Macroporous Polymer–Protein Hybrid Materials for Antibody Purification by Combination of Reactive Gelation and Click-Chemistry



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Synthesis of 4-Vinylbenzyl Azide (VBA)
2.3. Latex Synthesis
2.4. Latex Swelling
2.5. High-Shear Destabilization
2.6. Synthesis of Cluster-Epoxy
2.7. Synthesis of Cluster-NHS
2.8. Protein A Immobilization
2.9. Methods
2.10. Quantification of Immobilized Protein A and Static Binding Capacity
2.11. Breakthrough Curves & Dynamic Binding Capacity
3. Results and Discussion
3.1. Synthesis of Core-Shell Nanoparticles
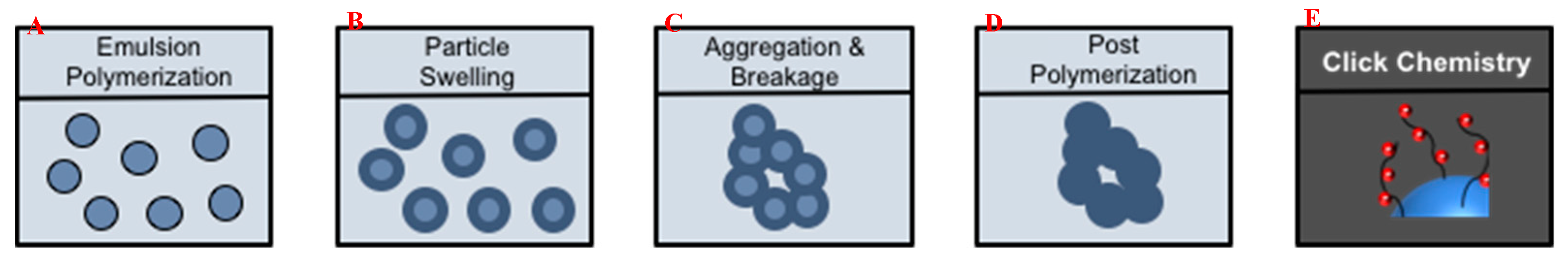
3.2. Fabrication of (Macro)Porous Microclusters from Polymer Nanoparticles
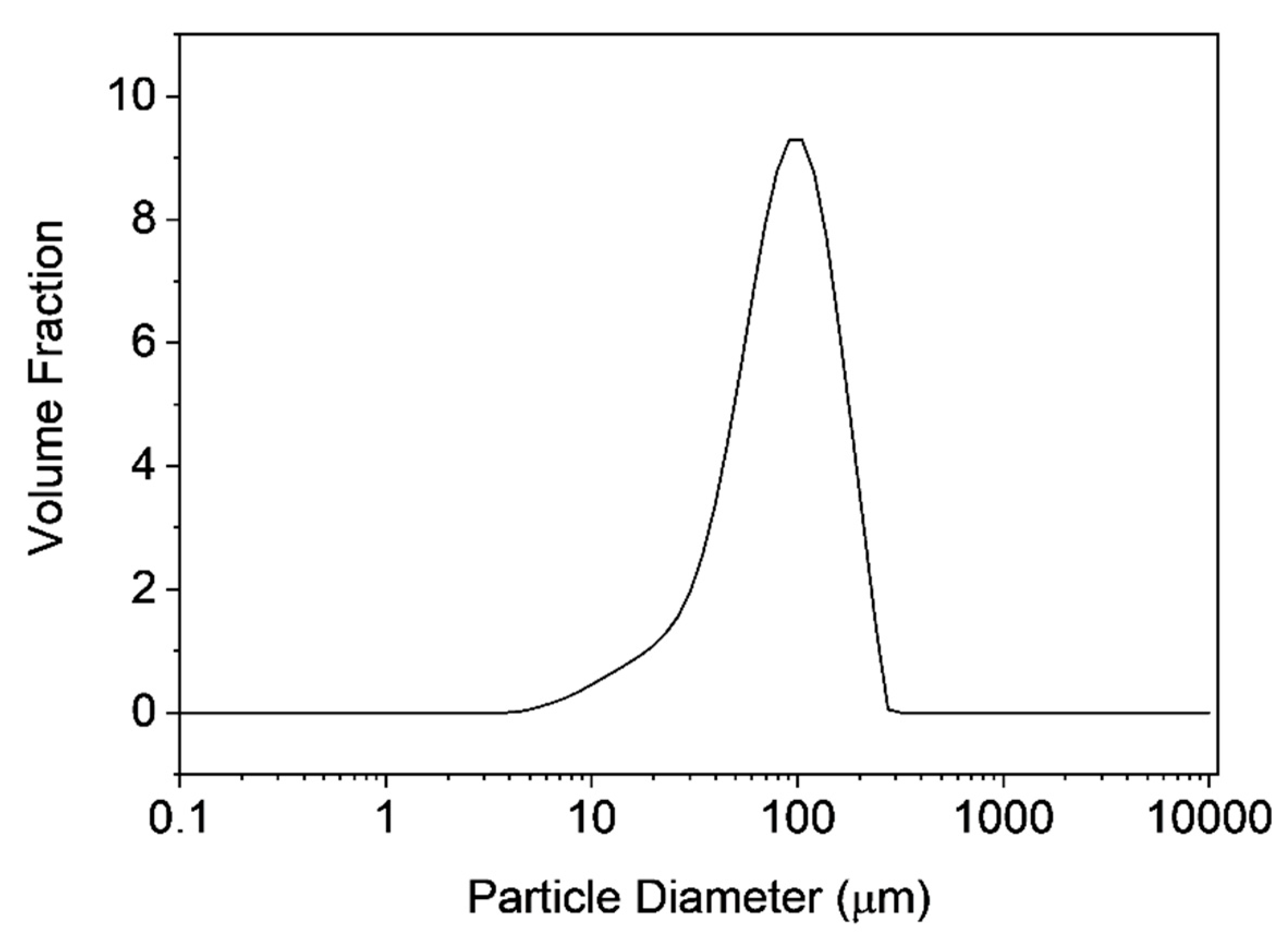
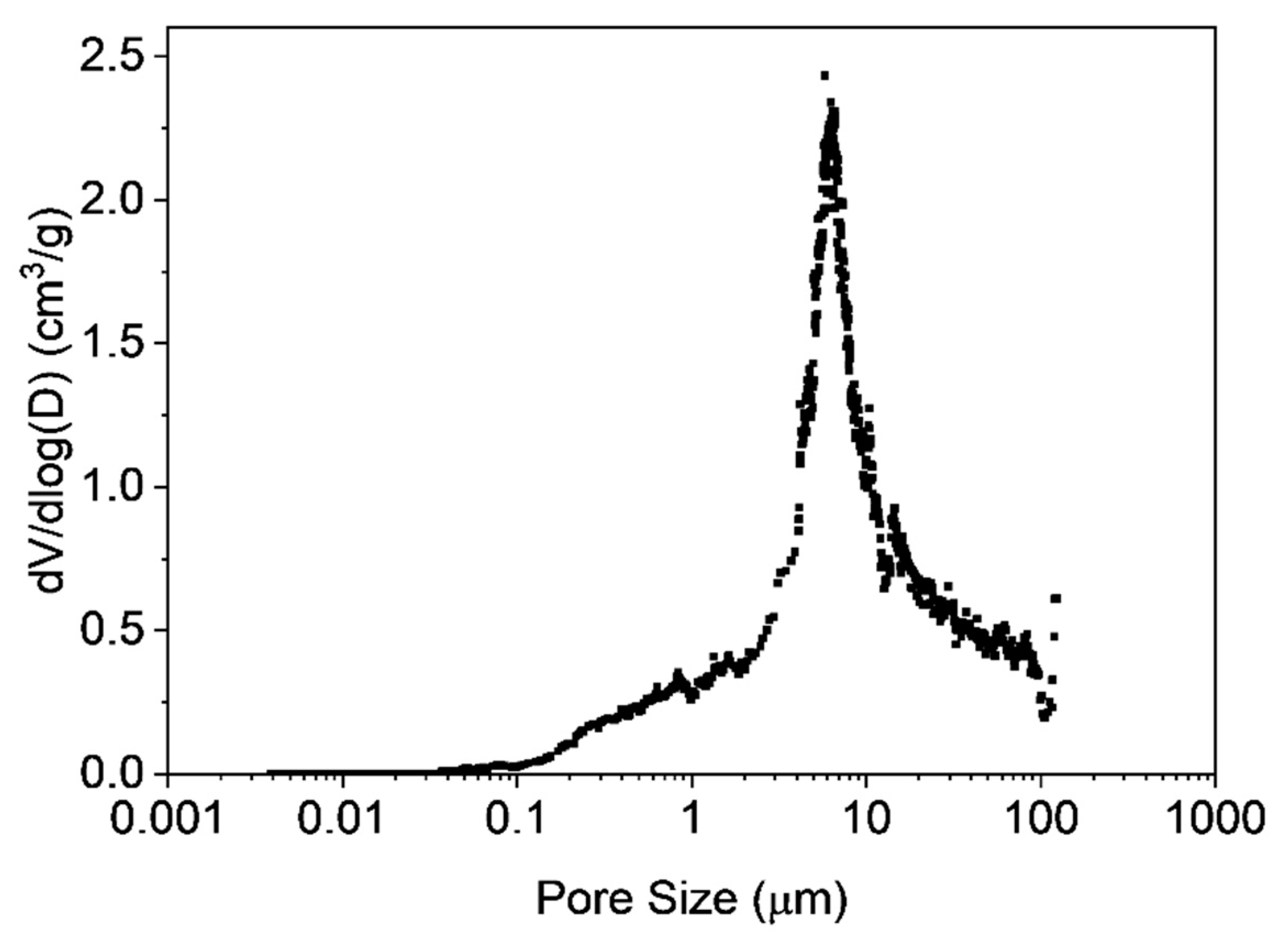
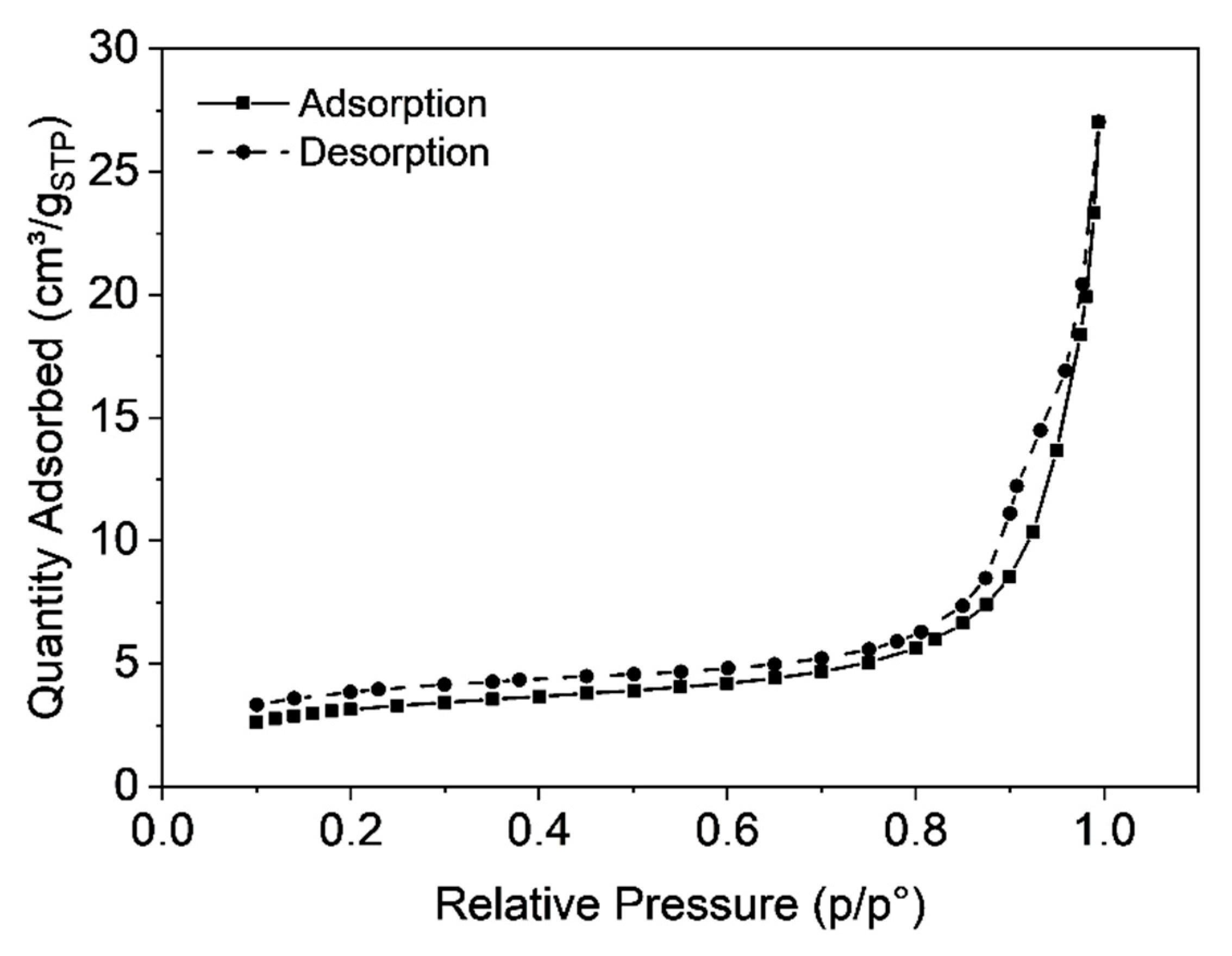
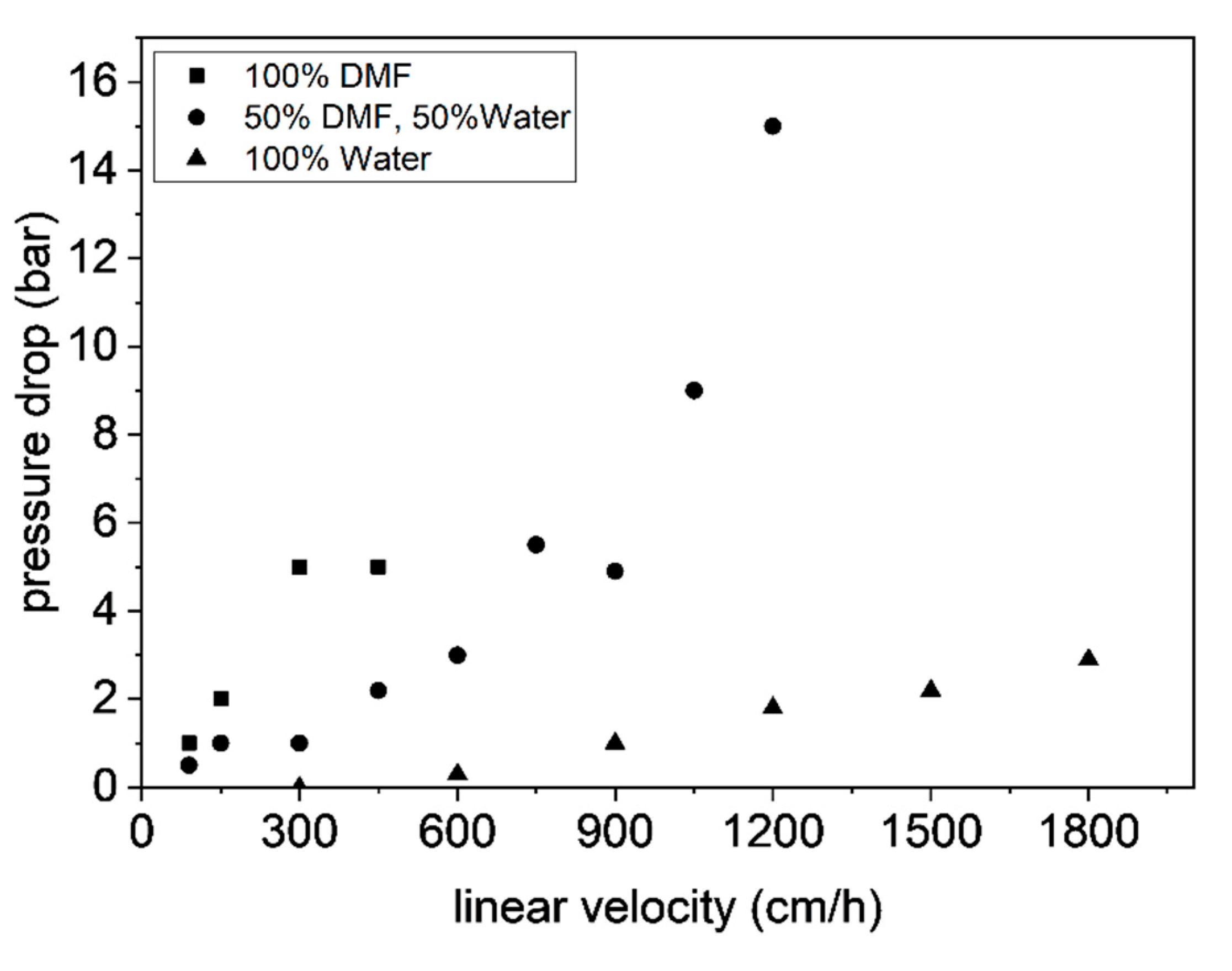
