Heat-Induced Discoloration of Chromophore Structures in Eucalyptus Lignin


Abstract
:1. Introduction
2. Materials and Methods
2.1. Wood Samples and Thermal Modification
2.2. Color Measurement
2.3. Lignin Analysis
3. Results
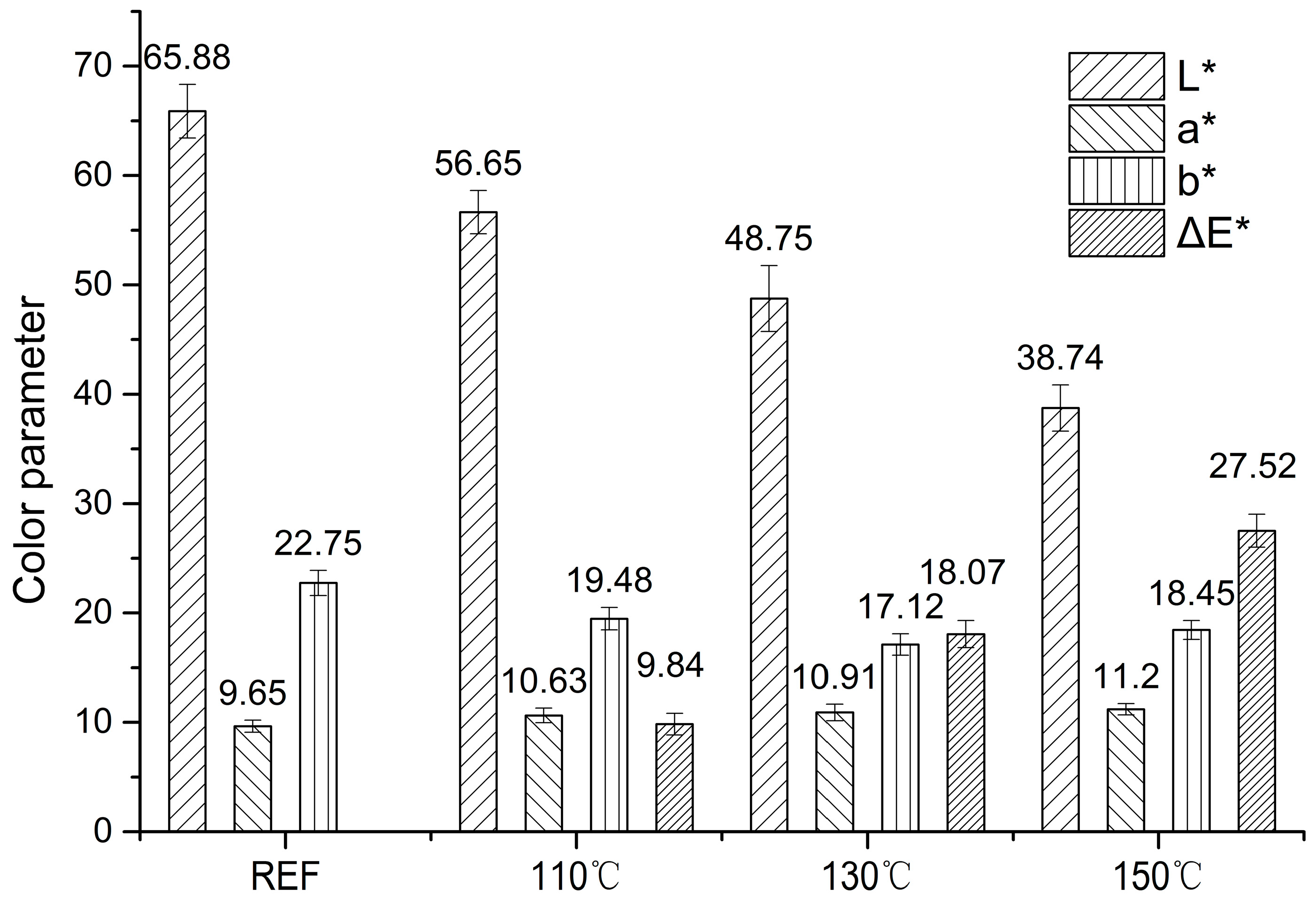
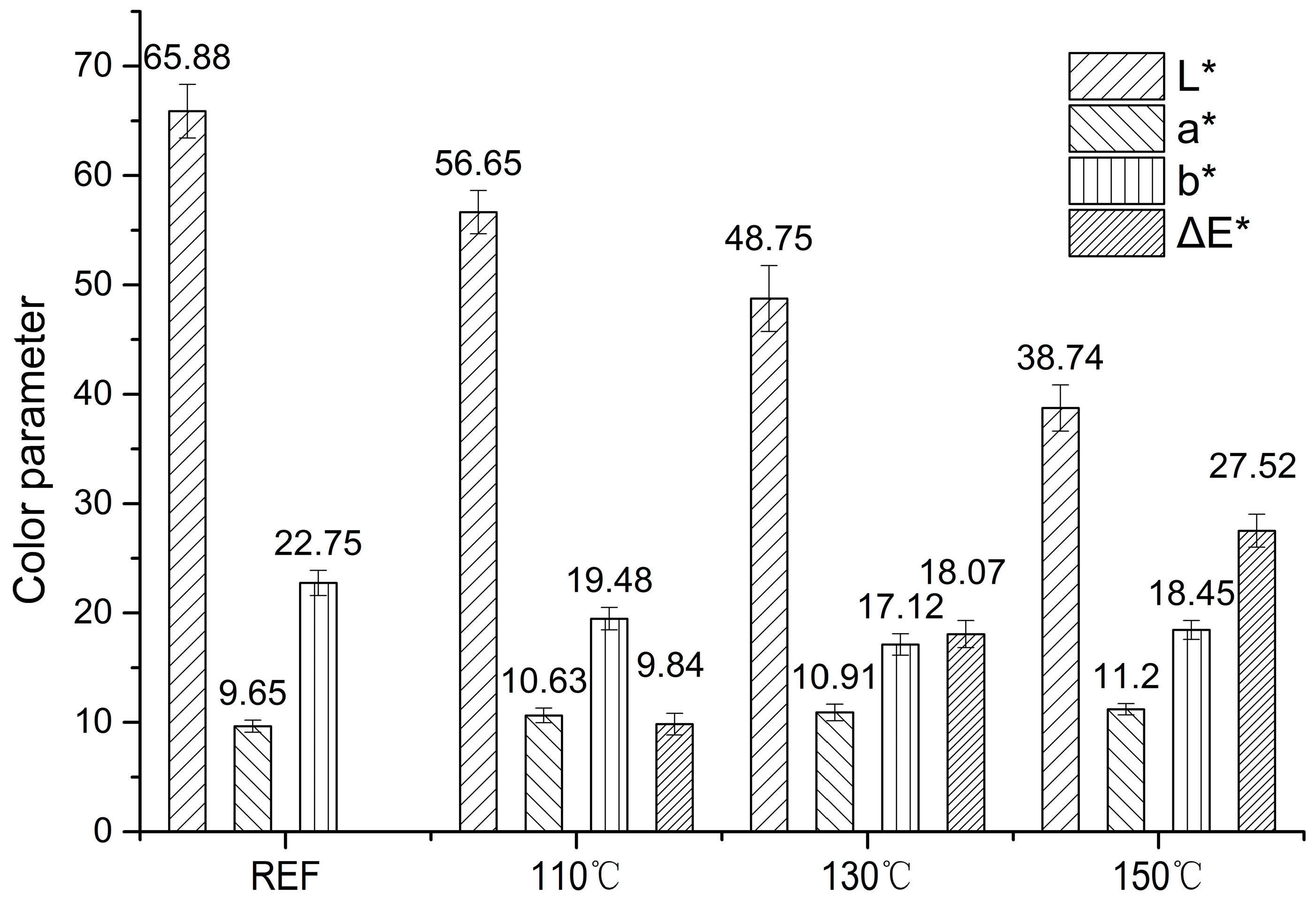
3.1. Heat Treatment Effects on Color Parameters of Wood Samples
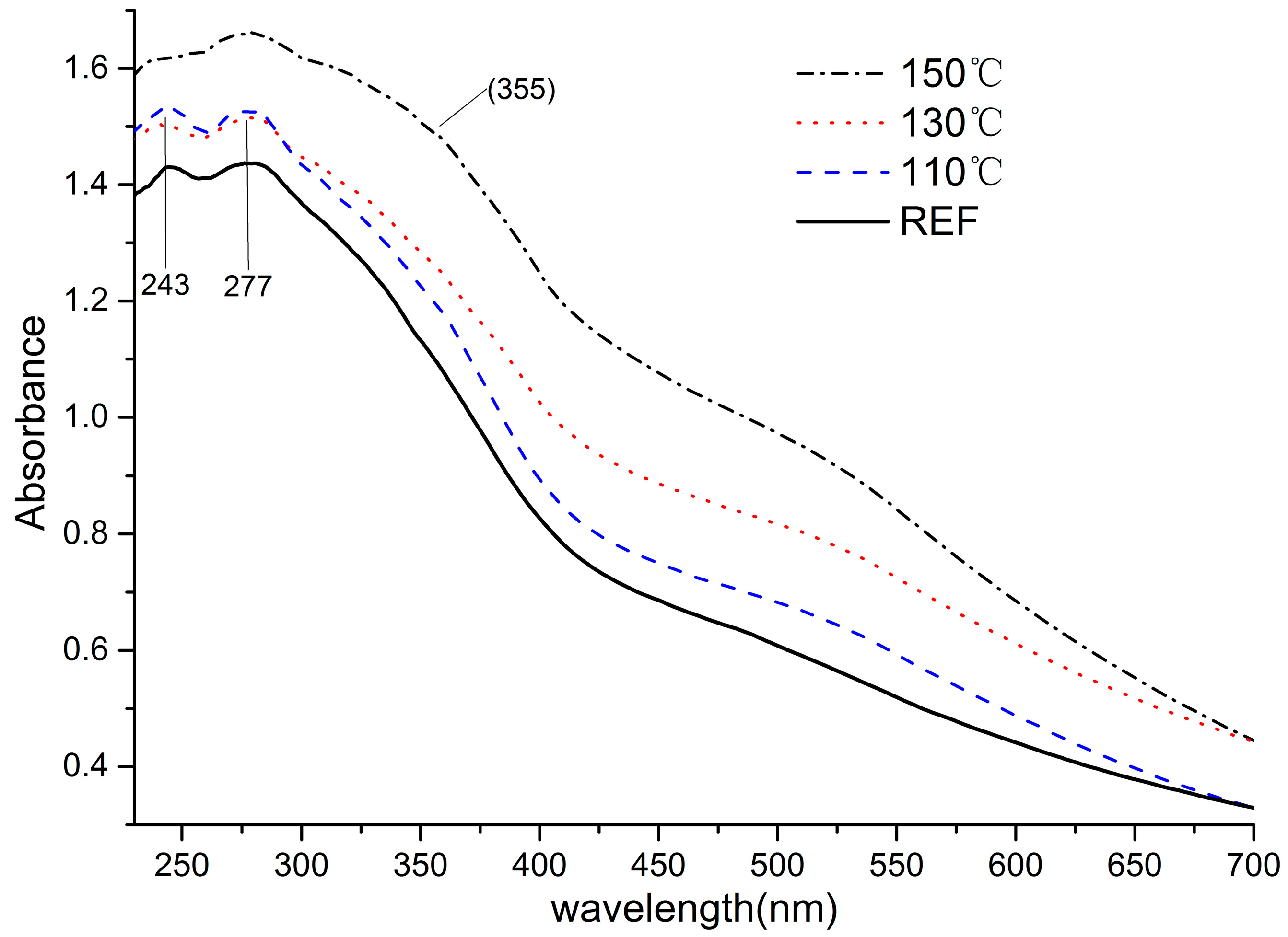
3.2. Content and Elemental Composition of Lignins
3.3. GPC Analysis of Molecular Weight and Polydispersity Index
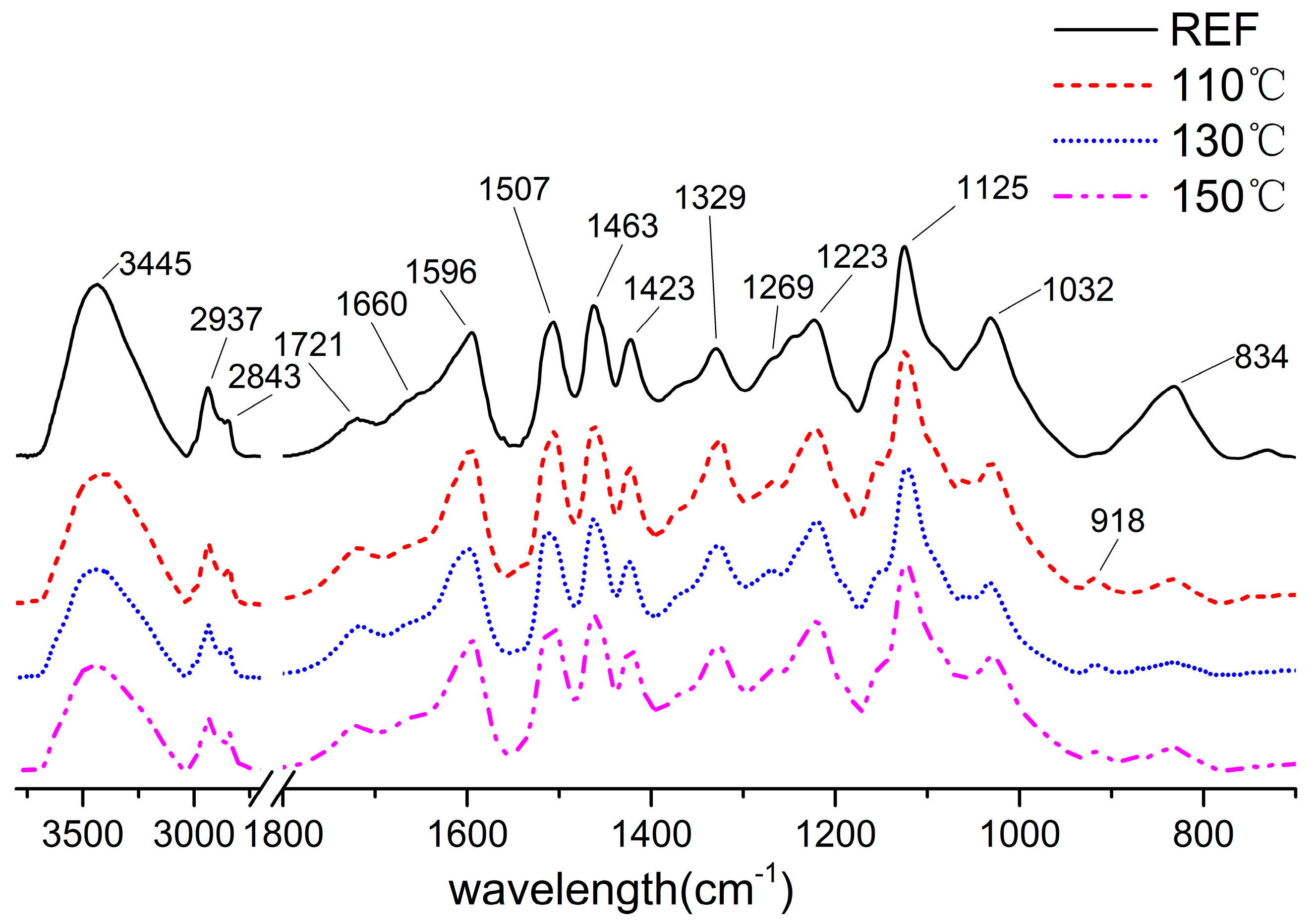
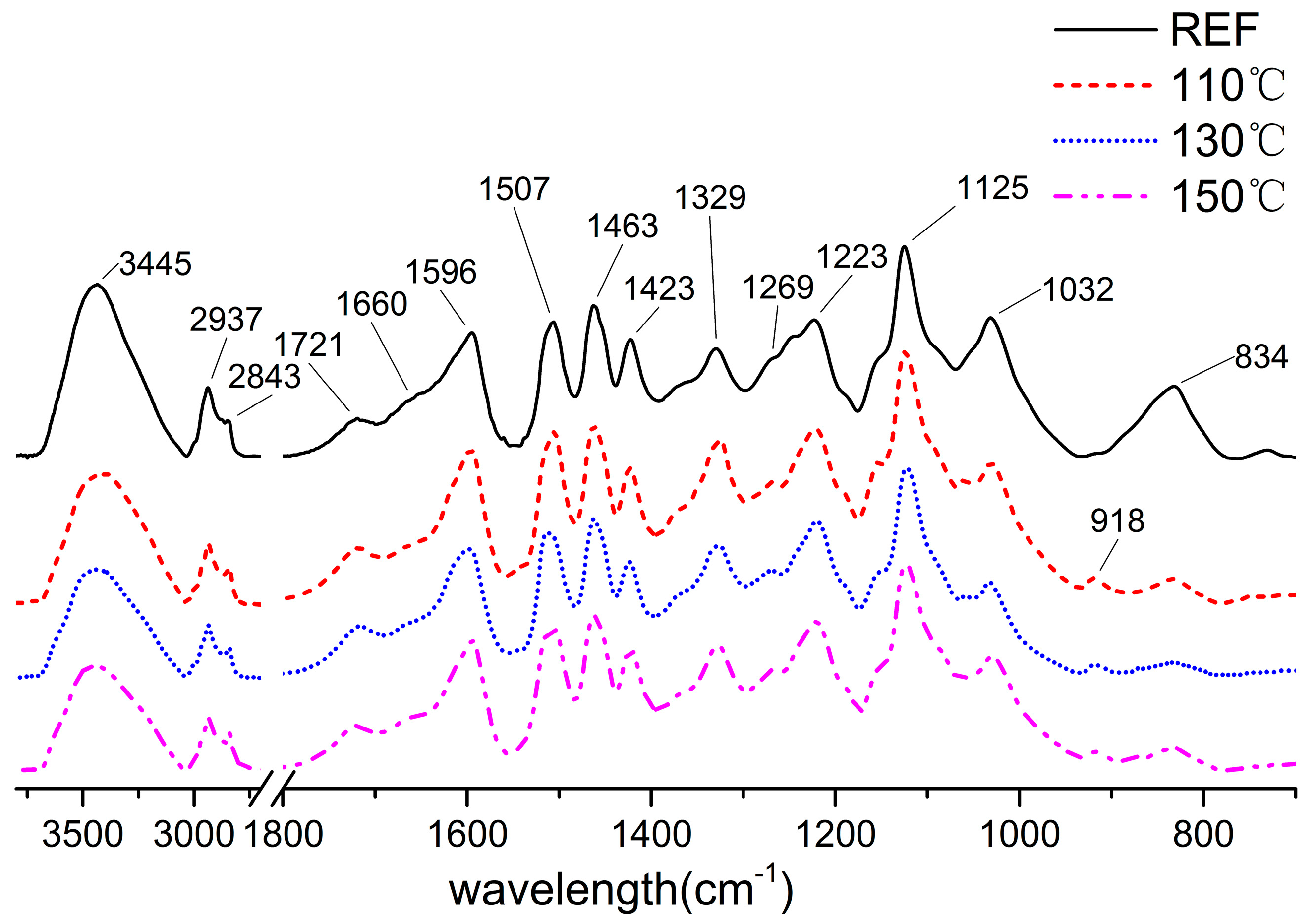
3.4. FTIR Spectra of the Lignin
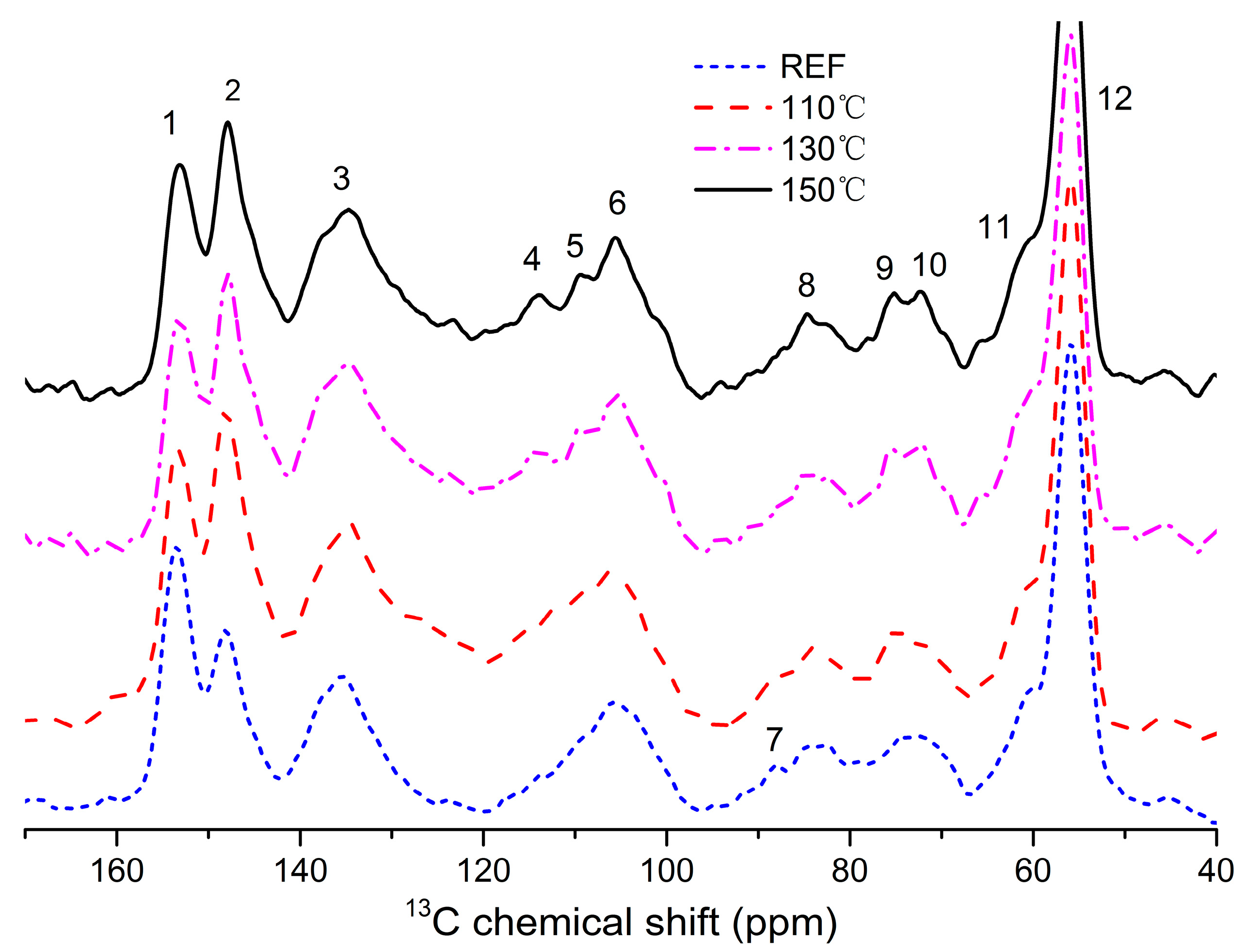
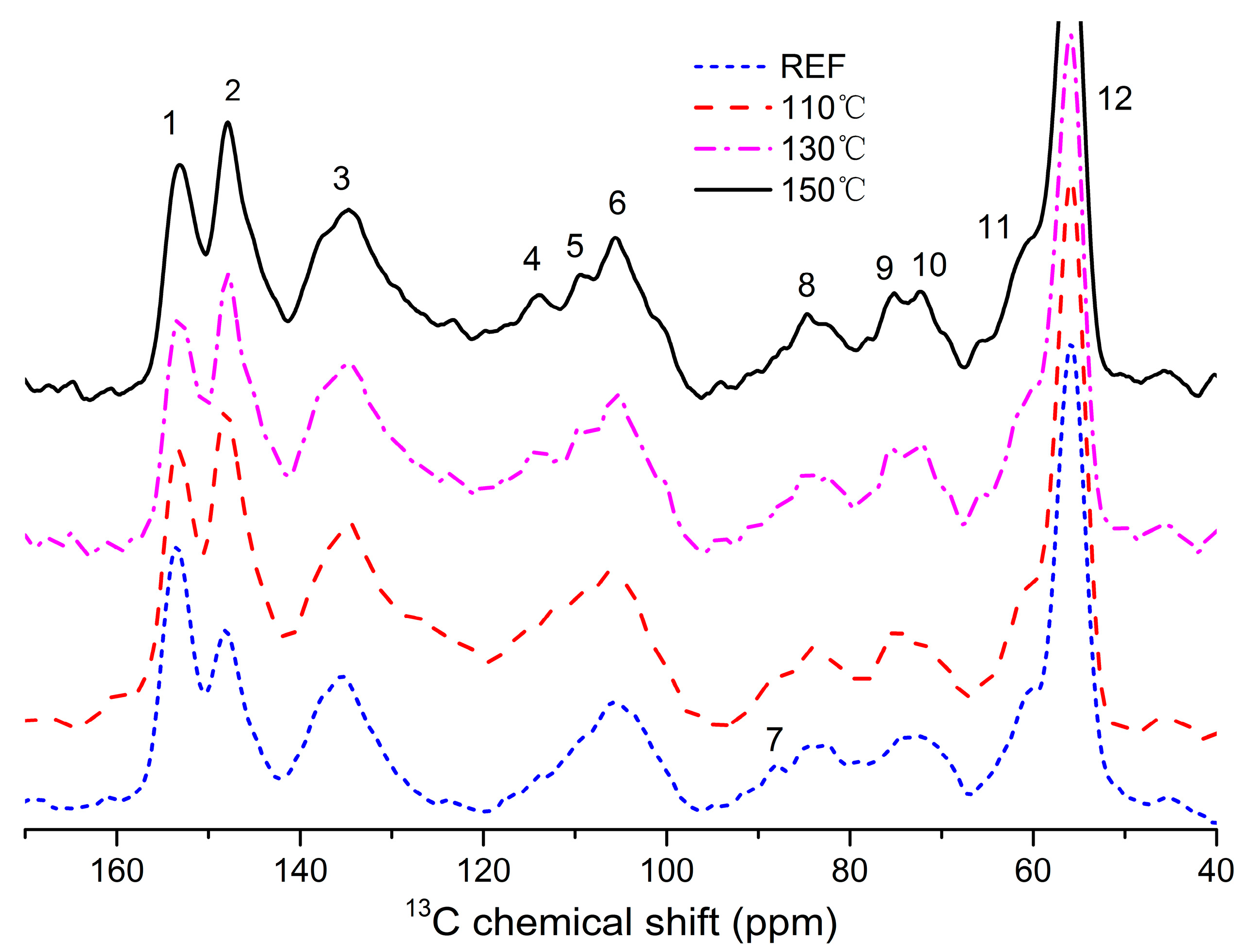
3.5. 13C NMR Spectroscopy
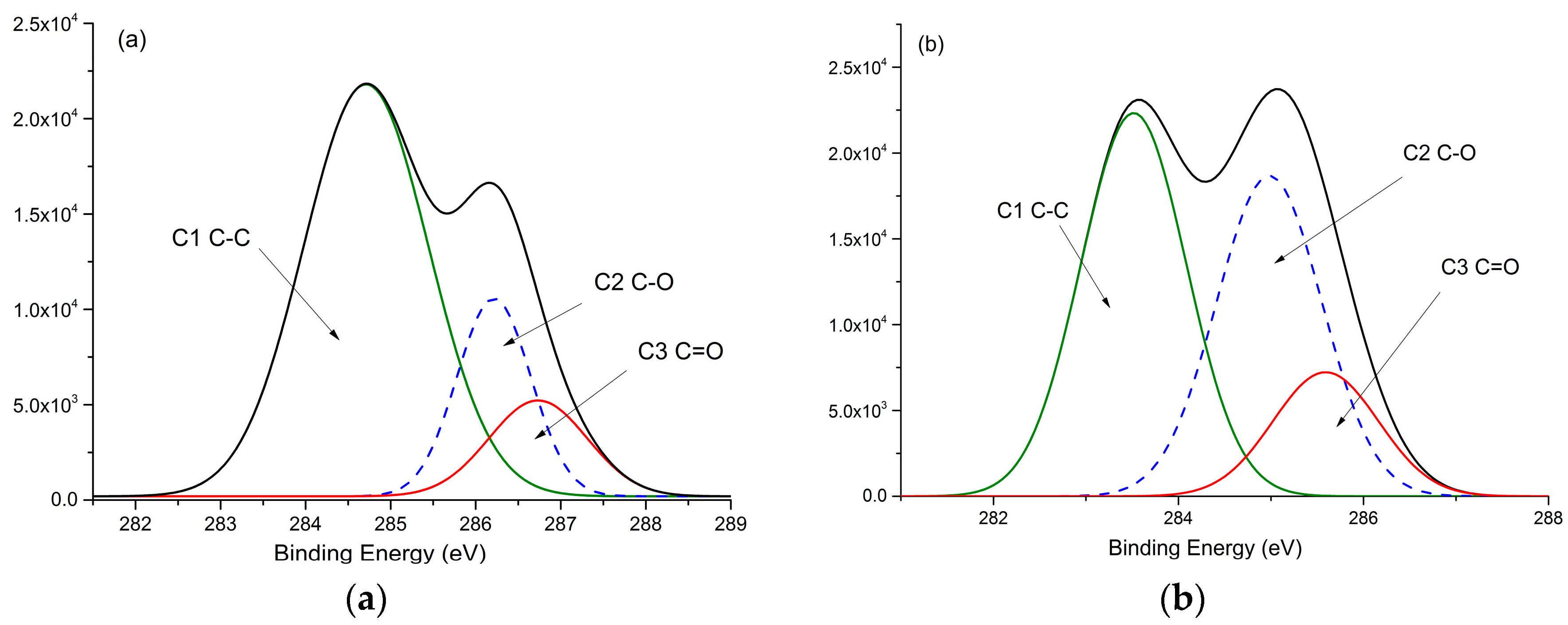
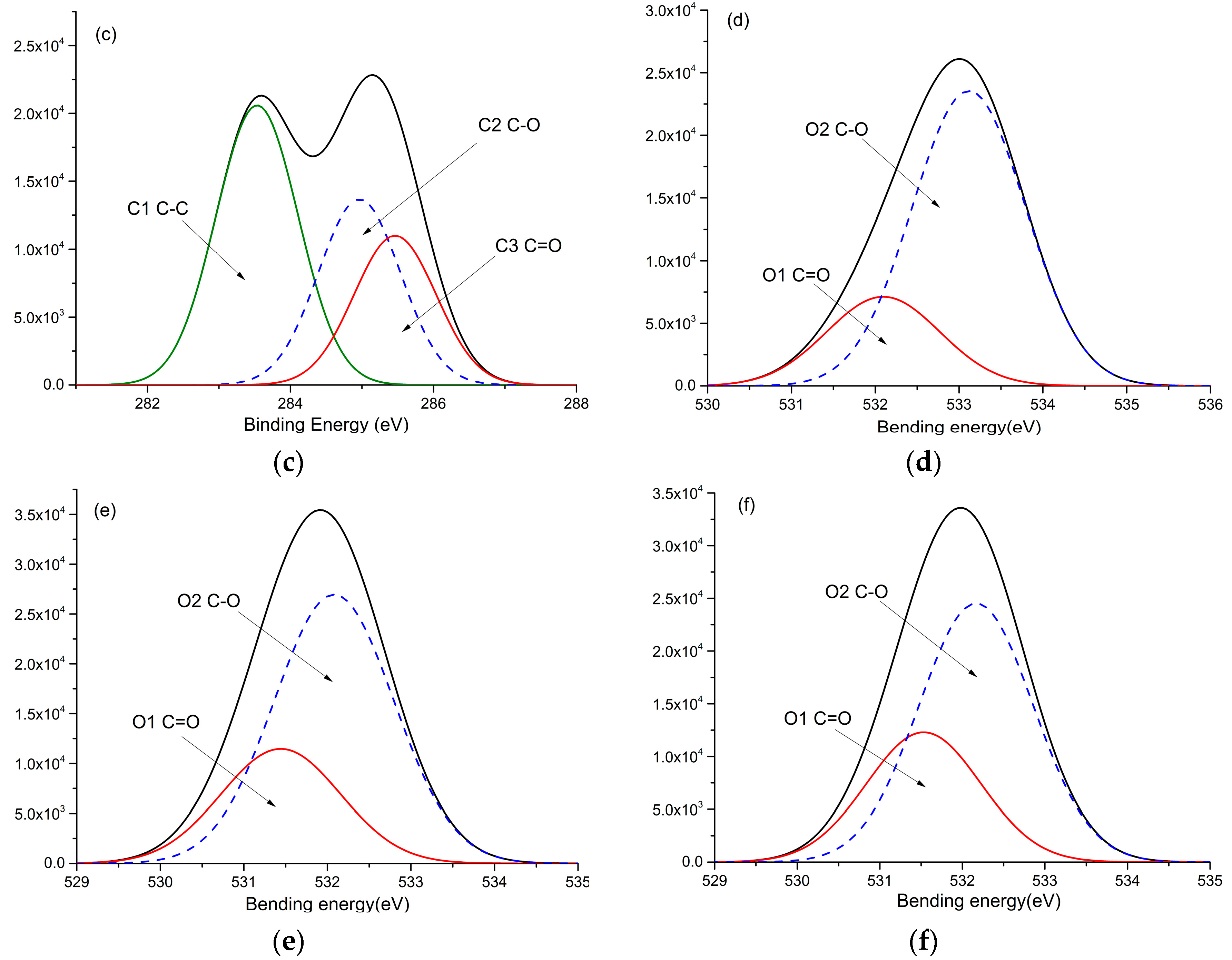
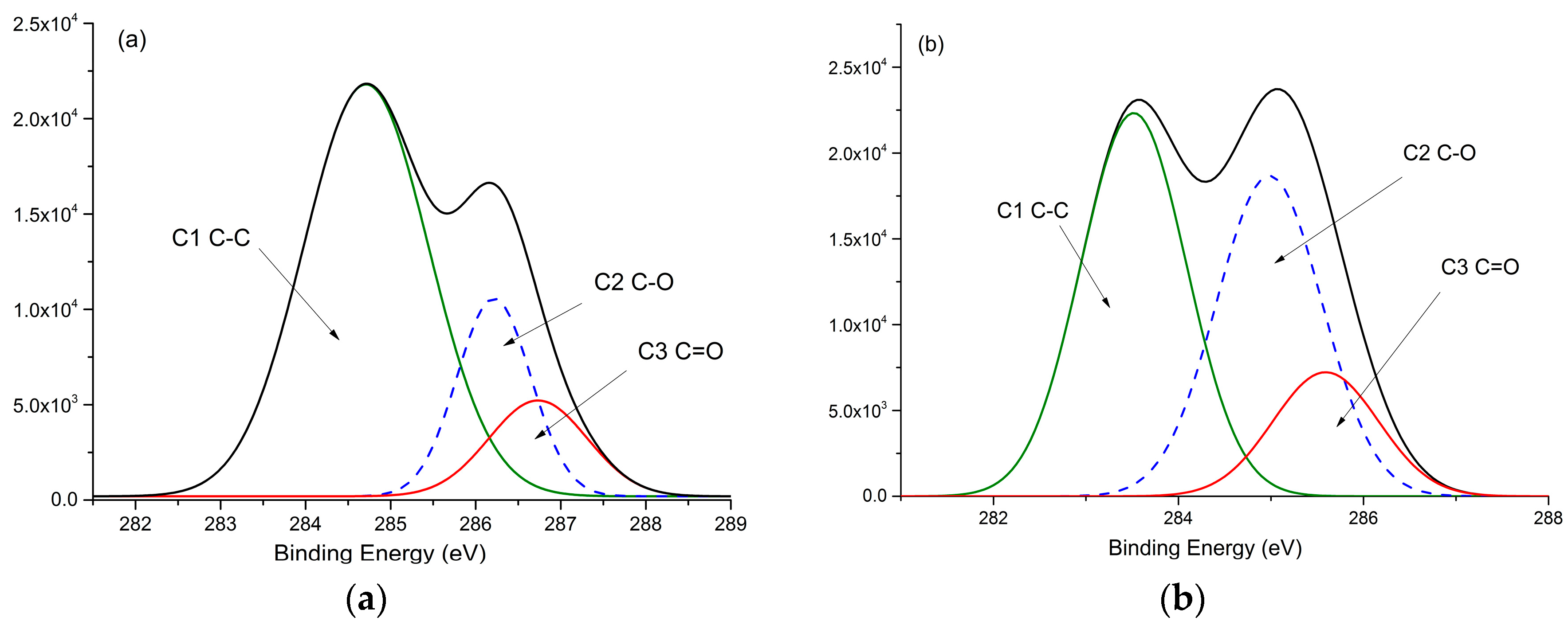
3.6. XPS Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fonseca, A. Environmental management in wood processing industries and the European legislation on voc emission control. In Proceedings of the 1st International Conference on Environmentally-Compatible Forest Products Oporto, Oporto, Portugal, 22–24 September 2004; pp. 313–324. [Google Scholar]
- Chen, Y.; Gao, J.M.; Fan, Y.M.; Tshabalala, M.A.; Stark, N.M. Heat-induced chemical and color changes of extractive-free black locust (Robinia Pseudoacacia) wood. Bioresources 2012, 7, 2236–2248. [Google Scholar] [CrossRef]
- Gonzalez-Pena, M.M.; Hale, M.D.C. Colour in thermally modified wood of beech, norway spruce and scots pine. Part 1: Colour evolution and colour changes. Holzforschung 2009, 63, 385–393. [Google Scholar] [CrossRef]
- Maunu, S. NMR studies of wood and wood products. Prog. Nucl. Mag. Res. Spectrosc. 2002, 40, 151–174. [Google Scholar] [CrossRef]
- Bekhta, P.; Niemz, P. Effect of high temperature on the change in color, dimensional stability and mechanical properties of spruce wood. Holzforschung 2003, 57, 539–546. [Google Scholar] [CrossRef]
