Biobased Functional Carbon Materials: Production, Characterization, and Applications—A Review



Abstract
1. Introduction
2. Carbonization Technologies
2.1. Pyrolysis
2.2. Hydrothermal Carbonization (HTC)
3. Activation Processes
3.1. Chemical Activation with KOH
3.2. Effect of the Carbonization Process on the Structural Properties of Microporous Carbon Materials
4. Characterization of Activated Carbons
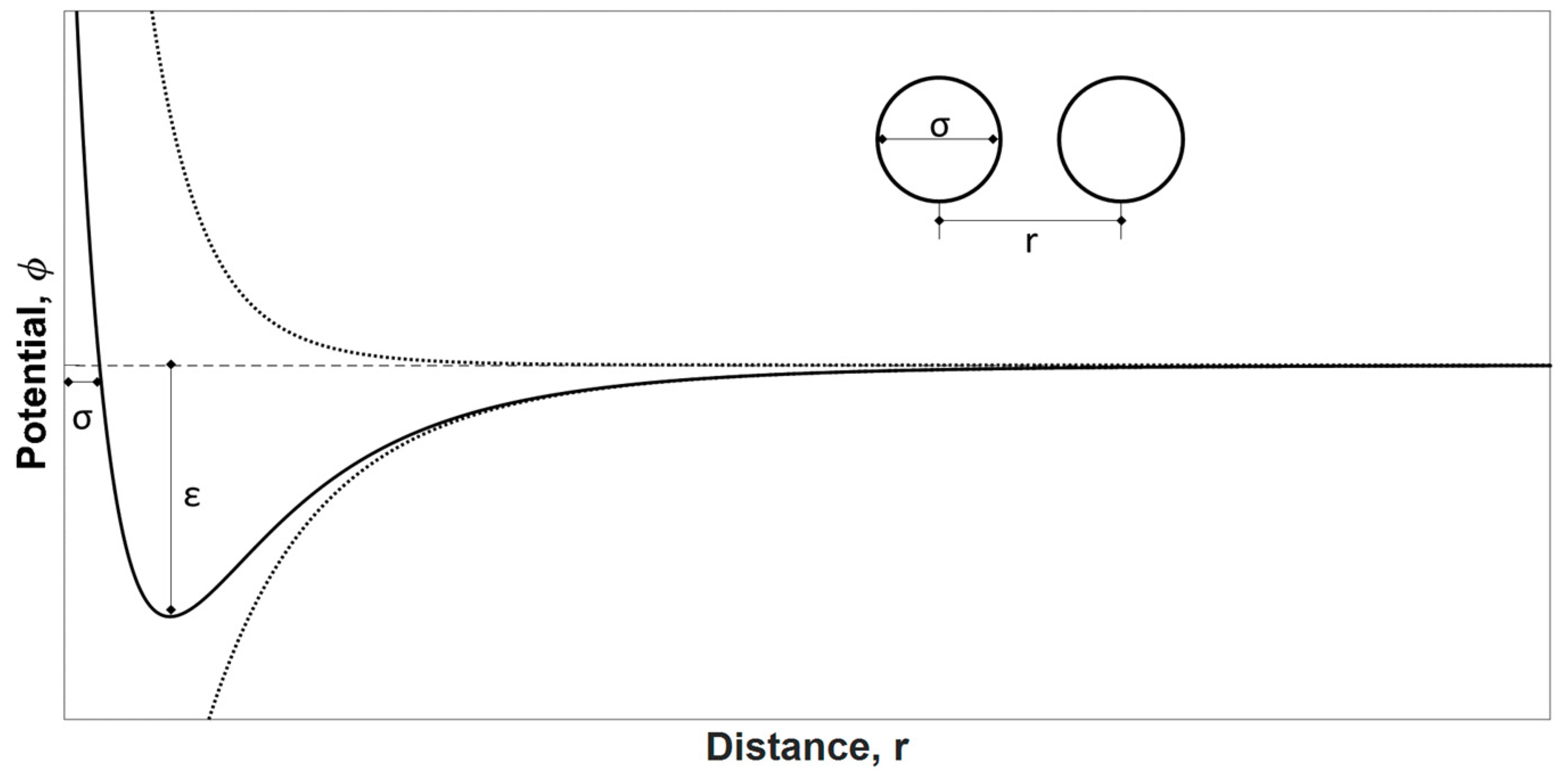
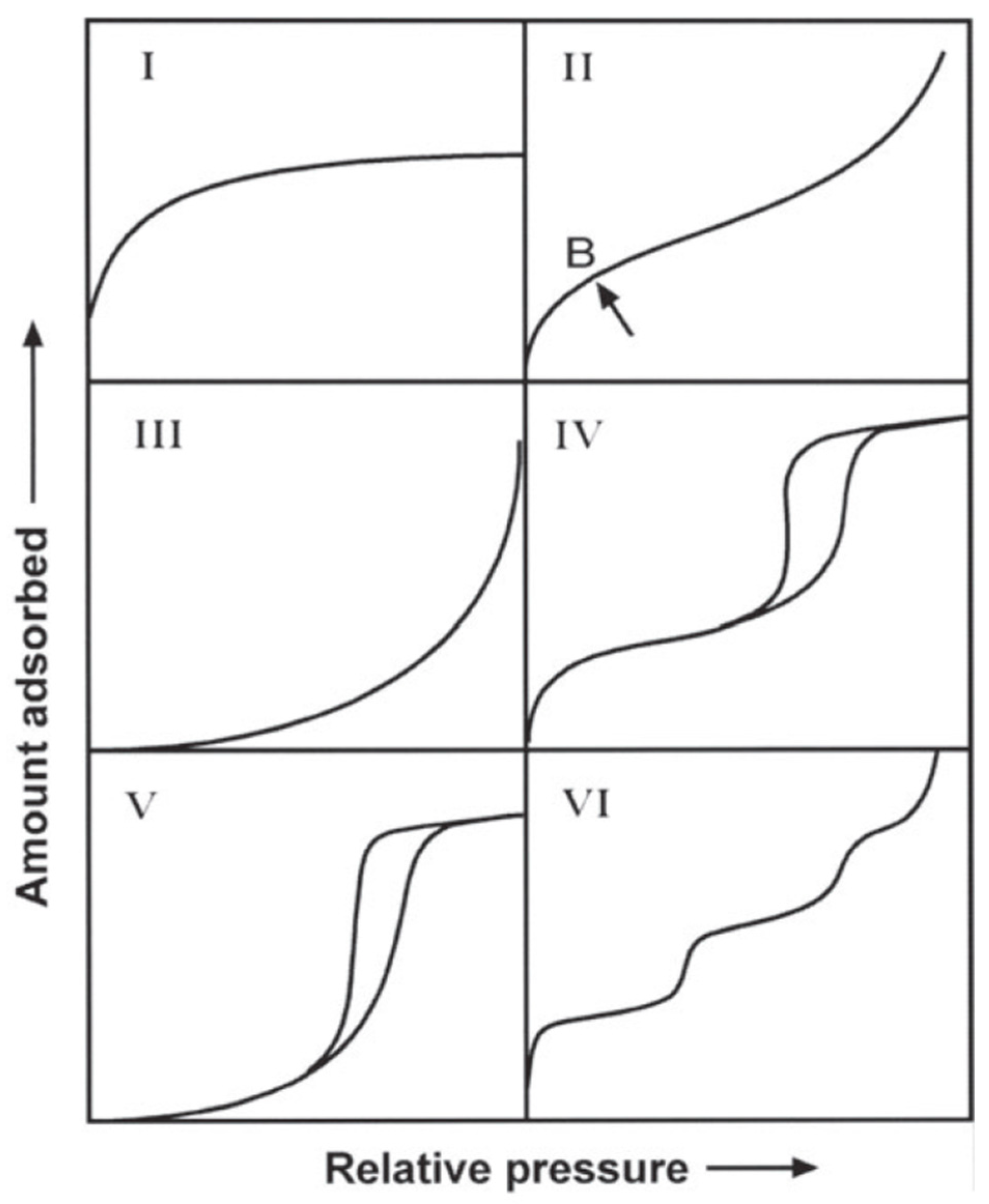
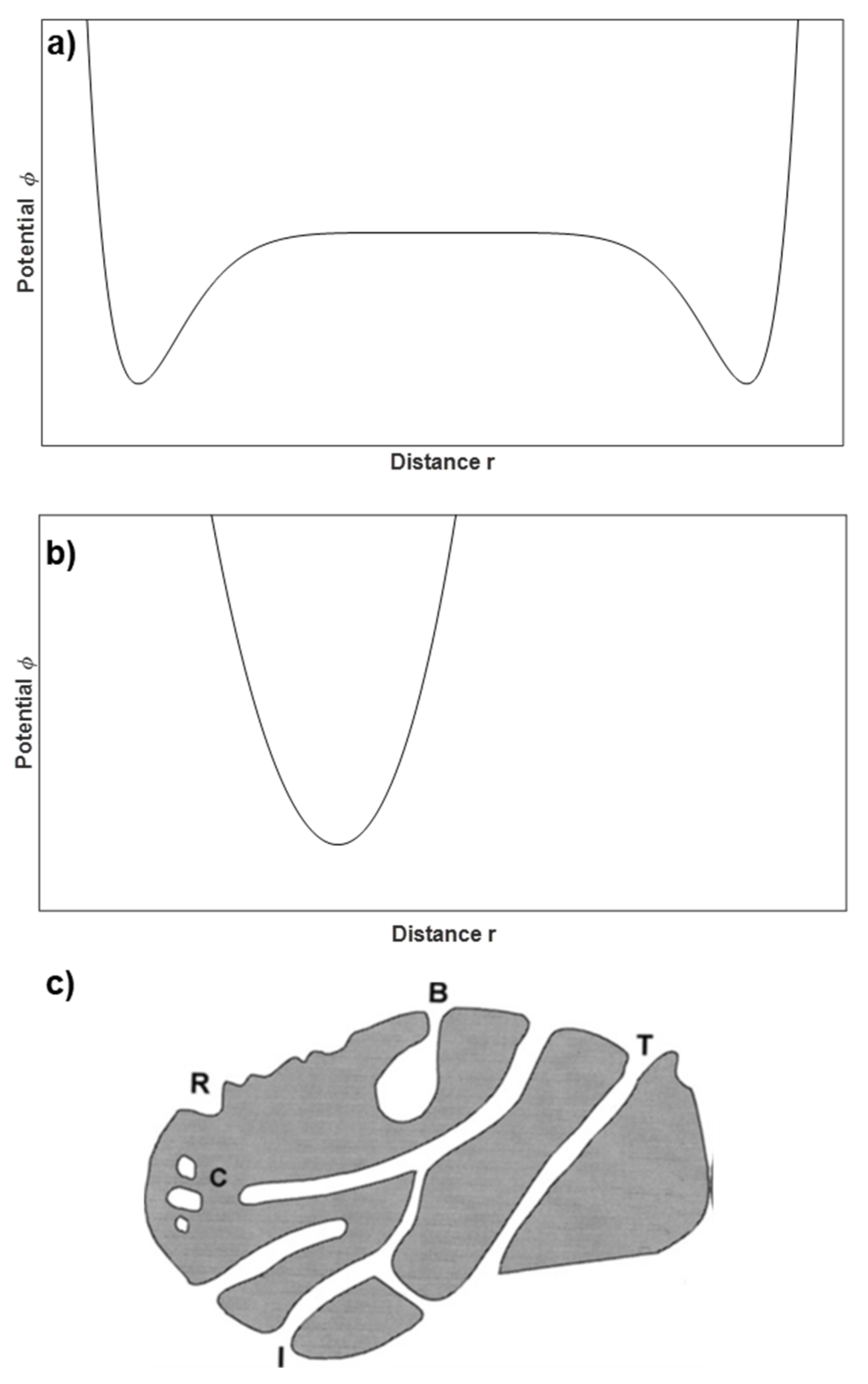
4.1. Textural Characterization
- The quantity C should be positive (i.e., a negative intercept on the ordinate of the BET plot is the first indication that one is outside the appropriate range).