3.3. Characterization of Microclusters
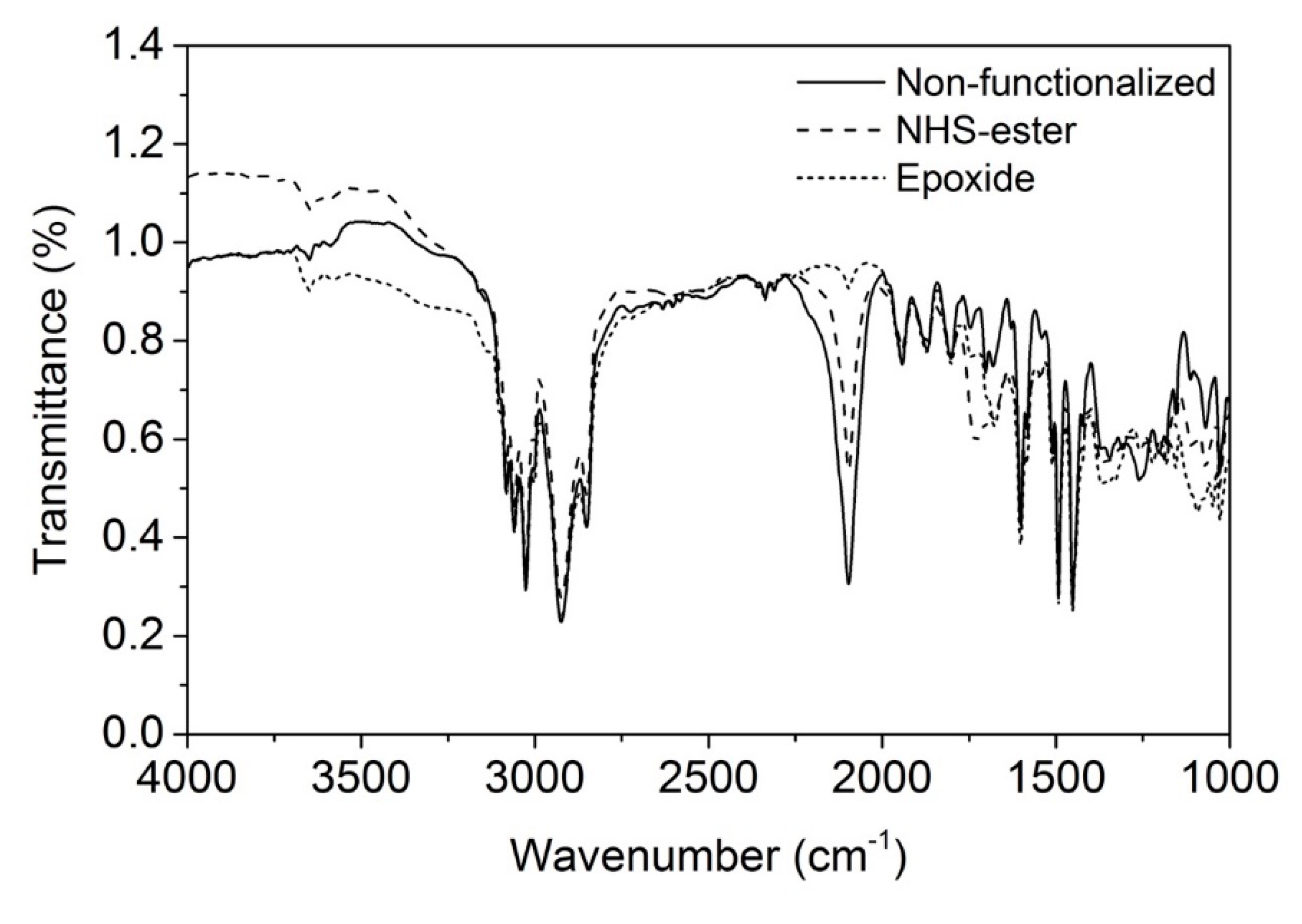
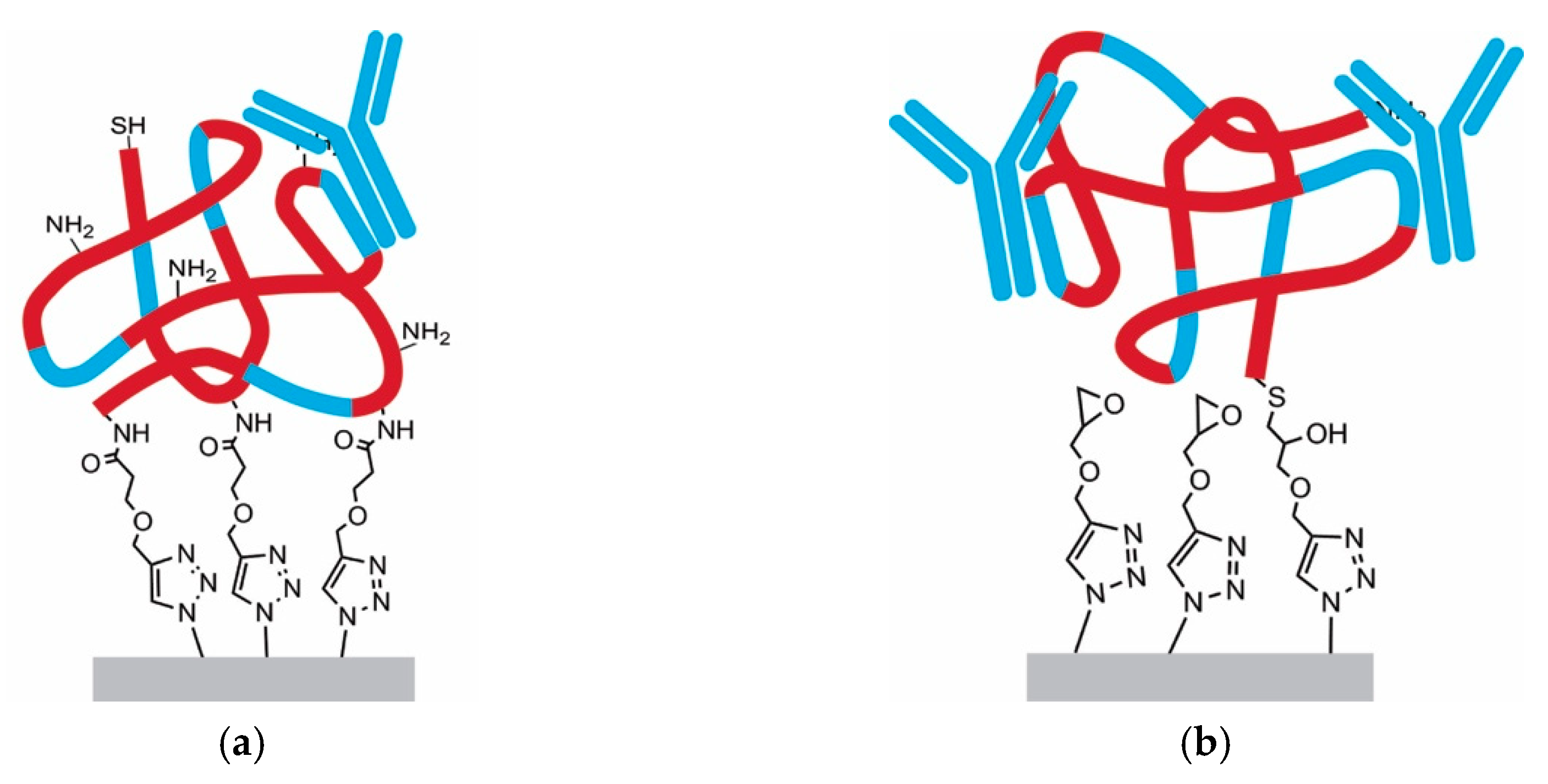
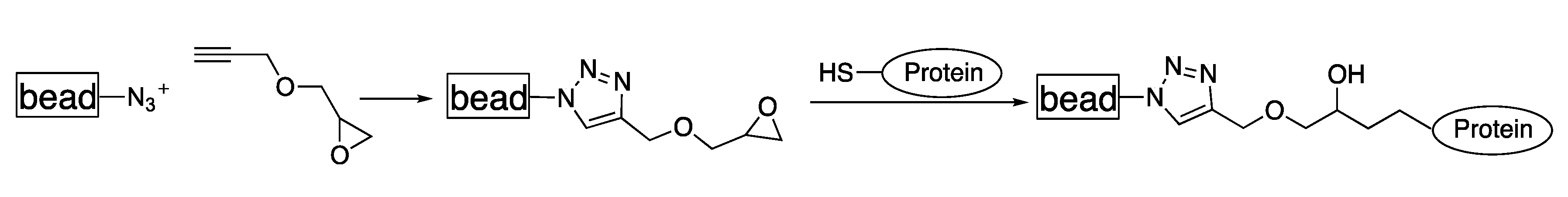
3.4. Functionalization by Click-Chemistry & Protein Immobilization
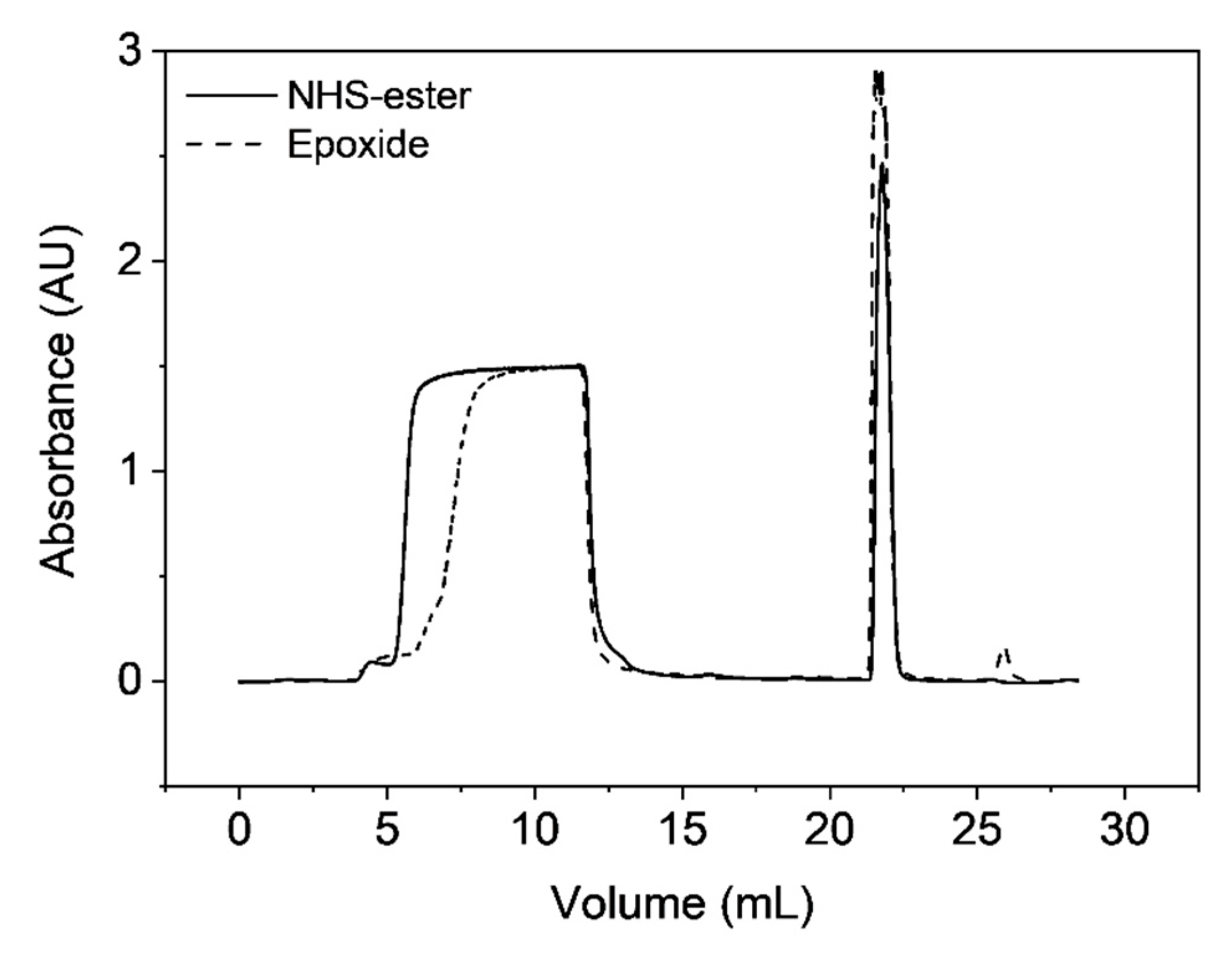
3.5. Single-Point Attachment by Thiol-Epoxide Click Chemistry
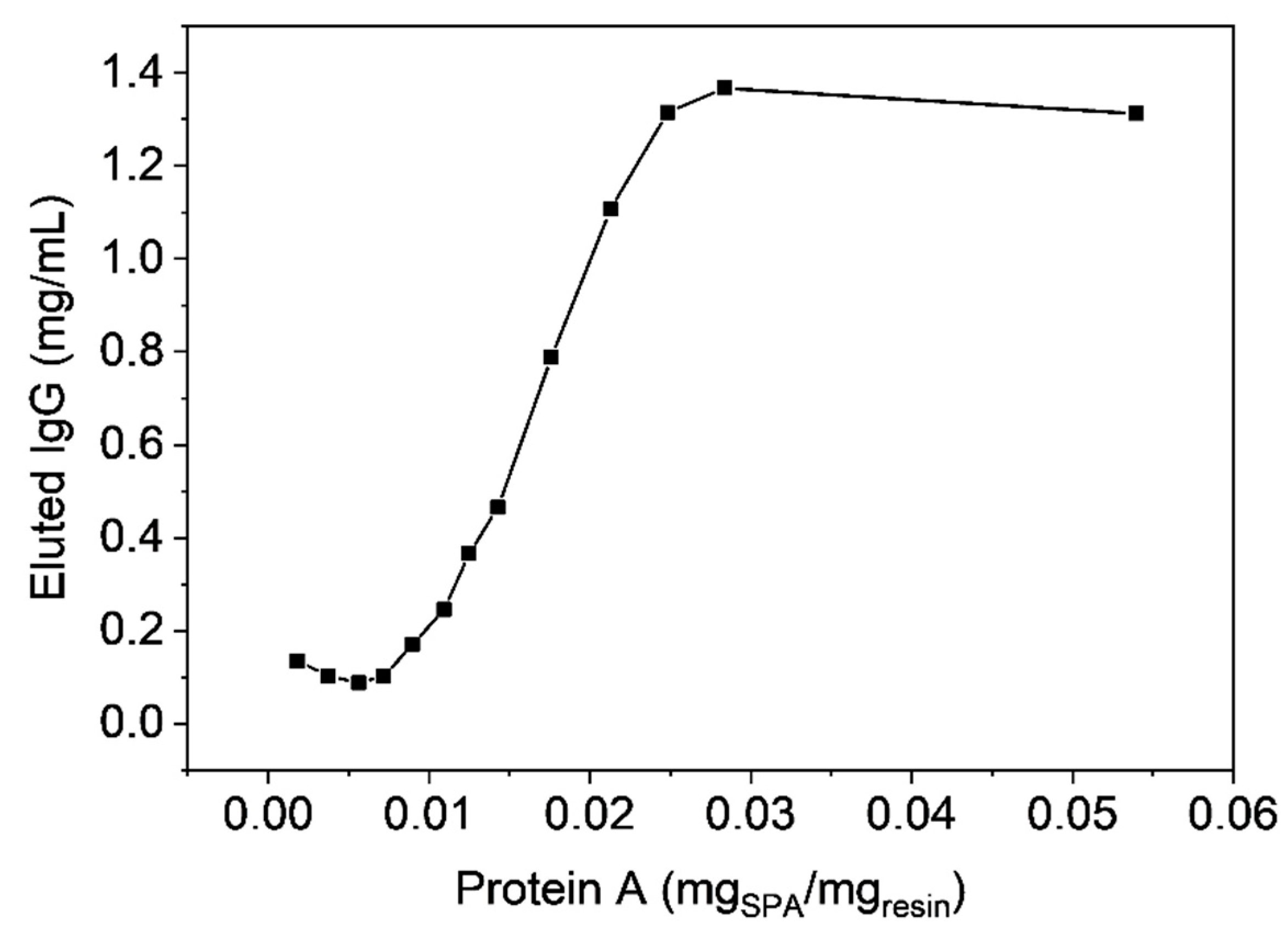
3.6. Antibody Chromatography
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abuchowski, A.; Mccoy, J.R.; Palczuk, N.C.; Vanes, T.; Davis, F.F. Effect of Covalent Attachment of Polyethylene-Glycol on Immunogenicity and Circulating Life of Bovine Liver Catalase. J. Biol. Chem. 1977, 252, 3582–3586. [Google Scholar] [PubMed]
- Bailon, P.; Berthold, W. Polyethylene glycol-conjugated pharmaceutical proteins. Pharm. Sci. Technol. Today 1998, 1, 352–356. [Google Scholar] [CrossRef]