- Sundqvist, B. Colour Changes and Acid Formation in Wood during Heating. Ph.D. Thesis, Luleå tekniska universitet, Luleå, Sweden, 2004. [Google Scholar]
- Kacikova, D.; Kacik, F.; Cabalova, I.; Durkovic, J. Effects of thermal treatment on chemical, mechanical and colour traits in norway spruce wood. Bioresour. Technol. 2013, 144, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Sivonen, H.; Maunu, S.L.; Sundholm, F.; Jämsä, S.; Viitaniemi, P. Magnetic resonance studies of thermally modified wood. Holzforschung 2002, 56, 648–654. [Google Scholar] [CrossRef]
- Weimer, P.; Hackney, J.; French, A. Effects of chemical treatments and heating on the crystallinity of celluloses and their implications for evaluating the effect of crystallinity on cellulose biodegradation. Biotechnol. Bioeng. 1995, 48, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Wikberg, H.; Maunu, S.L. Characterisation of thermally modified hard and softwoods by C-13 Cpmas NMR. Carbohyd. Polym. 2004, 58, 461–466. [Google Scholar] [CrossRef]
- Tjeerdsma, B.; Boonstra, M.; Pizzi, A.; Tekely, P.; Militz, H. Characterisation of thermally modified wood: Molecular reasons for wood performance improvement. Holz Roh Werkst 1998, 56, 149. [Google Scholar] [CrossRef]
- Sundqvist, B.; Karlsson, O.; Westermark, U. Determination of formic-acid and acetic acid concentrations formed during hydrothermal treatment of birch wood and its relation to colour, strength and hardness. Wood Sci. Technol. 2006, 40, 549–561. [Google Scholar] [CrossRef]
- Liu, X.Y.; Timar, M.C.; Varodi, A.M.; Sawyer, G. An investigation of accelerated temperature-induced ageing of four wood species: Colour and FTIR. Wood Sci. Technol. 2017, 51, 357–378. [Google Scholar] [CrossRef]
- Brito, J.O.; Silva, F.G.; Leao, M.M.; Almeida, G. Chemical composition changes in eucalyptus and pinus woods submitted to heat treatment. Bioresour. Technol. 2008, 99, 8545–8548. [Google Scholar] [CrossRef] [PubMed]