- Application of the BET equation should be restricted to the range where the term p/[n*(p0 − p)] continuously increases with p/p0.
- The p/p0 value corresponding to nm should be within the selected BET range.
- VN2 < VCO2: Activated carbons with very low burn-off (<5%). The differences are attributed to the restricted diffusion of N2. CO2 can penetrate very narrow microporosity and/or there are constrictions in the entry of the micropores.
- VN2 ≈ VCO2: Activated carbons with low-to-medium (<35%) burn-off. The microporosity is relatively narrow and homogeneous.
- VN2 > VCO2: Activated carbons with medium-to-high burn-off. The microporosity is wider and very heterogeneous.
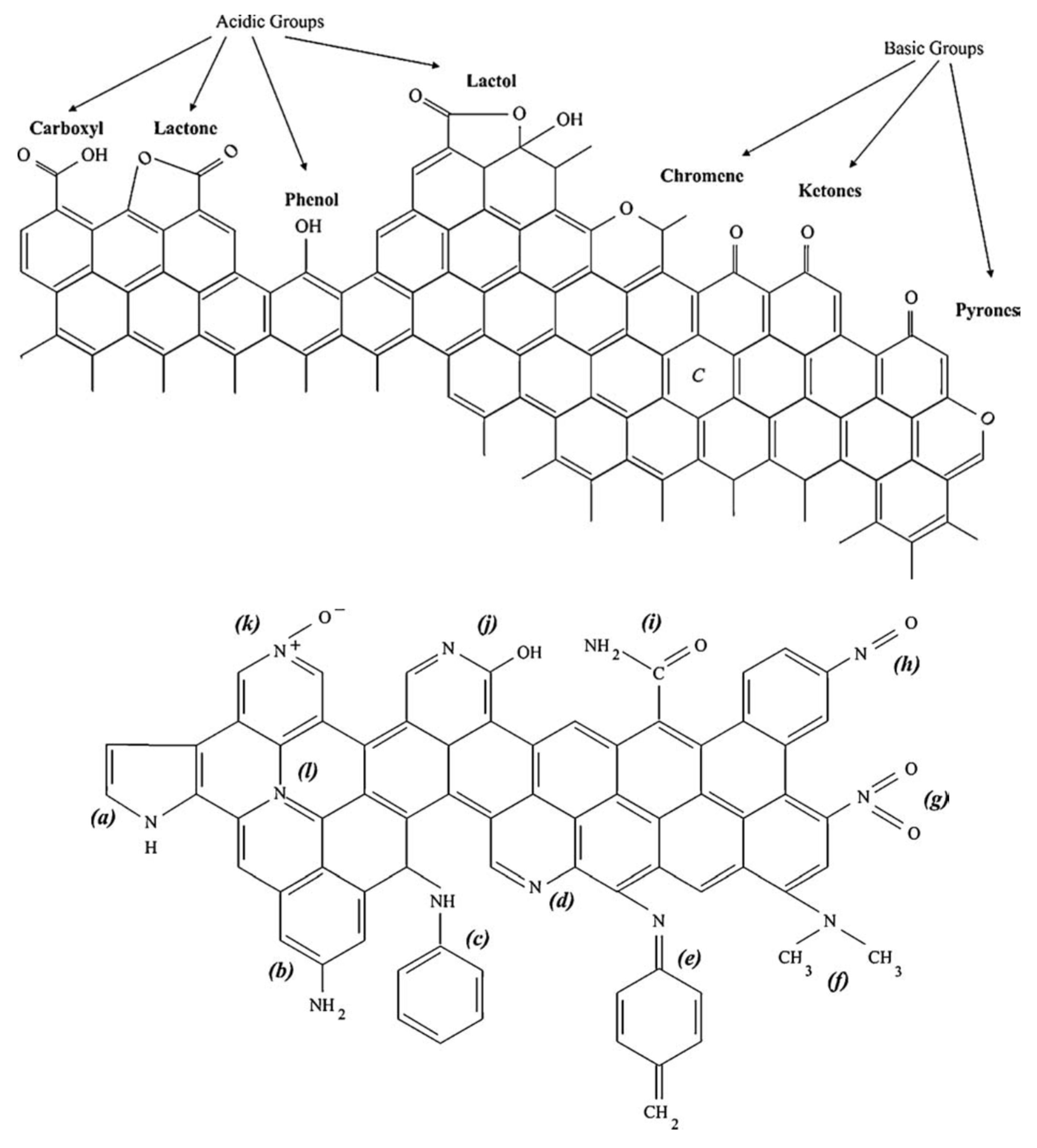
4.2. Surface Chemistry Characterization
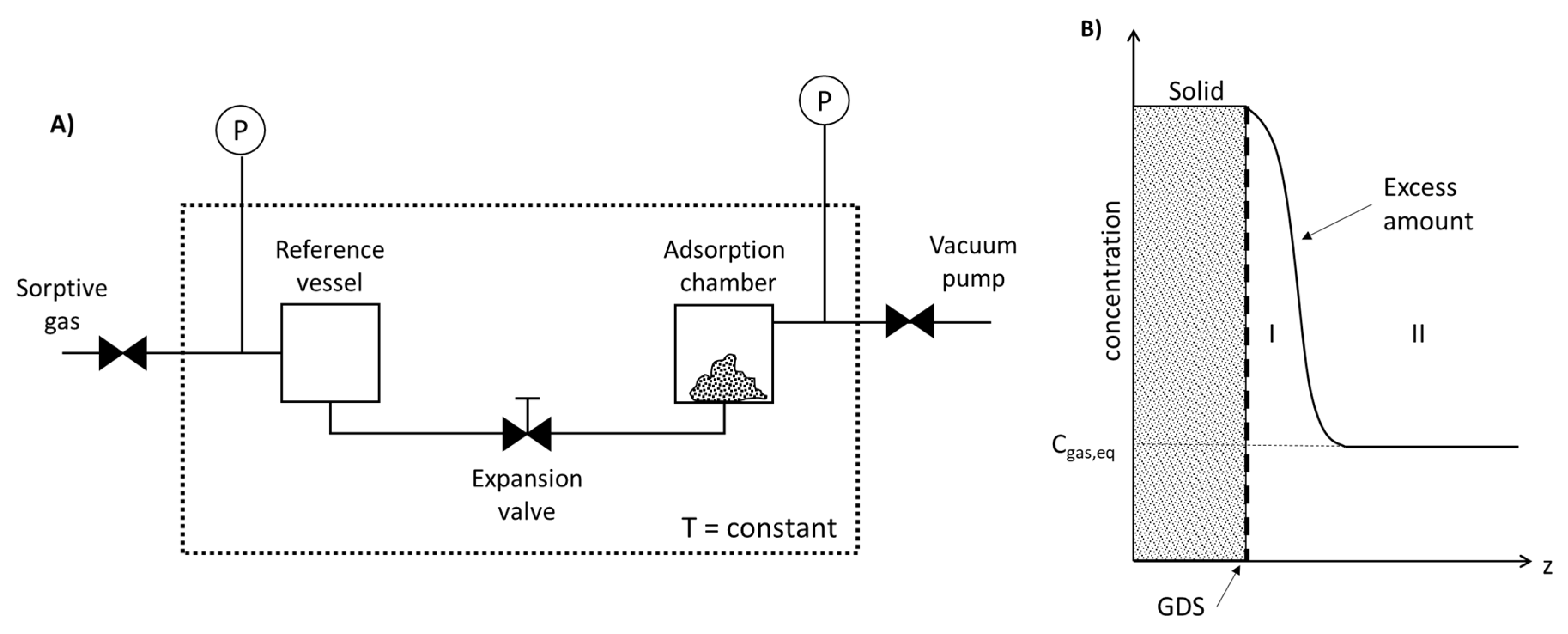
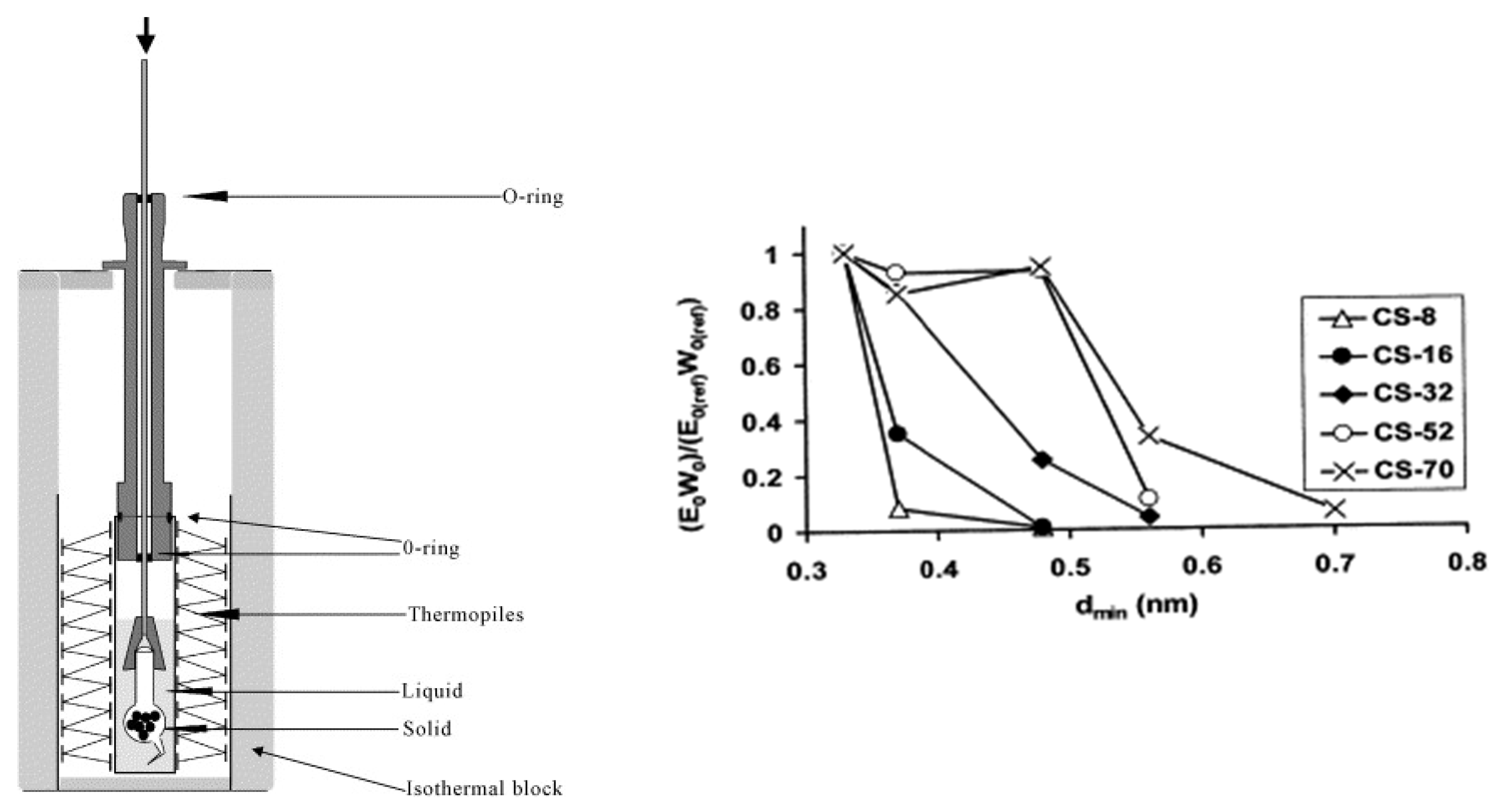
4.3. Adsorption in Gaseous Media
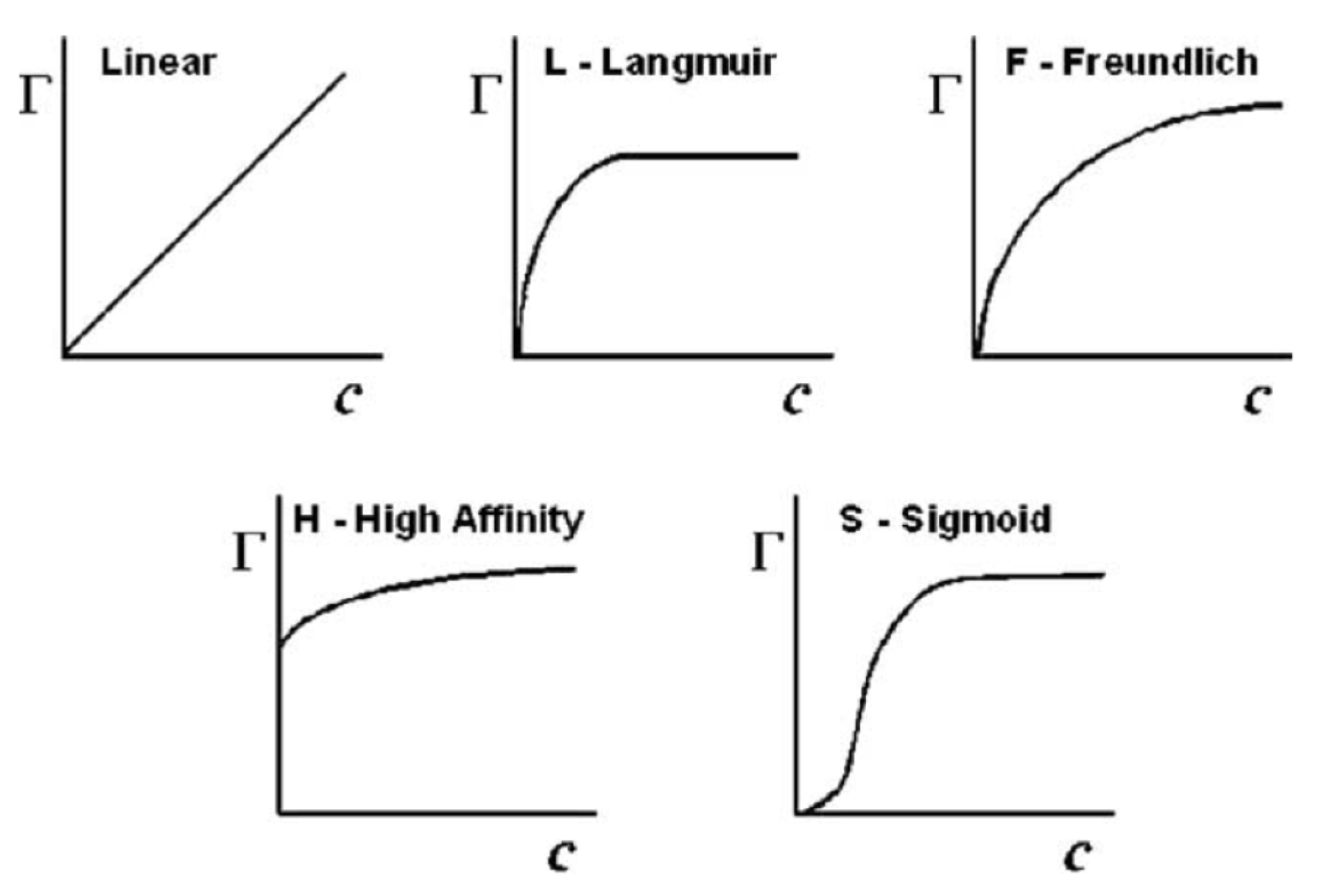
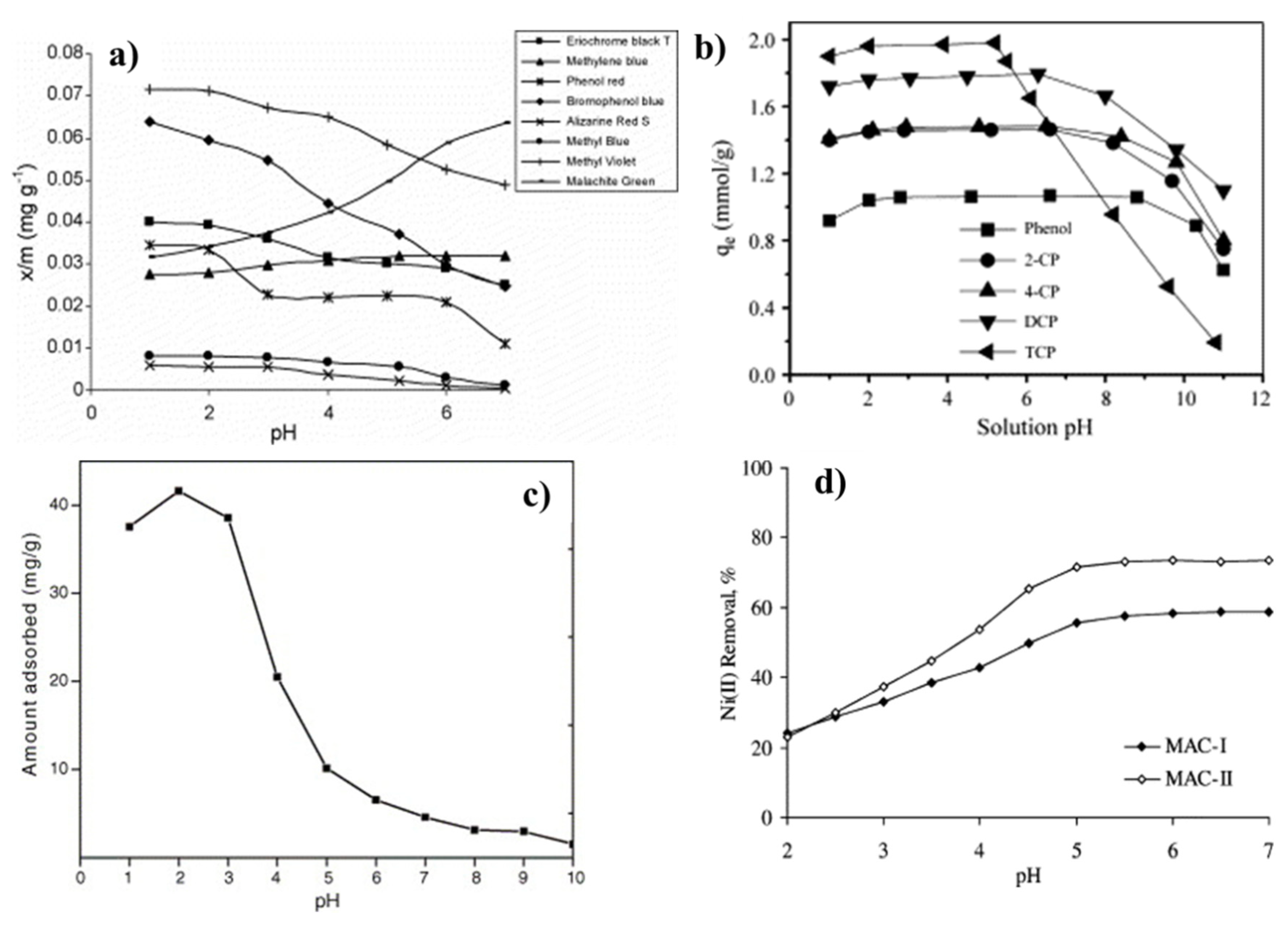
4.4. Adsorption in Liquid Media
5. Other Characterization Techniques
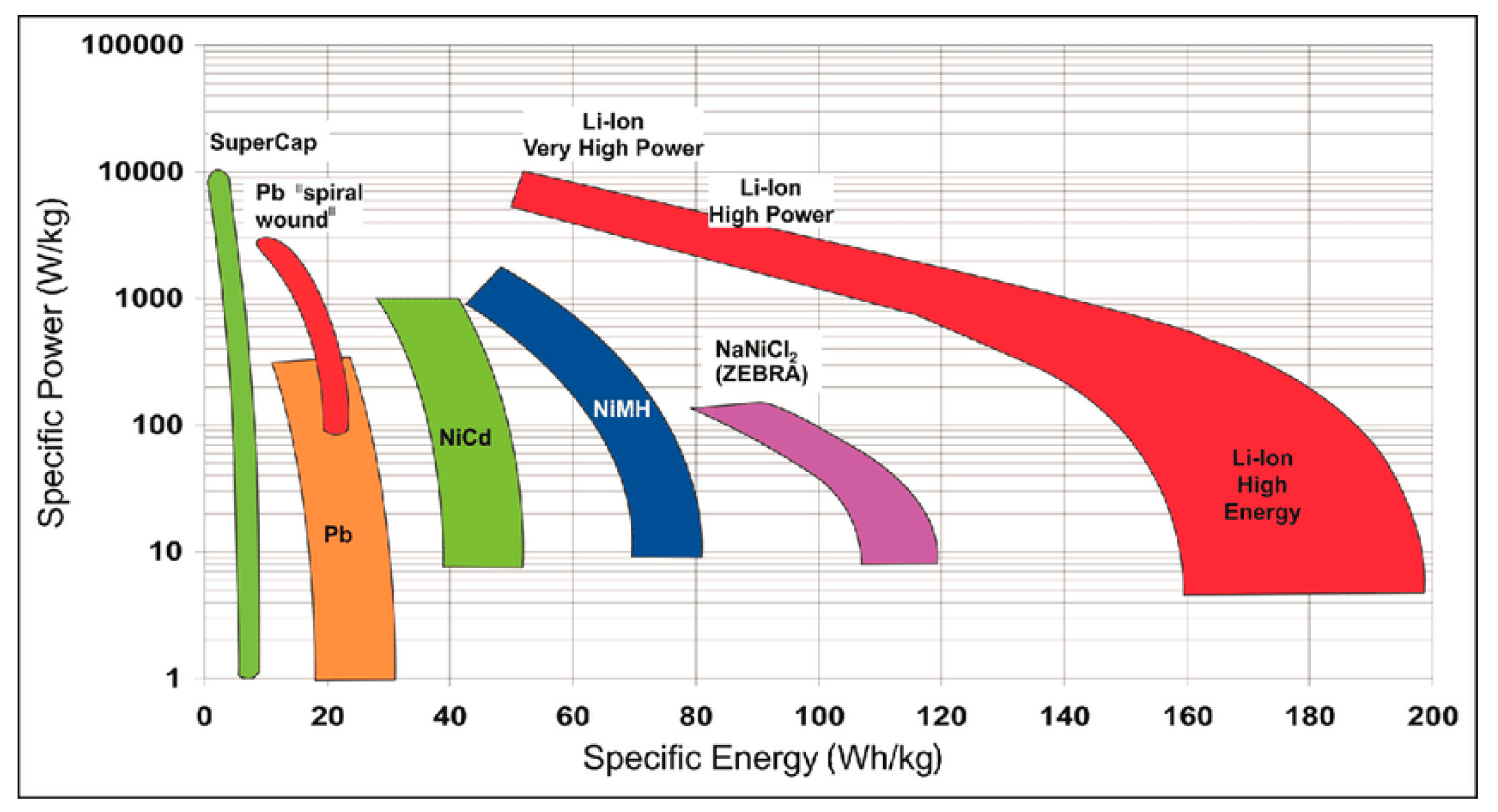
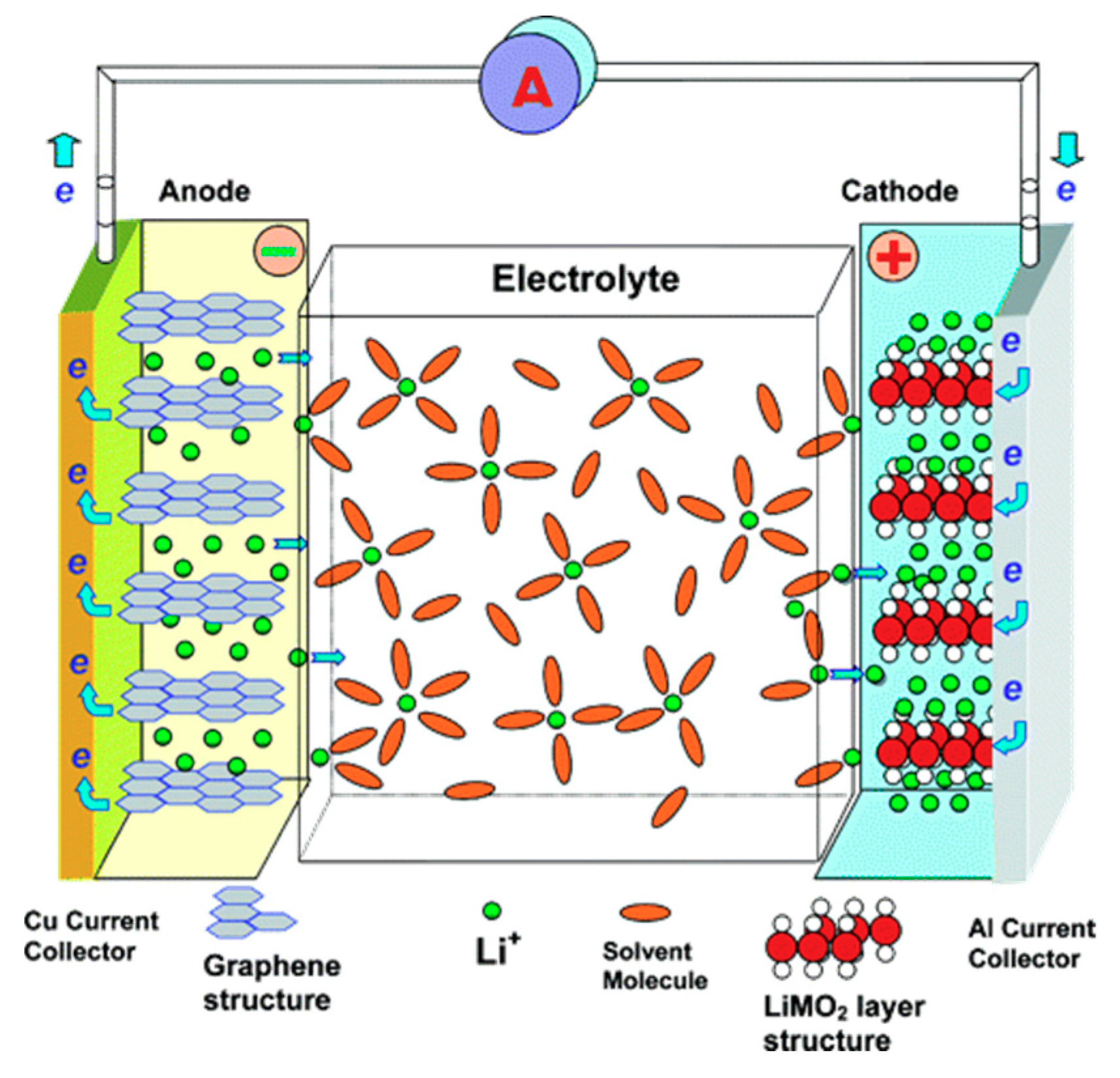
6. Microporous Carbon Materials in Energy Storage Systems
7. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- European Parliament Directive 2009/28/EC of the European Parliament and of the Council of 23 April 2009 on the promotion of the use of energy from renewable sources and amending and subsequently repealing Directives 2001/77/EC and 2003/30/EC. In Official Journal of the European Union; European Union, 2009; Volume 52, pp. 16–62. Available online: http://www.buildup.eu/en/practices/publications/directive-200928ec-european-parliament-and-council-23-april-2009-promotion (accessed on 31 August 2018).
