et al. All in the graphene family—A recommended nomenclature for two-dimensional carbon materials. Carbon 2013, 65, 1–6. [Google Scholar] [CrossRef]
- Justin, R.; Chen, B. Strong and conductive chitosan–reduced graphene oxide nanocomposites for transdermal drug delivery. J. Mater. Chem. B 2014, 2, 3759. [Google Scholar] [CrossRef]
- Kosowska, K.; Domalik-Pyzik, P.; Chłopek, J. Graphene oxide and green-synthesized reduced graphene oxide in chitosan-based nanocomposites. Eng. Biomater. 2017, 142, 11–16. [Google Scholar]
- Kosowska, K.; Domalik-Pyzik, P.; Nocuń, M.; Chłopek, J. Chitosan and graphene oxide/reduced graphene oxide hybrid nanocomposites—Evaluation of physicochemical properties. Mater. Chem. Phys. 2018, 216, 28–36. [Google Scholar] [CrossRef]
- Wang, L.; Lu, X.; Lei, S.; Song, Y. Graphene-based polyaniline nanocomposites: Preparation, properties and applications. J. Mater. Chem. A 2014, 2, 4491–4509. [Google Scholar] [CrossRef]
- Tabish, T.A.; Chabi, S.; Ali, M.; Xia, Y.; Jabeen, F.; Zhang, S. Tracing the Bioavailability of Three-Dimensional Graphene Foam in Biological Tissues. Materials 2017, 10, 336. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, S.; Baheiraei, N.; Mohseni, M.; Razavi, M.; Ghaderi, A.; Azizi, B.; Rabiee, N.; Karimi, M. Three-dimensional graphene foam as a conductive scaffold for cardiac tissue engineering. J. Biomater. Appl. 2019, 34, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Tabish, T.; Pranjol, M.Z.I.; Hayat, H.; Rahat, A.A.M.; Abdullah, T.M.; Whatmore, J.L.; Zhang, S. In vitro toxic effects of reduced graphene oxide nanosheets on lung cancer cells. Nanotechnology 2017, 28, 504001. [Google Scholar] [CrossRef] [PubMed]
- Lategan, K.; Alghadi, H.; Bayati, M.; De Cortalezzi, M.F.; Pool, E. Effects of Graphene Oxide Nanoparticles on the Immune System Biomarkers Produced by RAW 264.7 and Human Whole Blood Cell Cultures. Nanomaterials 2018, 8, 125. [Google Scholar] [CrossRef]
- Tabish, T.A.; Zhang, S.; Winyard, P.G. Developing the next generation of graphene-based platforms for cancer therapeutics: The potential role of reactive oxygen species. Redox Biol. 2018, 15, 34–40. [Google Scholar] [CrossRef]
- Ou, L.; Song, B.; Liang, H.; Liu, J.; Feng, X.; Deng, B.; Sun, T.; Shao, L. Toxicity of graphene-family nanoparticles: A general review of the origins and mechanisms. Part. Fibre Toxicol. 2016, 13, 57. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, F.; Wang, S.; Ma, J.; Xu, M.; Gao, M.; Liu, R.; Chen, W.; Liu, S. Reduction of graphene oxide alters its cyto-compatibility towards primary and immortalized macrophages. Nanoscale 2018, 10, 14637–14650. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, H.; Shen, G.; Cheng, P.; Zhang, J.; Guo, S. Reduction of graphene oxide via l -ascorbic acid. Chem. Commun. 2010, 46, 1112–1114. [Google Scholar] [CrossRef]
- Upadhyay, R.; Soin, N.; Bhattacharya, G.; Saha, S.; Barman, A.; Roy, S. Grape extract assisted green synthesis of reduced graphene oxide for water treatment application. Mater. Lett. 2015, 160, 355–358. [Google Scholar] [CrossRef]
- Liu, C.; Liu, H.; Zhang, K.; Dou, M.; Pan, B.; He, X.; Lu, C. Partly reduced graphene oxide aerogels induced by proanthocyanidins for efficient dye removal. Bioresour. Technol. 2019, 282, 148–155. [Google Scholar] [CrossRef]
- Muthoosamy, K.; Bai, R.G.; Abubakar, I.B.; Sudheer, S.M.; Lim, H.N.; Loh, H.-S.; Huang, N.M.; Chia, C.H.; Manickam, S.; Hongngee, L.; et al. Exceedingly biocompatible and thin-layered reduced graphene oxide nanosheets using an eco-friendly mushroom extract strategy. Int. J. Nanomed. 2015, 10, 1505–1519. [Google Scholar]
- Diekjürgen, D.; Grainger, D.W.; Diekjrgen, D. Polysaccharide matrices used in 3D in vitro cell culture systems. Biomaterials 2017, 141, 96–115. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Younes, I.; Rinaudo, M. Chitin and Chitosan Preparation from Marine Sources. Structure, Properties and Applications. Mar. Drugs 2015, 13, 1133–1174. [Google Scholar] [CrossRef] [PubMed]