- Melkior, T.; Jacob, S.; Gerbaud, G.; Hediger, S.; Le Pape, L.; Bonnefois, L.; Bardet, M. NMR analysis of the transformation of wood constituents by torrefaction. Fuel 2012, 92, 271–280. [Google Scholar] [CrossRef]
- Lee, S.H.; Ashaari, Z.; Lum, W.C.; Halip, J.A.; Ang, A.F.; Tan, L.P.; Chin, K.L.; Tahir, P.M. Thermal treatment of wood using vegetable oils: A review. Constr. Build. Mater. 2018, 181, 408–419. [Google Scholar] [CrossRef]
- Chen, Y.; Fan, Y.M.; Gao, J.M.; Li, H.K. Coloring characteristics of in situ lignin during heat treatment. Wood Sci. Technol. 2012, 46, 33–40. [Google Scholar] [CrossRef]
- Chen, Y.; Tshabalala, M.A.; Gao, J.; Stark, N.M.; Fan, Y. Color and surface chemistry changes of extracted wood flour after heating at 120 °C. Wood Sci. Technol. 2013, 48, 137–150. [Google Scholar] [CrossRef]
- Hiltunen, E.; Alvila, L.; Pakkanen, T.T. Characterization of brauns’ lignin from fresh and vacuum-dried birch (betula pendula) wood. Wood Sci. Technol. 2006, 40, 575. [Google Scholar] [CrossRef]
- Windeisen, E.; Wegener, G. Behaviour of lignin during thermal treatments of wood. Ind. Crop Prod. 2008, 27, 157–162. [Google Scholar] [CrossRef]
- Sandoval-Torres, S.; Jomaa, W.; Marc, F.; Puiggali, J.-R. Colour alteration and chemistry changes in oak wood (quercus pedunculata ehrh) during plain vacuum drying. Wood Sci. Technol. 2012, 46, 177–191. [Google Scholar] [CrossRef]
- Sandoval-Torres, S.; Jomaa, W.; Marc, F.; Puiggali, J.-R. Causes of color changes in wood during drying. For. Stud. China 2010, 12, 167–175. [Google Scholar] [CrossRef]
- Mononen, K.; Alvila, L.; Pakkanen, T.T. Ciel* a* b* measurements to determine the role of felling season, log storage and kiln drying on coloration of silver birch wood. J. For. Res. 2002, 17, 179–191. [Google Scholar] [CrossRef]
- Forsthuber, B.; Grüll, G. Prediction of wood surface discoloration for applications in the field of architecture. Wood Sci. Technol. 2018, 52, 1–19. [Google Scholar] [CrossRef]