- Hoffman, A.S.; Stayton, P.S. Conjugates of stimuli-responsive polymers and proteins. Prog. Polym. Sci. 2007, 32, 922–932. [Google Scholar] [CrossRef]
- Heredia, K.L.; Maynard, H.D. Synthesis of protein-polymer conjugates. Org. Biomol. Chem. 2007, 5, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef]
- Nicolas, J.; Mura, S.; Brambilla, D.; Mackiewicz, N.; Couvreur, P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem. Soc. Rev. 2013, 42, 1147–1235. [Google Scholar] [CrossRef]
- Stayton, P.S.; Shimoboji, T.; Long, C.; Chilkoti, A.; Chen, G.H.; Harris, J.M.; Hoffman, A.S. Control of Protein-Ligand Recognition Using a Stimuli-Responsive Polymer. Nature 1995, 378, 472–474. [Google Scholar] [CrossRef]
- Gu, Z.; Dang, T.T.; Ma, M.L.; Tang, B.C.; Cheng, H.; Jiang, S.; Dong, Y.Z.; Zhang, Y.L.; Anderson, D.G. Glucose-Responsive Microgels Integrated with Enzyme Nanocapsules for Closed-Loop Insulin Delivery. ACS Nano 2013, 7, 6758–6766. [Google Scholar] [CrossRef]