- Domalik-Pyzik, P.; Chłopek, J.; Pielichowska, K. Chitosan-Based Hydrogels: Preparation, Properties, and Applications. In Polymers and Polymeric Composites: A Reference Series; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2019; pp. 1665–1693. [Google Scholar]
- Croisier, F.; Jérôme, C. Chitosan-based biomaterials for tissue engineering. Eur. Polym. J. 2013, 49, 780–792. [Google Scholar] [CrossRef]
- Prasadh, S.; Suresh, S.; Wong, R. Osteogenic Potential of Graphene in Bone Tissue Engineering Scaffolds. Materials 2018, 11, 1430. [Google Scholar] [CrossRef] [PubMed]
- Bohner, M.; Lemaître, J. Can bioactivity be tested in vitro with SBF solution? Biomaterials 2009, 30, 2175–2179. [Google Scholar] [CrossRef] [PubMed]
- Mezzetta, A.; Guazzelli, L.; Chiappe, C. Access to cross-linked chitosans by exploiting CO 2 and the double solvent-catalytic effect of ionic liquids. Green Chem. 2017, 19, 1235–1239. [Google Scholar] [CrossRef]
- Posadowska, U.; Brzychczy-Wloch, M.; Pamula, E. Injectable gellan gum-based nanoparticles-loaded system for the local delivery of vancomycin in osteomyelitis treatment. J. Mater. Sci. Mater. Med. 2016, 27, 9. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Shi, X.; Ji, A.; Zhou, C.; Cui, Y. Fabrication and Characteristics of Reduced Graphene Oxide Produced with Different Green Reductants. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, Z.; Yin, J. Facile Synthesis of Soluble Graphene via a Green Reduction of Graphene Oxide in Tea Solution and Its Biocomposites. ACS Appl. Mater. Interfaces 2011, 3, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, J.; Liu, C.; Pan, B.; Kim, N.H.; Lee, J.H. Differently-charged graphene-based multilayer films by a layer-by-layer approach for oxygen gas barrier application. Compos. Part B: Eng. 2018, 155, 391–396. [Google Scholar] [CrossRef]
- Depan, D.; Girase, B.; Shah, J.; Misra, R. Structure–process–property relationship of the polar graphene oxide-mediated cellular response and stimulated growth of osteoblasts on hybrid chitosan network structure nanocomposite scaffolds. Acta Biomater. 2011, 7, 3432–3445. [Google Scholar] [CrossRef] [PubMed]
- Justin, R.; Chen, B. Characterisation and drug release performance of biodegradable chitosan–graphene oxide nanocomposites. Carbohydr. Polym. 2014, 103, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Gheisari, H.; Karamian, E.; Abdellahi, M. A novel hydroxyapatite–Hardystonite nanocomposite ceramic. Ceram. Int. 2015, 41, 5967–5975. [Google Scholar] [CrossRef]
- Rubentheren, V.; Ward, T.A.; Chee, C.Y.; Tang, C.K. Processing and analysis of chitosan nanocomposites reinforced with chitin whiskers and tannic acid as a crosslinker. Carbohydr. Polym. 2015, 115, 379–387. [Google Scholar] [CrossRef]
- Cervera, M.F.; Heinämäki, J.; de la Paz, N.; López, O.; Maunu, S.L.; Virtanen, T.; Hatanpää, T.; Antikainen, O.; Nogueira, A.; Fundora, J.; et al. Effects of Spray Drying on Physicochemical Properties of Chitosan Acid Salts. AAPS PharmSciTech 2011, 12, 637–649. [Google Scholar] [CrossRef]
- Ogawa, K.; Yui, T.; Okuyama, K. Three D structures of chitosan. Int. J. Biol. Macromol. 2004, 34, 1–8. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosowska, K.; Domalik-Pyzik, P.; Krok-Borkowicz, M.; Chłopek, J. Synthesis and Characterization of Chitosan/Reduced Graphene Oxide Hybrid Composites. Materials 2019, 12, 2077. https://doi.org/10.3390/ma12132077
Kosowska K, Domalik-Pyzik P, Krok-Borkowicz M, Chłopek J. Synthesis and Characterization of Chitosan/Reduced Graphene Oxide Hybrid Composites. Materials. 2019; 12(13):2077. https://doi.org/10.3390/ma12132077
Chicago/Turabian StyleKosowska, Karolina, Patrycja Domalik-Pyzik, Małgorzata Krok-Borkowicz, and Jan Chłopek. 2019. "Synthesis and Characterization of Chitosan/Reduced Graphene Oxide Hybrid Composites" Materials 12, no. 13: 2077. https://doi.org/10.3390/ma12132077
APA StyleKosowska, K., Domalik-Pyzik, P., Krok-Borkowicz, M., & Chłopek, J. (2019). Synthesis and Characterization of Chitosan/Reduced Graphene Oxide Hybrid Composites. Materials, 12(13), 2077. https://doi.org/10.3390/ma12132077