- Marsh, H.; Rodríguez-Reinoso, F. Applicability of Activated Carbon. In Activated Carbon; Elsevier: Oxford, UK, 2006; pp. 383–453. ISBN 9780080444635. [Google Scholar]
- Chen, J.Y. Activated Carbon Fiber and Textiles; Woodhead Publishing: Oxford, UK, 2017; ISBN 9780081006603. [Google Scholar]
- Burns, D.T.; Müller, R.K.; Salzer, R.; Werner, G. Important Figures of Analytical Chemistry from Germany in Brief Biographies; Springer Briefs in Molecular Science; Springer International Publishing: Cham, Switherland, 2014; ISBN 978-3-319-12150-5. [Google Scholar]
- Leicester, H.M. Tobias Lowitz—Discoverer of basic laboratory methods. J. Chem. Educ. 1945, 22, 149. [Google Scholar] [CrossRef]
- Hertzog, E.S.; Broderick, S.J. Activated Carbon for Sugar Decolorization. Ind. Eng. Chem. 1941, 33, 1192–1198. [Google Scholar] [CrossRef]
- Szinicz, L. History of chemical and biological warfare agents. Toxicology 2005, 214, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Peel, R.G.; Benedek, A.; Crowe, C.M. A branched pore kinetic model for activated carbon adsorption. AIChE J. 1981, 27, 26–32. [Google Scholar] [CrossRef]
- Kruse, A.; Funke, A.; Titirici, M.-M. Hydrothermal conversion of biomass to fuels and energetic materials. Curr. Opin. Chem. Biol. 2013, 17, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Dieguez-Alonso, A.; Anca-Couce, A.; Zobel, N.; Behrendt, F. Understanding the primary and secondary slow pyrolysis mechanisms of holocellulose, lignin and wood with laser-induced fluorescence. Fuel 2015, 153, 102–109. [Google Scholar] [CrossRef]
- Funke, A.; Ziegler, F. Hydrothermal carbonization of biomass: A summary and discussion of chemical mechanisms for process engineering. Biofuels Bioprod. Biorefin. 2010, 4, 160–177. [Google Scholar] [CrossRef]
- Hagemann, N.; Spokas, K.; Schmidt, H.-P.; Kägi, R.; Böhler, M.; Bucheli, T. Activated Carbon, Biochar and Charcoal: Linkages and Synergies across Pyrogenic Carbon’s ABCs. Water 2018, 10, 182. [Google Scholar] [CrossRef]
- Jacobi Carbons AB Activated Carbons—General Price Increase Notice. Available online: http://www.jacobi.net/general-price-increase-notice/ (accessed on 7 September 2017).
- Franklin, R.E. Crystallite Growth in Graphitizing and Non-Graphitizing Carbons. Proc. R. Soc. A Math. Phys. Eng. Sci. 1951, 209, 196–218. [Google Scholar] [CrossRef]
- Mohamad Nor, N.; Lau, L.C.; Lee, K.T.; Mohamed, A.R. Synthesis of activated carbon from lignocellulosic biomass and its applications in air pollution control—A review. J. Environ. Chem. Eng. 2013, 1, 658–666. [Google Scholar] [CrossRef]
- Rodríguez-Reinoso, F.; Molina-Sabio, M. Activated carbons from lignocellulosic materials by chemical and/or physical activation: An overview. Carbon N. Y. 1992, 30, 1111–1118. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, Y.; Wang, Z.; Li, Y.; Wang, L.; Ding, L.; Gao, X.; Ma, Y.; Guo, Y. Application studies of activated carbon derived from rice husks produced by chemical-thermal process—A review. Adv. Colloid Interface Sci. 2011, 163, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Correa, C.; Otto, T.; Kruse, A. Influence of the biomass components on the pore formation of activated carbon. Biomass Bioenergy 2017, 97, 53–64. [Google Scholar] [CrossRef]
- Bourke, J.; Manley-Harris, M.; Fushimi, C.; Dowaki, K.; Nunoura, T.; Antal, M.J. Do All Carbonized Charcoals Have the Same Chemical Structure? 2. A Model of the Chemical Structure of Carbonized Charcoal. Ind. Eng. Chem. Res. 2007, 46, 5954–5967. [Google Scholar] [CrossRef]
- Emmerich, F.G. Evolution with heat treatment of crystallinity in carbons. Carbon N. Y. 1995, 33, 1709–1715. [Google Scholar] [CrossRef]
- Anca-Couce, A. Reaction mechanisms and multi-scale modelling of lignocellulosic biomass pyrolysis. Prog. Energy Combust. Sci. 2016, 53, 41–79. [Google Scholar] [CrossRef]
- Han, S.W.; Jung, D.W.; Jeong, J.H.; Oh, E.S. Effect of pyrolysis temperature on carbon obtained from green tea biomass for superior lithium ion battery anodes. Chem. Eng. J. 2014, 254, 597–604. [Google Scholar] [CrossRef]
- Thompson, E.; Danks, A.E.; Bourgeois, L.; Schnepp, Z. Iron-catalyzed graphitization of biomass. Green Chem. 2014, 17, 551–556. [Google Scholar] [CrossRef]
- Antal, M.J.; Allen, S.G.; Dai, X.; Shimizu, B.; Tam, M.S.; Grønli, M. Attainment of the Theoretical Yield of Carbon from Biomass. Ind. Eng. Chem. Res. 2000, 39, 4024–4031. [Google Scholar] [CrossRef]
- Wang, L.; Skreiberg, Ø.; Gronli, M.; Specht, G.P.; Antal, M.J. Is Elevated Pressure Required to Achieve a High Fixed-Carbon Yield of Charcoal from Biomass? Part 2: The Importance of Particle Size. Energy Fuels 2013, 27, 2146–2156. [Google Scholar] [CrossRef]
- Wang, L.; Trninic, M.; Skreiberg, Ø.; Gronli, M.; Considine, R.; Antal, M.J. Is Elevated Pressure Required To Achieve a High Fixed-Carbon Yield of Charcoal from Biomass? Part 1: Round-Robin Results for Three Different Corncob Materials. Energy Fuels 2011, 25, 3251–3265. [Google Scholar] [CrossRef]
- Banyasz, J.L.; Li, S.; Lyons-Hart, J.; Shafer, K.H. Gas evolution and the mechanism of cellulose pyrolysis. Fuel 2001, 80, 1757–1763. [Google Scholar] [CrossRef]
- Klason, P.V; Heidenstam, G.; Norlin, E. Untersuchungen zur Holzverkohlung. Z. Angew. Chem. 1909, 22, 1205–1214. [Google Scholar] [CrossRef]
- Fu, P.; Hu, S.; Xiang, J.; Sun, L.; Su, S.; Wang, J. Evaluation of the porous structure development of chars from pyrolysis of rice straw: Effects of pyrolysis temperature and heating rate. J. Anal. Appl. Pyrolysis 2012, 98, 177–183. [Google Scholar] [CrossRef]
- Henning, K.; von Kienle, H. Carbon, 5. Activated Carbon. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 23–44. ISBN 9783527306732. [Google Scholar]
- Bergius, F. Production of hydrogen from water and coal from cellulose at high temperatures and pressures. J. Soc. Chem. Ind. 1913, 32, 462–467. [Google Scholar] [CrossRef]
- He, C.; Giannis, A.; Wang, J.-Y. Conversion of sewage sludge to clean solid fuel using hydrothermal carbonization: Hydrochar fuel characteristics and combustion behavior. Appl. Energy 2013, 111, 257–266. [Google Scholar] [CrossRef]
- Rodriguez Correa, C.; Bernardo, M.; Ribeiro, R.P.P.L.; Esteves, I.A.A.C.; Kruse, A. Evaluation of hydrothermal carbonization as a preliminary step for the production of functional materials from biogas digestate. J. Anal. Appl. Pyrolysis 2017, 124, 461–474. [Google Scholar] [CrossRef]
- Heilmann, S.M.; Molde, J.S.; Timler, J.G.; Wood, B.M.; Mikula, A.L.; Vozhdayev, G.V.; Colosky, E.C.; Spokas, K.A.; Valentas, K.J. Phosphorus reclamation through hydrothermal carbonization of animal manures. Environ. Sci. Technol. 2014, 48, 10323–10329. [Google Scholar] [CrossRef] [PubMed]
- Demirbas, A. Effects of temperature and particle size on bio-char yield from pyrolysis of agricultural residues. J. Anal. Appl. Pyrolysis 2004, 72, 243–248. [Google Scholar] [CrossRef]
- Antal, M.J.; Mok, W.S.L.; Richards, G.N. Mechanism of formation of 5-(hydroxymethyl)-2-furaldehyde from d-fructose and sucrose. Carbohydr. Res. 1990, 199, 91–109. [Google Scholar] [CrossRef]
- Poerschmann, J.; Weiner, B.; Koehler, R.; Kopinke, F.-D. Hydrothermal Carbonization of Glucose, Fructose, and Xylose—Identification of Organic Products with Medium Molecular Masses. ACS Sustain. Chem. Eng. 2017, 5, 6420–6428. [Google Scholar] [CrossRef]
- Weingarten, R.; Conner, W.C.; Huber, G.W. Production of levulinic acid from cellulose by hydrothermal decomposition combined with aqueous phase dehydration with a solid acid catalyst. Energy Environ. Sci. 2012, 5, 7559–7574. [Google Scholar] [CrossRef]
- Körner, P.; Jung, D.; Kruse, A. The effect of different Brønsted acids on the hydrothermal conversion of fructose to HMF. Green Chem. 2018, 20, 2231–2241. [Google Scholar] [CrossRef]
- Karayıldırım, T.; Sınağ, A.; Kruse, A. Char and Coke Formation as Unwanted Side Reaction of the Hydrothermal Biomass Gasification. Chem. Eng. Technol. 2008, 31, 1561–1568. [Google Scholar] [CrossRef]
- Kruse, A.; Bernolle, P.; Dahmen, N.; Dinjus, E.; Maniam, P. Hydrothermal gasification of biomass: Consecutive reactions to long-living intermediates. Energy Environ. Sci. 2010, 3, 136–143. [Google Scholar] [CrossRef]
- Rodríguez Correa, C.; Stollovsky, M.; Hehr, T.; Rauscher, Y.; Rolli, B.; Kruse, A. Influence of the Carbonization Process on Activated Carbon Properties from Lignin and Lignin-Rich Biomasses. ACS Sustain. Chem. Eng. 2017, 5, 8222–8233. [Google Scholar] [CrossRef]