- ASTM D1106-96. Standard Test Method for Acid-Insoluble Lignin in Wood; ASTM International: West Conshohocken, PA, USA, 2013. [Google Scholar]
- Adler, E. Lignin chemistry—past, present and future. Wood Sci. Technol. 1977, 11, 169–218. [Google Scholar] [CrossRef]
- Lin, S.Y.; Dence, C.W. Methods in Lignin Chemistry; Springer: Berlin, Germany, 2012. [Google Scholar]
- Evtuguin, D.V.; Neto, C.P.; Silva, A.M.; Domingues, P.M.; Amado, F.M.; Robert, D.; Faix, O. Comprehensive study on the chemical structure of dioxane lignin from plantation eucalyptus globulus wood. J. Agric. Food Chem. 2001, 49, 4252–4261. [Google Scholar] [CrossRef] [PubMed]
- Gabov, K.; Gosselink, R.J.; Smeds, A.I.; Fardim, P. Characterization of lignin extracted from birch wood by a modified hydrotropic process. J. Agric. Food Chem. 2014, 62, 10759–10767. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.J.; Du, X.Y.; Liu, J.; Chang, H.M.; Jameel, H. Structural characterization of pine kraft lignin: Biochoice lignin vs. Indulin AT. J. Wood Chem. Technol. 2016, 36, 432–446. [Google Scholar] [CrossRef]
- Wang, J.; Wu, B.; Li, S.; He, Y. Nir light and enzyme dual stimuli-responsive amphiphilic diblock copolymer assemblies. J. Polym. Sci. Pol. Chem. 2017, 55, 2450–2457. [Google Scholar] [CrossRef]
- Chen, Y.; Fan, Y.; Gao, J.; Tshabalala, M.A.; Stark, N.M. Spectroscopic analysis of the role of extractives on heat-induced discoloration of black locust (Robinia pseudoacacia). Wood Mater. Sci. Eng. 2012, 7, 209–216. [Google Scholar] [CrossRef]
- Charrier, B.; Haluk, J.; Janin, G. Prevention of brown discoloration in European oakwood occurring during kiln drying by a vacuum process: Colorimetric comparative study with a traditional process. Holz Roh Werkst 1992, 50, 433–437. [Google Scholar] [CrossRef]
- Burtin, P.; Jay-Allemand, C.; Charpentier, J.-P.; Janin, G. Modifications of hybrid walnut (juglans nigra 23 x juglans regia) wood colour and phenolic composition under various steaming conditions. Holzforschung 2000, 54, 33–38. [Google Scholar] [CrossRef]