- Velonia, K.; Rowan, A.E.; Nolte, R.J.M. Lipase polystyrene giant amphiphiles. J. Am. Chem. Soc. 2002, 124, 4224–4225. [Google Scholar] [CrossRef]
- Hannink, J.M.; Cornelissen, J.J.L.M.; Farrera, J.A.; Foubert, P.; De Schryver, F.C.; Sommerdijk, N.A.J.M.; Nolte, R.J.M. Protein-polymer hybrid amphiphiles. Angew. Chem. Int. Ed. 2001, 40, 4732–4734. [Google Scholar] [CrossRef]
- Heredia, K.L.; Maynard, H.D. In-situ preparation of bovine serum albumin-poly(PEG methacrylate) conjugates. Polym. Mater. Sci. Eng. 2006, 94, 354–355. [Google Scholar]
- Rocchitta, G.; Spanu, A.; Babudieri, S.; Latte, G.; Madeddu, G.; Galleri, G.; Nuvoli, S.; Bagella, P.; Demartis, M.I.; Fiore, V.; et al. Enzyme Biosensors for Biomedical Applications: Strategies for Safeguarding Analytical Performances in Biological Fluids. Sensors 2016, 16, 780. [Google Scholar] [CrossRef]
- Li, R.X.; Dowd, V.; Stewart, D.J.; Burton, S.J.; Lowe, C.R. Design, synthesis, and application of a Protein A mimetic. Nat. Biotechnol. 1998, 16, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.Y.; Zhang, Y.; Zhao, H.Y. Fabrication of Polymer-Protein Hybrids. Macromol. Rapid Commun. 2018, 39, e1700737. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.V.; Anderson, K.W.; Bachas, L.G. Oriented immobilization of proteins. Mikrochim. Acta 1998, 128, 127–143. [Google Scholar] [CrossRef]
- Bailon, P.; Palleroni, A.; Schaffer, C.A.; Spence, C.L.; Fung, W.J.; Porter, J.E.; Ehrlich, G.K.; Pan, W.; Xu, Z.X.; Modi, M.W.; et al. Rational design of a potent, long-lasting form of interferon: A 40 kDa branched polyethylene glycol-conjugated interferon alpha-2a for the treatment of hepatitis C. Bioconjugate Chem. 2001, 12, 195–202. [Google Scholar] [CrossRef]