- Liu, F.; Guo, M. Comparison of the characteristics of hydrothermal carbons derived from holocellulose and crude biomass. J. Mater. Sci. 2015, 50, 1624–1631. [Google Scholar] [CrossRef]
- Sevilla, M.; Fuertes, A.B. The production of carbon materials by hydrothermal carbonization of cellulose. Carbon N. Y. 2009, 47, 2281–2289. [Google Scholar] [CrossRef]
- Garlapalli, R.K.; Wirth, B.; Reza, M.T. Pyrolysis of hydrochar from digestate: Effect of hydrothermal carbonization and pyrolysis temperatures on pyrochar formation. Bioresour. Technol. 2016, 220, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Liu, Y.; Qian, F.; Zhang, S.; Chen, J. Investigation on the Physical and Chemical Properties of Hydrochar and Its Derived Pyrolysis Char for Their Potential Application: Influence of Hydrothermal Carbonization Conditions. Energy Fuels 2015, 29, 5222–5230. [Google Scholar] [CrossRef]
- Bhat, V.V.; Contescu, C.I.; Gallego, N.C. The role of destabilization of palladium hydride in the hydrogen uptake of Pd-containing activated carbons. Nanotechnology 2009, 20, 204011. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.M.; Alvim-Ferraz, M.C.M.; Almeida, M.F.; Rivera-Utrilla, J.; Sánchez-Polo, M. Waste materials for activated carbon preparation and its use in aqueous-phase treatment: A review. J. Environ. Manag. 2007, 85, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Hui, T.S.; Zaini, M.A.A. Potassium hydroxide activation of activated carbon: A commentary. Carbon Lett. 2015, 16, 275–280. [Google Scholar] [CrossRef]
- Maciá-Agulló, J.A.; Moore, B.C.; Cazorla-Amorós, D.; Linares-Solano, A. Activation of coal tar pitch carbon fibres: Physical activation vs. chemical activation. Carbon N. Y. 2004, 42, 1367–1370. [Google Scholar] [CrossRef]
- Jagtoyen, M.; Derbyshire, F. Activated carbons from yellow poplar and white oak by H3PO4 activation. Carbon N. Y. 1998, 36, 1085–1097. [Google Scholar] [CrossRef]
- United Nations Environment Programme. UNEP Year Book 2011: Emerging Issues in Our Global Environment; United Nations Environment Programme: Nairobi, Kenya, 2011; ISBN 978-92-807-3101-9. [Google Scholar]
- Chae, H.K.; Siberio-Perez, D.Y.; Kim, J.; Go, Y.; Eddaoudi, M.; Matzger, A.J.; O’Keeffe, M.; Yaghi, O.M. A route to high surface area, porosity and inclusion of large molecules in crystals. Nature 2004, 427, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Otowa, T.; Tanibata, R.; Itoh, M. Production and adsorption characteristics of MAXSORB: High-surface-area active carbon. Gas Sep. Purif. 1993, 7, 241–245. [Google Scholar] [CrossRef]
- Wen, W. Mechanisms of alkali metal catalysis in the gasification of coal, char, or graphite. Catal. Rev. 1980, 22, 1–28. [Google Scholar] [CrossRef]
- McKee, D.W.; Spiro, C.L.; Kosky, P.G.; Lamby, E.J. Catalysis of coal char gasification by alkali metal salts. Fuel 1983, 62, 217–220. [Google Scholar] [CrossRef]
- Mckee, D.W.; Spiro, C.L.; Kosky, P.G; Lamby, E.J. Catalytic Effects of Alkali Metal Salts in the Gasification of Coal Char. In Symposium on Coal Gasification; 1982; pp. 74–86. Available online: https://www.osti.gov/biblio/5568073 (accessed on 31 August 2018).
- Spiro, C.L.; Mckee, D.W.; Kosky, P.G.; Lamby, E.J. Catalytic CO2-gasification of graphite versus coal char. Fuel 1983, 62, 180–184. [Google Scholar] [CrossRef]
- Tromp, P.J.J.; Cordfunke, E.H.P. A thermochemical study of the reactive intermediate in the alkali-catalyzed carbon gasification. II. Alkali metal vapour pressures. Thermochim. Acta 1984, 81, 113–123. [Google Scholar] [CrossRef]
- Tromp, P.J.J.; Cordfunke, E.H.P. A thermochemical study of the reactive intermediate in the alkali-catalyzed carbon gasification. I. X-ray diffraction results on the alkali-carbon interaction. Thermochim. Acta 1984, 77, 49–58. [Google Scholar] [CrossRef]
- Mims, C.A.; Linden, N.; Pabst, J.K. Alkali-catalyzed carbon gasification. I. Nature of the catalytic sites. Am. Chem. Soc. Div. Fuel Chem. Prepr. 1980, 25, 258–262. [Google Scholar]
- Mims, C.A.; Pabst, J.K. Alkali Catalyzed Carbon Gasification—II. Kinetics and Mechanism. Am. Chem. Soc. Div. Fuel Chem. Prepr. 1980, 25, 263–268. [Google Scholar]
- Lillo-Ródenas, M.; Cazorla-Amorós, D.; Linares-Solano, A. Understanding chemical reactions between carbons and NaOH and KOH: An insight into the chemical activation mechanism. Carbon N. Y. 2003, 41, 267–275. [Google Scholar] [CrossRef]
- Lillo-Ródenas, M.A.; Juan-Juan, J.; Cazorla-Amorós, D.; Linares-Solano, A. About reactions occurring during chemical activation with hydroxides. Carbon N. Y. 2004, 42, 1365–1369. [Google Scholar] [CrossRef]
- Alcañiz-Monge, J.; Illán-Gómez, M.J. Insight into hydroxides-activated coals: Chemical or physical activation? J. Colloid Interface Sci. 2008, 318, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Laine, J.; Calafat, A. Factors affecting the preparation of activated carbons from coconut shell catalized by potassium. Carbon N. Y. 1991, 29, 949–953. [Google Scholar] [CrossRef]
- Sevilla, M.; Ferrero, G.A.; Fuertes, A.B. Beyond KOH activation for the synthesis of superactivated carbons from hydrochar. Carbon 2017, 114, 50–58. [Google Scholar] [CrossRef]
- Tsai, W.T.; Chang, C.Y.; Wang, S.Y.; Chang, C.F.; Chien, S.F.; Sun, H.F. Preparation of activated carbons from corn cob catalyzed by potassium salts and subsequent gasification with CO2. Bioresour. Technol. 2001, 78, 203–208. [Google Scholar] [CrossRef]
- Hayashi, J.; Kazehaya, A.; Muroyama, K.; Watkinson, A.P. Preparation of activated carbon from lignin by chemical activation. Carbon N. Y. 2000, 38, 1873–1878. [Google Scholar] [CrossRef]
- Hayashi, J.; Horikawa, T.; Takeda, I.; Muroyama, K.; Nasir Ani, F. Preparing activated carbon from various nutshells by chemical activation with K2CO3. Carbon N. Y. 2002, 40, 2381–2386. [Google Scholar] [CrossRef]
- Adianta, D.; Wandaud, W.; Aroua, M. Preparation and characterization of activated carbon from palm shell by chemical activation with K2CO3. Bioresour. Technol. 2007, 98, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.B.; Halliop, E.; MacDonald, J.A.F. The production of chemically-activated carbon. Carbon N. Y. 1999, 37, 269–274. [Google Scholar] [CrossRef]
- Lillo-Ródenas, M.A.; Marco-Lozar, J.P.; Cazorla-Amorós, D.; Linares-Solano, A. Activated carbons prepared by pyrolysis of mixtures of carbon precursor/alkaline hydroxide. J. Anal. Appl. Pyrolysis 2007, 80, 166–174. [Google Scholar] [CrossRef]
- Khezami, L.; Chetouani, A.; Taouk, B.; Capart, R. Production and characterisation of activated carbon from wood components in powder: Cellulose, lignin, xylan. Powder Technol. 2005, 157, 48–56. [Google Scholar] [CrossRef]
- Rodríguez Correa, C.; Ngamying, C.; Klank, D.; Kruse, A. Investigation of the textural and adsorption properties of activated carbon from HTC and pyrolysis carbonizates. Biomass Convers. Biorefin. 2017. [Google Scholar] [CrossRef]
- Liou, Y.-J.; Huang, W.-J. A Process for Preparing High Graphene Sheet Content Carbon Materials from Biochar Materials. In Nanotechnology and Nanomaterials “Electroplating of Nanostructures”; InTech: London, UK, 2015; pp. 175–186. ISBN 978-953-51-2213-5. [Google Scholar]
- Lowell, S.; Shields, J.E.; Thomas, M.A.; Thommes, M. Characterization of Porous Solids and Powders-Surface Area, Pore Size and Density; Particle Technology Series; Springer Netherlands: Dordrecht, The Netherlands, 2004; Volume 16, ISBN 978-90-481-6633-6. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Bansal, R.C.; Goyal, M. Activated Carbon Adsorption; Taylor & Francis: Abingdon, UK, 2005; ISBN 9780824753443. [Google Scholar]
- Rouquerol, J.; Llewellyn, P.; Rouquerol, F. Is the bet equation applicable to microporous adsorbents? Stud. Surf. Sci. Catal. 2007, 160, 49–56. [Google Scholar] [CrossRef]
- Thommes, M.; Cychosz, K.A. Physical adsorption characterization of nanoporous materials: Progress and challenges. Adsorption 2014, 20, 233–250. [Google Scholar] [CrossRef]
- Rodríguez-Reinoso, F.; Garrido, J.; Martín-Martínez, J.M.; Molina-Sabio, M.; Torregrosa, R. The combined use of different approaches in the characterization of microporous carbons. Carbon N. Y. 1989, 27, 23–32. [Google Scholar] [CrossRef]