- Grekin, M. Color and color uniformity variation of scots pine wood in the air-dry condition. Wood Fiber Sci. 2007, 39, 279–290. [Google Scholar]
- Hon, D.N.S. Photooxidative degradation of cellulose: Reactions of the cellulosic free radicals with oxygen. J. Polym. Sci. Pol. Chem. 1979, 17, 441–454. [Google Scholar] [CrossRef]
- Johansson, M. Formation of Chromophores and Leucochromophores during Manufacturing of Mechanical Pulp. Licentiate Thesis, Royal Institute of Technology, Stockholm, Sweden, 2000. [Google Scholar]
- Sun, J.X.; Sun, X.F.; Sun, R.C.; Fowler, P.; Baird, M.S. Inhomogeneities in the chemical structure of sugarcane bagasse lignin. J. Agric. Food Chem. 2003, 51, 6719–6725. [Google Scholar] [CrossRef] [PubMed]
- Nguila Inari, G.; Petrissans, M.; Gerardin, P. Chemical reactivity of heat-treated wood. Wood Sci. Technol. 2006, 41, 157–168. [Google Scholar] [CrossRef]
- Rousset, P.; Lapierre, C.; Pollet, B.; Quirino, W.; Perre, P. Effect of severe thermal treatment on spruce and beech wood lignin. Ann. For. Sci. 2009, 66, 1. [Google Scholar] [CrossRef]
- Park, J.; Meng, J.J.; Lim, K.H.; Rojas, O.J.; Park, S. Transformation of lignocellulosic biomass during torrefaction. J. Anal. Appl. Pyrolysis 2013, 100, 199–206. [Google Scholar] [CrossRef]
- Lin, B.J.; Colin, B.; Chen, W.H.; Pétrissans, A.; Rousset, P.; Pétrissans, M. Thermal degradation and compositional changes of wood treated in a semi-industrial scale reactor in vacuum. J. Anal. Appl. Pyrolysis 2018, 130, 8–18. [Google Scholar] [CrossRef]
- Gosselink, R.J.A.; Abacherli, A.; Semke, H.; Malherbe, R.; Kauper, P.; Nadif, A.; van Dam, J.E.G. Analytical protocols for characterisation of sulphur-free lignin. Ind. Crop Prod. 2004, 19, 271–281. [Google Scholar] [CrossRef]