- Kochendoerfer, G.G. Site-specific polymer modification of therapeutic proteins. Curr. Opin. Chem. Biol. 2005, 9, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Thordarson, P.; Le Droumaguet, B.; Velonia, K. Well-defined protein-polymer conjugates-synthesis and potential applications. Appl. Microbiol. Biotechnol. 2006, 73, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Cobo, I.; Li, M.; Sumerlin, B.S.; Perrier, S. Smart hybrid materials by conjugation of responsive polymers to biomacromolecules. Nat. Mater. 2015, 14, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Stenzel, M.H. Bioconjugation Using Thiols: Old Chemistry Rediscovered to Connect Polymers with Nature’s Building Blocks. ACS Macro Lett. 2013, 2, 14–18. [Google Scholar] [CrossRef]
- Huisgen, R.; Szeimies, G.; Mobius, L. 1.3-Dipolare Cycloadditionen, 32. Kinetik Der Additionen Organischer Azide an Cc-Mehrfachbindungen. Chemische Berichte 1967, 100, 2494–2507. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A strain-promoted [3+2] azide-alkyne cycloaddition for covalent modification of blomolecules in living systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef]
- Marti, N.; Quattrini, F.; Butte, A.; Morbidelli, M. Production of polymeric materials with controlled pore structure: The “Reactive gelation” process. Macromol. Mater. Eng. 2005, 290, 221–229. [Google Scholar] [CrossRef]
- Lorenz, M.S.C.; Finkelstein, P.; Cingolani, A.; Storti, G.; Morbidelli, M. Template-free synthesis of porous polymeric microclusters from core-shell nanoparticles by reactive gelation and their post-functionalization via click chemistry. submitted.