- Rodriguez-Reinoso, F. Preparation and characterization of activated carbons. In Carbon and Coal Gasification; Springer Netherlands: Dordrecht, The Netherlands, 1986; pp. 601–642. [Google Scholar]
- Garrido, J.; Linares-Solano, A.; Martin-Martinez, J.M.; Molina-Sabio, M.; Rodriguez-Reinoso, F.; Torregrosa, R. Use of N2 vs. CO2 in the Characterization of Activated Carbons. Langmuir 1987, 3, 76–81. [Google Scholar] [CrossRef]
- Rios, R.V.R.A.; Silvestre-Albero, J.; Sepúlveda-Escribano, A.; Molina-Sabio, M.; Rodríguez-Reinoso, F. Kinetic Restrictions in the Characterization of Narrow Microporosity in Carbon Materials. J. Phys. Chem. C 2007, 111, 3803–3805. [Google Scholar] [CrossRef]
- Silvestre-Albero, J.; Silvestre-Albero, A.; Rodríguez-Reinoso, F.; Thommes, M. Physical characterization of activated carbons with narrow microporosity by nitrogen (77.4 K), carbon dioxide (273 K) and argon (87.3 K) adsorption in combination with immersion calorimetry. Carbon N. Y. 2012, 50, 3128–3133. [Google Scholar] [CrossRef]
- Rouquerol, J. Porous or divided solids: A special state of matter; 1990. Impact Sci. Soc. 1990, 157, 5–16. [Google Scholar]
- Lippens, B.C.; de Boer, J.H. Studies on Pore Systems in Catalysts III. Pore-Size Distribution Curves in Aluminum Oxide Systems. J. Catal. 1964, 3, 44–49. [Google Scholar] [CrossRef]
- Lippens, B.C.; de Boer, J.H. Studies on pore systems in catalysts: V. The t method. J. Catal. 1965, 4, 319–323. [Google Scholar] [CrossRef]
- De Boer, J.H.; Lippens, B.C.; Linsen, B.G.; Broekhoff, J.C.P.; van den Heuvel, A.; Osinga, T.J. The t-curve of multimolecular N2-adsorption. J. Colloid Interface Sci. 1966, 21, 405–414. [Google Scholar] [CrossRef]
- Polanyi, M. The Potential Theory of Adsorption. Science (80-) 1963, 141, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Ritter, J.A.; Yang, R.T. On the Dubinin-Radushkevich equation for adsorption in microporous solids in the Henry’s law region. Langmuir 1989, 5, 1118–1121. [Google Scholar] [CrossRef]
- Seaton, N.A.; Walton, J.P.R.B.; Quirke, N. A new analysis method for the determination of the pore size distribution of porous carbons from nitrogen adsorption measurements. Carbon N. Y. 1989, 27, 853–861. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Rouquerol, F.; Llewellyn, P.; Rouquerol, J. Assessment of Microporosity. In Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, 2nd ed.; Elsevier: London, UK, 2013; pp. 303–320. ISBN 9780080970356. [Google Scholar]
- Liou, Y.-J.; Huang, W.-J. Quantitative Analysis of Graphene Sheet Content in Wood Char Powders during Catalytic Pyrolysis. J. Mater. Sci. Technol. 2013, 29, 406–410. [Google Scholar] [CrossRef]
- Bandosz, T.J.; Ania, C.O. Surface chemistry of activated carbons and its characterization. In Interface Science and Technology; 2006; Chapter 4; Volume 7, pp. 159–229. ISBN 9780123705365. Available online: https://www.sciencedirect.com/science/article/pii/S157342850680013X (accessed on 31 August 2018).
- Darmstadt, H.; Roy, C. Surface spectroscopic study of basic sites on carbon blacks. Carbon N. Y. 2003, 41, 2662–2665. [Google Scholar] [CrossRef]
- Ilnicka, A.; Gauden, P.A.; Terzyk, A.P.; Lukaszewicz, J.P. Nano-Structured Carbon Matrixes Obtained from Chitin and Chitosan by a Novel Method. J. Nanosci. Nanotechnol. 2016, 16, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, Z.; Zhang, X.; Ren, G.; Lai, Y.; Liu, Y.; Li, J. Highly ordered nitrogen-rich mesoporous carbon derived from biomass waste for high-performance lithium–sulfur batteries. Carbon N. Y. 2015, 84, 399–408. [Google Scholar] [CrossRef]
- Studebaker, M.L. The Chemistry of Carbon Black and Reinforcement. Rubber Chem. Technol. 1957, 30, 1400–1483. [Google Scholar] [CrossRef]
- Montes-Morán, M.A.; Suárez, D.; Menéndez, J.A.; Fuente, E. On the nature of basic sites on carbon surfaces: An overview. Carbon N. Y. 2004, 42, 1219–1224. [Google Scholar] [CrossRef]
- Pietrzak, R. XPS study and physico-chemical properties of nitrogen-enriched microporous activated carbon from high volatile bituminous coal. Fuel 2009, 88, 1871–1877. [Google Scholar] [CrossRef]
- Stuart, B.H. Infrared Spectroscopy: Fundamentals and Applications; Analytical Techniques in the Sciences; John Wiley & Sons, Ltd.: Chichester, UK, 2004; ISBN 9780470011140. [Google Scholar]
- Boehm, H.-P.; Diehl, E.; Heck, W.; Sappok, R. Surface Oxides of Carbon. Angew. Chemie Int. Ed. Engl. 1964, 3, 669–677. [Google Scholar] [CrossRef]
- Lopez-Ramon, M.V.; Stoeckli, F.; Moreno-Castilla, C.; Carrasco-Marin, F. On the characterization of acidic and basic surface sites on carbons by various techniques. Carbon N. Y. 1999, 37, 1215–1221. [Google Scholar] [CrossRef]
- Yang, W.; Wu, D.; Fu, R. Effect of surface chemistry on the adsorption of basic dyes on carbon aerogels. Colloids Surf. A Physicochem. Eng. Asp. 2008, 312, 118–124. [Google Scholar] [CrossRef]
- Tsechansky, L.; Graber, E.R. Methodological limitations to determining acidic groups at biochar surfaces via the Boehm titration. Carbon N. Y. 2014, 66, 730–733. [Google Scholar] [CrossRef]
- Rouquerol, J.; Rouquerol, F.; Llewellyn, P.; Denoyel, R. Surface excess amounts in high-pressure gas adsorption: Issues and benefits. Colloids Surf. A Physicochem. Eng. Asp. 2016, 496, 3–12. [Google Scholar] [CrossRef]
- Llewellyn, P. Adsorption by Ordered Mesoporous Materials. In Adsorption by Powders and Porous Solids; Elsevier: Cambridge, MA, USA, 2014; pp. 529–564. ISBN 9780080970356. [Google Scholar]
- Sevilla, M.; Fuertes, A.B.; Mokaya, R. High density hydrogen storage in superactivated carbons from hydrothermally carbonized renewable organic materials. Energy Environ. Sci. 2011, 4, 1400–1410. [Google Scholar] [CrossRef]
- Lozano-Castelló, D.; Cazorla-Amorós, D.; Linares-Solano, A.; Quinn, D.F. Activated carbon monoliths for methane storage: Influence of binder. Carbon N. Y. 2002, 40, 2817–2825. [Google Scholar] [CrossRef]
- Ribeiro, R.P.; Sauer, T.P.; Lopes, F.V.; Moreira, R.F.; Grande, C.A.; Rodrigues, A.E. Adsorption of CO2, CH4, and N2 in activated carbon honeycomb monolith. J. Chem. Eng. Data 2008, 53, 2311–2317. [Google Scholar] [CrossRef]
- Giraldo, L.; Moreno-Piraján, J.C. Novel Activated Carbon Monoliths for Methane Adsorption Obtained from Coffee Husks. Mater. Sci. Appl. 2011, 2, 331–339. [Google Scholar] [CrossRef]
- Do, D.D. Adsorption Analysis: Equilibria and Kinetics; Series on Chemical Engineering; Imperial College Press: London, UK, 1998; Volume 2, ISBN 1860941303. [Google Scholar]
- Moreno-Castilla, C. Adsorption of organic molecules from aqueous solutions on carbon materials. Carbon N. Y. 2004, 42, 83–94. [Google Scholar] [CrossRef]
- Menéndez, J.A.; Illán-Gómez, M.J.; y León, C.A.L.; Radovic, L.R. On the difference between the isoelectric point and the point of zero charge of carbons. Carbon N. Y. 1995, 33, 1655–1657. [Google Scholar] [CrossRef]
- Noh, J.S.; Schwarz, J.A. Estimation of the point of zero charge of simple oxides by mass titration. J. Colloid Interface Sci. 1989, 130, 157–164. [Google Scholar] [CrossRef]
- Noh, J.S.; Schwarz, J.A. Effect of HNO3 treatment on the surface acidity of activated carbons. Carbon N. Y. 1990, 28, 675–682. [Google Scholar] [CrossRef]
- Hasar, H. Adsorption of nickel(II) from aqueous solution onto activated carbon prepared from almond husk. J. Hazard. Mater. 2003, 97, 49–57. [Google Scholar] [CrossRef]
- Iqbal, M.J.; Ashiq, M.N. Adsorption of dyes from aqueous solutions on activated charcoal. J. Hazard. Mater. 2007, 139, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.-S.; Zheng, T.; Wang, P.; Jiang, J.-P.; Li, N. Adsorption isotherm, kinetic and mechanism studies of some substituted phenols on activated carbon fibers. Chem. Eng. J. 2010, 157, 348–356. [Google Scholar] [CrossRef]