- Kim, J.Y.; Hwang, H.; Oh, S.; Kim, Y.S.; Kim, U.J.; Choi, J.W. Investigation of structural modification and thermal characteristics of lignin after heat treatment. Int. J. Biol. Macromol. 2014, 66, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xu, W.; Wang, X.; Wang, C. Modeling and predicting of the color changes of wood surface during CO2 laser modification. J. Clean. Prod. 2018, 183, 818–823. [Google Scholar] [CrossRef]
- Nakano, J. Chemistry of Lignin—Basis and Application; Gao, J.; Bao, H.; Li, Z.Z., Translators; Light Industry Press: Beijing, China, 1988. [Google Scholar]
- Faix, O. Classification of lignins from different botanical origins by FT-IR spectroscopy. Holzforschung 1991, 45, 21–28. [Google Scholar] [CrossRef]
- She, D.A.; Xu, F.; Geng, Z.C.; Sun, R.C.; Jones, G.L.; Baird, M.S. Physicochemical characterization of extracted lignin from sweet sorghum stem. Ind. Crop Prod. 2010, 32, 21–28. [Google Scholar] [CrossRef]
- Nimz, H.; Robert, D.; Faix, O.; Nemr, M. Carbon-13 NMR spectra of lignins, 8. Structural differences between lignins of hardwoods, softwoods, grasses and compression wood. Holzforschung-Int. J. Biol. Chem. Phys. Technol. Wood 1981, 35, 16–26. [Google Scholar] [CrossRef]
- Capanema, E.A.; Balakshin, M.Y.; Kadla, J.F. A comprehensive approach for quantitative lignin characterization by nmr spectroscopy. J. Agric. Food Chem. 2004, 52, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L. Characterization of milled wood lignins and dehydrogenative polymerisates from monolignols by carbon-13 NMR spectroscopy. Lignin Lignan Biosynth. 1998, 697, 1947–5918. [Google Scholar]
- Brosse, N.; El Hage, R.; Chaouch, M.; Petrissans, M.; Dumarcay, S.; Gerardin, P. Investigation of the chemical modifications of beech wood lignin during heat treatment. Polym. Degrad. Stabil. 2010, 95, 1721–1726. [Google Scholar] [CrossRef]
- Haw, J.F.; Schultz, T.P. Carbon-13 cp/mas NMR and FT-IR study of low-temperature lignin pyrolysis. Holzforschung-Int. J. Biol. Chem. Phys. Technol. Wood 1985, 39, 289–296. [Google Scholar] [CrossRef]