- Bechtle, M.; Butte, A.; Storti, G.; Morbidelli, M. Preparation of macroporous methacrylate-based monoliths for chromatographic applications by the Reactive Gelation Process. J. Chromatogr. A 2010, 1217, 4675–4681. [Google Scholar] [CrossRef]
- Brand, B.; Krattli, M.; Storti, G.; Morbidelli, M. Strong cation exchange monoliths for HPLC by Reactive Gelation. J. Sep. Sci. 2011, 34, 2159–2163. [Google Scholar] [CrossRef]
- Brand, B.; Morbidelli, M.; Soos, M. Shear-Induced Reactive Gelation. Langmuir 2015, 31, 12727–12735. [Google Scholar] [CrossRef]
- Cingolani, A.; Baur, D.; Caimi, S.; Storti, G.; Morbidelli, M. Preparation of perfusive chromatographic materials via shear-induced reactive gelation. J. Chromatogr. A 2018, 1538, 25–33. [Google Scholar] [CrossRef]
- Lamprou, A.; Kose, I.; Aguirre, Z.P.; Storti, G.; Morbidelli, M.; Soos, M. Macroporous Polymer Particles via Reactive Gelation under Shear: Effect of Primary Particle Properties and Operating Parameters. Langmuir 2014, 30, 13970–13978. [Google Scholar] [CrossRef]
- Lamprou, A.; Kose, I.; Storti, G.; Morbidelli, M.; Soos, M. Synthesis of Macroporous Polymer Particles Using Reactive Gelation under Shear. Langmuir 2014, 30, 6946–6953. [Google Scholar] [CrossRef] [PubMed]
- Albuszis, M.; Roth, P.J.; Exnowitz, F.; Wong, D.L.; Pauer, W.; Moritz, H.U. Synthesis and in-depth characterization of reactive, uniform, crosslinked microparticles based on free radical copolymerization of 4-vinylbenzyl azide. Polym. Chem. 2016, 7, 1168–1180. [Google Scholar] [CrossRef]
- Rouquerol, J.; Avnir, D.; Fairbridge, C.W.; Everett, D.H.; Haynes, J.H.; Pernicone, N.; Ramsay, J.D.F.; Sing, K.S.W.; Unger, K.K. Recommendations for the Characterization of Porous Solids. Pure Appl. Chem. 1994, 66, 1739–1758. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Reporting Physisorption Data for Gas Solid Systems with Special Reference to the Determination of Surface-Area and Porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Garcia-Diego, C.; Cuellar, J. Synthesis of macroporous poly(styrene-co-divinylbenzene) microparticles using n-heptane as the porogen: Quantitative effects of the DVB concentration and the monomeric fraction on their structural characteristics. Ind. Eng. Chem. Res. 2005, 44, 8237–8247. [Google Scholar] [CrossRef]
- Cha, T.; Guo, A.; Zhu, X.Y. Enzymatic activity on a chip: The critical role of protein orientation. Proteomics 2005, 5, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Paganini, C.; Vogg, S.; Storti, G.; Morbidelli, M. Convective Protein A resin for high-throughput capture of monoclonal antibodies. 2018. Submitted. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Core Synthesis | Shell Synthesis | ||||
|---|---|---|---|---|---|
| Initial Charge [g] | Initiator Shot [g] | Feed [g] | Feed [g] | Initiator Shot [g] | |
| water | 650.0 | 100.0 | 45.0 | 12.0 | |
| styrene | 32.0 | 17.3 | |||
| DVB | 8.0 | 0.6 | |||
| VBA | 11.3 | ||||
| SDS | 4.1 | 1.3 | |||
| KPS | 1.5 | 0.3 | |||
| Time [min] | Diameter [nm] | PDI | Conversion [%] | Dry Content [%] |
|---|---|---|---|---|
| 60 | 24 | 0.12 | 97 | 2.5 |
| 120 | 27 | 0.06 | 98 | 4 |
| 180 | 30 | 0.03 | 99 | 5.5 |
| 240 | 31 | 0.06 | 99 | 6.5 |
| 300 | 34 | 0.04 | 97 | 7.4 |
| 360 | 38 | 0.06 | 99 | 8.6 |
| C | H | N |
|---|---|---|
| [%] | [%] | [%] |
| 84.2 | 7.5 | 3.4 |
| Reaction 1 | Reaction 2 | |||
| Click Reaction (CuAAC) | Thiol-Epoxide Ring Opening Reaction | |||
| S-1 | 50% DMF 50% Water | 10% yield | Aq. Buffer pH 7.5 | 15.2 mgIgG/mLpolymer |
| S-2 | 100% DMF | 98% yield | Aq. Buffer pH 7.5 | 14.3 mgIgG/mLpolymer |
| Thiol-Epoxide Ring Opening Reaction | Click Reactions (CuAAC) | |||
| S-3 | Aq. Buffer pH 7.5 | 9.6 mgIgG/mLpolymer | Aq. Buffer pH 7.5 | 10% yield |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorenz, M.; Paganini, C.; Storti, G.; Morbidelli, M. Macroporous Polymer–Protein Hybrid Materials for Antibody Purification by Combination of Reactive Gelation and Click-Chemistry. Materials 2019, 12, 1580. https://doi.org/10.3390/ma12101580
Lorenz M, Paganini C, Storti G, Morbidelli M. Macroporous Polymer–Protein Hybrid Materials for Antibody Purification by Combination of Reactive Gelation and Click-Chemistry. Materials. 2019; 12(10):1580. https://doi.org/10.3390/ma12101580
Chicago/Turabian StyleLorenz, Marcel, Carolina Paganini, Giuseppe Storti, and Massimo Morbidelli. 2019. "Macroporous Polymer–Protein Hybrid Materials for Antibody Purification by Combination of Reactive Gelation and Click-Chemistry" Materials 12, no. 10: 1580. https://doi.org/10.3390/ma12101580
APA StyleLorenz, M., Paganini, C., Storti, G., & Morbidelli, M. (2019). Macroporous Polymer–Protein Hybrid Materials for Antibody Purification by Combination of Reactive Gelation and Click-Chemistry. Materials, 12(10), 1580. https://doi.org/10.3390/ma12101580