- Karthikeyan, T.; Rajgopal, S.; Miranda, L.R. Chromium(VI) adsorption from aqueous solution by Hevea Brasilinesis sawdust activated carbon. J. Hazard. Mater. 2005, 124, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Parshetti, G.K.; Chowdhury, S.; Balasubramanian, R. Hydrothermal conversion of urban food waste to chars for removal of textile dyes from contaminated waters. Bioresour. Technol. 2014, 161, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meas, A.; Shan, S.; Yang, R.; Gai, X.; Wang, H.; Tsend, N. Hydrochars from bamboo sawdust through acid assisted and two-stage hydrothermal carbonization for removal of two organics from aqueous solution. Bioresour. Technol. 2018, 261, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Ro, K.S.; Sun, K.; Sun, H.; Wang, Z.; Libra, J.A.; Xing, B. New Evidence for High Sorption Capacity of Hydrochar for Hydrophobic Organic Pollutants. Environ. Sci. Technol. 2016, 50, 13274–13282. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Sun, H.; Ro, K.S.; Sun, K.; Libra, J.A.; Xing, B. Removal of antimony (III) and cadmium (II) from aqueous solution using animal manure-derived hydrochars and pyrochars. Bioresour. Technol. 2017, 234, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, T.; Ren, H.; Kruse, A.; Cui, R. Polyethylene imine modified hydrochar adsorption for chromium (VI) and nickel (II) removal from aqueous solution. Bioresour. Technol. 2018, 247, 370–379. [Google Scholar] [CrossRef] [PubMed]
- American Society for Testing and Materials. D4607-94 Standard Test Method for Determination of Iodine Number of Activated Carbon. ASTM Int. 2011, 94, 1–5. [Google Scholar] [CrossRef]
- Hill, A.; Marsh, H. A study of the adsorption of iodine and acetic acid from aqueous solutions on characterized porous carbons. Carbon N. Y. 1968, 6, 31–39. [Google Scholar] [CrossRef]
- Denoyel, R.; Beurroies, I.; Lefevre, B. Thermodynamics of wetting: Information brought by microcalorimetry. J. Pet. Sci. Eng. 2004, 45, 203–212. [Google Scholar] [CrossRef]
- Silvestre-Albero, J.; Gómez de Salazar, C.; Sepúlveda-Escribano, A.; Rodríguez-Reinoso, F. Characterization of microporous solids by immersion calorimetry. Colloids Surf. A Physicochem. Eng. Asp. 2001, 187, 151–165. [Google Scholar] [CrossRef]
- Madou, M.J.; Perez-Gonzalez, V.H.; Pramanick, B. Carbon: The Next Silicon? Book 1, Fundamentals; Momentum Press: New York, NY, USA, 2016; ISBN 9781606507247. [Google Scholar]
- Tian, X.; Zhou, M.; Tan, C.; Li, M.; Liang, L.; Li, K.; Su, P. KOH activated N-doped novel carbon aerogel as efficient metal-free oxygen reduction catalyst for microbial fuel cells. Chem. Eng. J. 2018, 348, 775–785. [Google Scholar] [CrossRef]
- Chen, W.; Feng, H.; Shen, D.; Jia, Y.; Li, N.; Ying, X.; Chen, T.; Zhou, Y.; Guo, J.; Zhou, M. Carbon materials derived from waste tires as high-performance anodes in microbial fuel cells. Sci. Total Environ. 2018, 618, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Paraknowitsch, J.P.; Thomas, A.; Antonietti, M. Carbon Colloids Prepared by Hydrothermal Carbonization as Efficient Fuel for Indirect Carbon Fuel Cells. Chem. Mater. 2009, 21, 1170–1172. [Google Scholar] [CrossRef]
- Cao, D.; Sun, Y.; Wang, G. Direct carbon fuel cell: Fundamentals and recent developments. J. Power Sources 2007, 167, 250–257. [Google Scholar] [CrossRef]
- Cai, W.; Zhou, Q.; Xie, Y.; Liu, J.; Long, G.; Cheng, S.; Liu, M. A direct carbon solid oxide fuel cell operated on a plant derived biofuel with natural catalyst. Appl. Energy 2016, 179, 1232–1241. [Google Scholar] [CrossRef]
- Choudhary, N.; Li, C.; Moore, J.; Nagaiah, N.; Zhai, L.; Jung, Y.; Thomas, J. Asymmetric Supercapacitor Electrodes and Devices. Adv. Mater. 2017, 29, 1605336. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Fan, T.; Zhang, D. Biotemplated Materials for Sustainable Energy and Environment: Current Status and Challenges. ChemSusChem 2011, 4, 1344–1387. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Wang, K.; Wu, L.; Yu, S.-H.; Antonietti, M.; Titirici, M.-M. Engineering Carbon Materials from the Hydrothermal Carbonization Process of Biomass. Adv. Mater. 2010, 22, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, G.A.; Fuertes, A.B.; Sevilla, M.; Titirici, M.-M. Efficient metal-free N-doped mesoporous carbon catalysts for ORR by a template-free approach. Carbon N. Y. 2016, 106, 179–187. [Google Scholar] [CrossRef]
- Calvo, E.G.; Rey-Raap, N.; Arenillas, A.; Menéndez, J.A. The effect of the carbon surface chemistry and electrolyte pH on the energy storage of supercapacitors. RSC Adv. 2014, 4, 32398–32404. [Google Scholar] [CrossRef]
- Chun, S.-E.; Whitacre, J.F. The evolution of electrochemical functionality of carbons derived from glucose during pyrolysis and activation. Electrochim. Acta 2012, 60, 392–400. [Google Scholar] [CrossRef]
- Deheryan, S.; Cott, D.J.; Mertens, P.W.; Heyns, M.; Vereecken, P.M. Direct correlation between the measured electrochemical capacitance, wettability and surface functional groups of CarbonNanosheets. Electrochim. Acta 2014, 132, 574–582. [Google Scholar] [CrossRef]
- Budde-Meiwes, H.; Drillkens, J.; Lunz, B.; Muennix, J.; Rothgang, S.; Kowal, J.; Sauer, D.U. A review of current automotive battery technology and future prospects. Proc. Inst. Mech. Eng. Part D J. Automob. Eng. 2013, 227, 761–776. [Google Scholar] [CrossRef]
- Larcher, D.; Tarascon, J.-M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2015, 7, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Kummer, J.T.; Weber, N. A Sodium-Sulfur Secondary Battery. SAE Trans. 1968, 76, 1003–1007. [Google Scholar] [CrossRef]
- Luo, W.; Shen, F.; Bommier, C.; Zhu, H.; Ji, X.; Hu, L. Na-Ion Battery Anodes: Materials and Electrochemistry. Acc. Chem. Res. 2016, 49, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Xu, K. Nonaqueous Liquid Electrolytes for Lithium-Based Rechargeable Batteries. Chem. Rev. 2004, 104, 4303–4418. [Google Scholar] [CrossRef] [PubMed]
- Moseley, P.T.; Garche, J. Electrochemical Energy Storage for Renewable Sources and Grid Balancing; Elsevier: Amsterdam, The Netherlands, 2015; ISBN 9780444626165. [Google Scholar]
- Zheng, F.; Liu, D.; Xia, G.; Yang, Y.; Liu, T.; Wu, M.; Chen, Q. Biomass waste inspired nitrogen-doped porous carbon materials as high-performance anode for lithium-ion batteries. J. Alloys Compd. 2017, 693, 1197–1204. [Google Scholar] [CrossRef]
- Wen, Y.; He, K.; Zhu, Y.; Han, F.; Xu, Y.; Matsuda, I.; Ishii, Y.; Cumings, J.; Wang, C. Expanded graphite as superior anode for sodium-ion batteries. Nat. Commun. 2014, 5, 4033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, L.; Meng, Y.; Xie, J.; Guo, Y.; Xiao, D. Lithium and sodium storage in highly ordered mesoporous nitrogen-doped carbons derived from honey. J. Power Sources 2016, 335, 20–30. [Google Scholar] [CrossRef]
- Izanzar, I.; Dahbi, M.; Kiso, M.; Doubaji, S.; Komaba, S.; Saadoune, I. Hard carbons issued from date palm as efficient anode materials for sodium-ion batteries. Carbon N. Y. 2018, 137, 165–173. [Google Scholar] [CrossRef]
- Komaba, S.; Murata, W.; Ishikawa, T.; Yabuuchi, N.; Ozeki, T.; Nakayama, T.; Ogata, A.; Gotoh, K.; Fujiwara, K. Electrochemical Na Insertion and Solid Electrolyte Interphase for Hard-Carbon Electrodes and Application to Na-Ion Batteries. Adv. Funct. Mater. 2011, 21, 3859–3867. [Google Scholar] [CrossRef]
- Bommier, C.; Ji, X. Electrolytes, SEI Formation, and Binders: A Review of Nonelectrode Factors for Sodium-Ion Battery Anodes. Small 2018, 14, 1703576. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stoichiometric Composition | Temperature (°C) | Mole% of K | wt. % of K |
|---|---|---|---|
| C8K | 250–318 | 11 | 29 |
| C24K | 356–420 | 4.0 | 12 |
| C36K | 420–487 | 2.7 | 8.3 |
| C48K | 479–508 | 2.0 | 6.4 |
| C60K | Above 500 | 1.6 | 5.1 |
| Gas | Temperature (K) | Vapor Pessure (kPa) | Cross-Sectional Area (nm2/molecule) | Application |
|---|---|---|---|---|
| N2 | 77.4 | 101.35 | 0.162 | Surface area determination |
| Ar | 87.3 | 101.35 | 0.142 | Micropore analysis |
| CO2 | 273.1 | 3485.2 | 0.21 | Pore size distribution for pores <1 nm |
| Kr | 77 | 0.35 | 0.205 | Materials with low surface areas |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correa, C.R.; Kruse, A. Biobased Functional Carbon Materials: Production, Characterization, and Applications—A Review. Materials 2018, 11, 1568. https://doi.org/10.3390/ma11091568
Correa CR, Kruse A. Biobased Functional Carbon Materials: Production, Characterization, and Applications—A Review. Materials. 2018; 11(9):1568. https://doi.org/10.3390/ma11091568
Chicago/Turabian StyleCorrea, Catalina Rodriguez, and Andrea Kruse. 2018. "Biobased Functional Carbon Materials: Production, Characterization, and Applications—A Review" Materials 11, no. 9: 1568. https://doi.org/10.3390/ma11091568
APA StyleCorrea, C. R., & Kruse, A. (2018). Biobased Functional Carbon Materials: Production, Characterization, and Applications—A Review. Materials, 11(9), 1568. https://doi.org/10.3390/ma11091568