- Haw, J.F.; Maciel, G.E.; Biermann, C.J. Carbon-13 nuclear magnetic resonance study of the rapid steam hydrolysis of read oak. Holzforschung-Int. J. Biol. Chem. Phys. Technol. Wood 1984, 38, 327–331. [Google Scholar] [CrossRef]
- Zhang, F.; Lin, J.; Zhao, G. Preparation and characterization of modified soda lignin with polyethylene glycol. Materials 2016, 9, 822. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Liu, J.; Du, X.Y.; Hu, Z.J.; Chang, H.M.; Jameel, H. Phenolation to improve lignin reactivity toward thermosets application. ACS Sustain. Chem. Eng. 2018, 6, 5504–5512. [Google Scholar] [CrossRef]
- Jiang, X.; Savithri, D.; Du, X.; Pawar, S.N.; Jameel, H.; Chang, H.M.; Zhou, X. Fractionation and characterization of kraft lignin by sequential precipitation with various organic solvents. ACS Sustain. Chem. Eng. 2016, 5, 835–842. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (°C) | Klason Lignin (%) | Dioxane Lignin (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Content | Content | C | H | O | N | S | O/C | H/C | |
| REF | 22.66 (±0.34) | 6.06 (±0.41) | 57.25 | 5.75 | 36.79 | 0.08 | 0.131 | 0.48 | 1.20 |
| 110 | 24.17 (±0.21) | 11.24 (±1.1) | 56.06 | 5.35 | 38.29 | 0.10 | 0.193 | 0.51 | 1.15 |
| 130 | 25.4 (±0.52) | 12.2 (±1.8) | 55.43 | 5.19 | 38.33 | 0.17 | 0.884 | 0.52 | 1.12 |
| 150 | 21.18 (±0.65) | 8.08 (±0.13) | 59.02 | 5.77 | 34.90 | 0.12 | 0.195 | 0.44 | 1.17 |
| Group | Mw (g/mol) | Mn (g/mol) | PDI (Mw/Mn) |
|---|---|---|---|
| REF | 7025 | 3822 | 1.838 |
| 110 | 3962 | 1483 | 2.671 |
| 130 | 4513 | 1687 | 2.675 |
| 150 | 8777 | 4332 | 2.026 |
| Wave Number | REF | 110 | 130 | 150 |
|---|---|---|---|---|
| 1660(cm−1) | 0.44 | 0.34 | 0.39 | 0.37 |
| 1721(cm−1) | 0.29 | 0.32 | 0.35 | 0.31 |
| ppm | Assignment | Amount (Per Ar) | ||||
|---|---|---|---|---|---|---|
| REF | 110 | 130 | 150 | |||
| 1 | 153.18 | C-3, S etherified | 1.71 | 1.05 | 1.02 | 0.91 |
| 2 | 148.05 | C-3/5, S non-etherified; C-4, G etherified | 1.21 | 1.27 | 1.27 | 1.28 |
| 3 | 134.88 | C-1/4, S/G etherified | 1.47 | 1.32 | 1.35 | 1.44 |
| 4 | 114.2 | C-3/5, H etherified | 0.23 | 0.40 | 0.39 | 0.40 |
| 5 | 109.61 | C-2, G | 0.43 | 0.42 | 0.42 | 0.42 |
| 6 | 105.7 | C-5/6, G etherified; C-2/6, S etherified | 0.79 | 0.58 | 0.57 | 0.66 |
| 7 | 88.16 | C-α, phenylcoumarans | 0.21 | 0.13 | 0.13 | 0.12 |
| 8 | 85.02 | C-β, β-O-4 | 0.56 | 0.33 | 0.35 | 0.36 |
| 9 | 75.15 | C-a, β-O-4 | 0.31 | 0.22 | 0.24 | 0.27 |
| 10 | 72.28 | C-γ, pinoresinols | 0.39 | 0.22 | 0.21 | 0.25 |
| 11 | 60.83 | C-γ in β-O-4 | 0.67 | 0.45 | 0.43 | 0.58 |
| 12 | 56 | methoxyl groups OCH3 | 2.53 | 1.49 | 1.41 | 1.65 |
| Sample Description | Relative Area of C1s Peaks (%) | Relative Area of O1s Peaks (%) | |||||
|---|---|---|---|---|---|---|---|
| C1 (C-C/C=C) (284.7 eV) | C2(C-O) (285.9 eV) | C3(C=O) (286.4 eV) | C3/C2 | O1(C=O) (531.7 eV) | O2(C-O) (532.8 eV) | O1/O2 | |
| REF | 57.32 | 32.84 | 9.83 | 0.30 | 23.20 | 76.79 | 0.30 |
| 130 | 46.83 | 38.20 | 14.97 | 0.39 | 29.83 | 70.17 | 0.43 |
| 150 | 45.39 | 37.40 | 17.21 | 0.46 | 31.80 | 68.20 | 0.47 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Wei, Y.; Liu, Y.; Gao, J.; Chen, Y.; Fan, Y. Heat-Induced Discoloration of Chromophore Structures in Eucalyptus Lignin. Materials 2018, 11, 1686. https://doi.org/10.3390/ma11091686
Zhang P, Wei Y, Liu Y, Gao J, Chen Y, Fan Y. Heat-Induced Discoloration of Chromophore Structures in Eucalyptus Lignin. Materials. 2018; 11(9):1686. https://doi.org/10.3390/ma11091686
Chicago/Turabian StyleZhang, Peng, Yanxia Wei, Yang Liu, Jianmin Gao, Yao Chen, and Yongming Fan. 2018. "Heat-Induced Discoloration of Chromophore Structures in Eucalyptus Lignin" Materials 11, no. 9: 1686. https://doi.org/10.3390/ma11091686
APA StyleZhang, P., Wei, Y., Liu, Y., Gao, J., Chen, Y., & Fan, Y. (2018). Heat-Induced Discoloration of Chromophore Structures in Eucalyptus Lignin. Materials, 11(9), 1686. https://doi.org/10.3390/ma11091686