CO Oxidation at 20 °C on Au Catalysts Supported on Mesoporous Silica: Effects of Support Structural Properties and Modifiers

 ,
, 

Abstract

1. Introduction
2. Experimental
2.1. Synthesis of Mesoporous Silica and Their Modification with Additives
2.2. Catalyst Preparation
2.3. Catalyst Characterization
2.3.1. N2 Adsorption-Desorption Isotherms
2.3.2. Determination of Metal Content
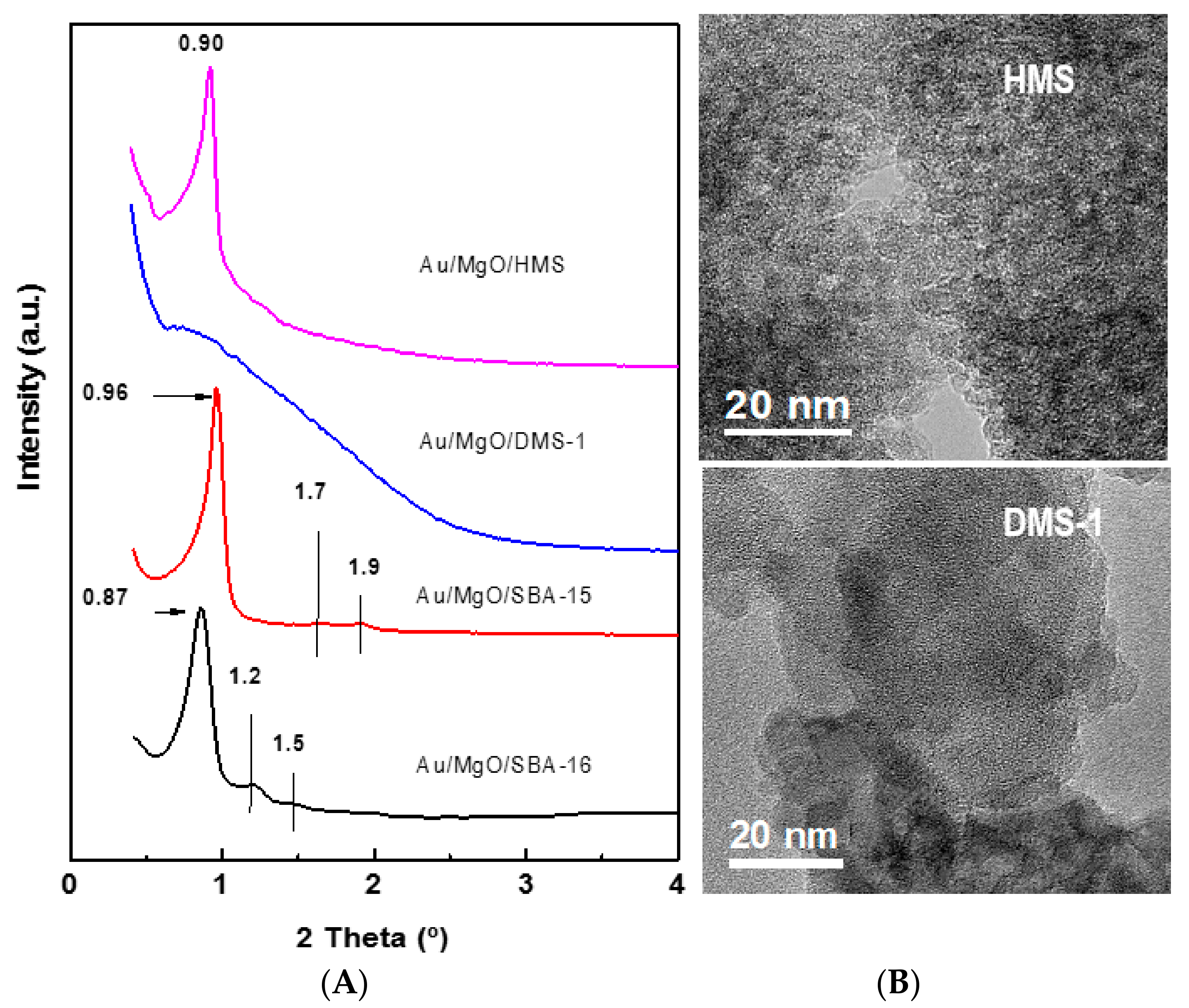
2.3.3. Small-Angle X-ray Diffraction
2.3.4. Powder X-ray Diffraction (XRD)
2.3.5. DRIFT Spectroscopy in the OH Region
2.3.6. DRIFT Spectroscopy of Adsorbed CO (DRIFTS-CO)
2.3.7. Oxygen Storage Capacity (OSC)
2.3.8. UV-vis Diffuse Reflectance Spectra (DRS UV-vis)
2.3.9. X-ray Photoelectron Spectroscopy (XPS)
2.3.10. High Resolution Transmission Electron Microscopy (HRTEM)
2.4. Catalytic Activity Measurements
3. Results
3.1. Catalyst Characterization
3.1.1. Structural Characterization of Pure Supports and Fresh Catalysts
3.1.2. N2 Adsorption-Desorption Isotherms
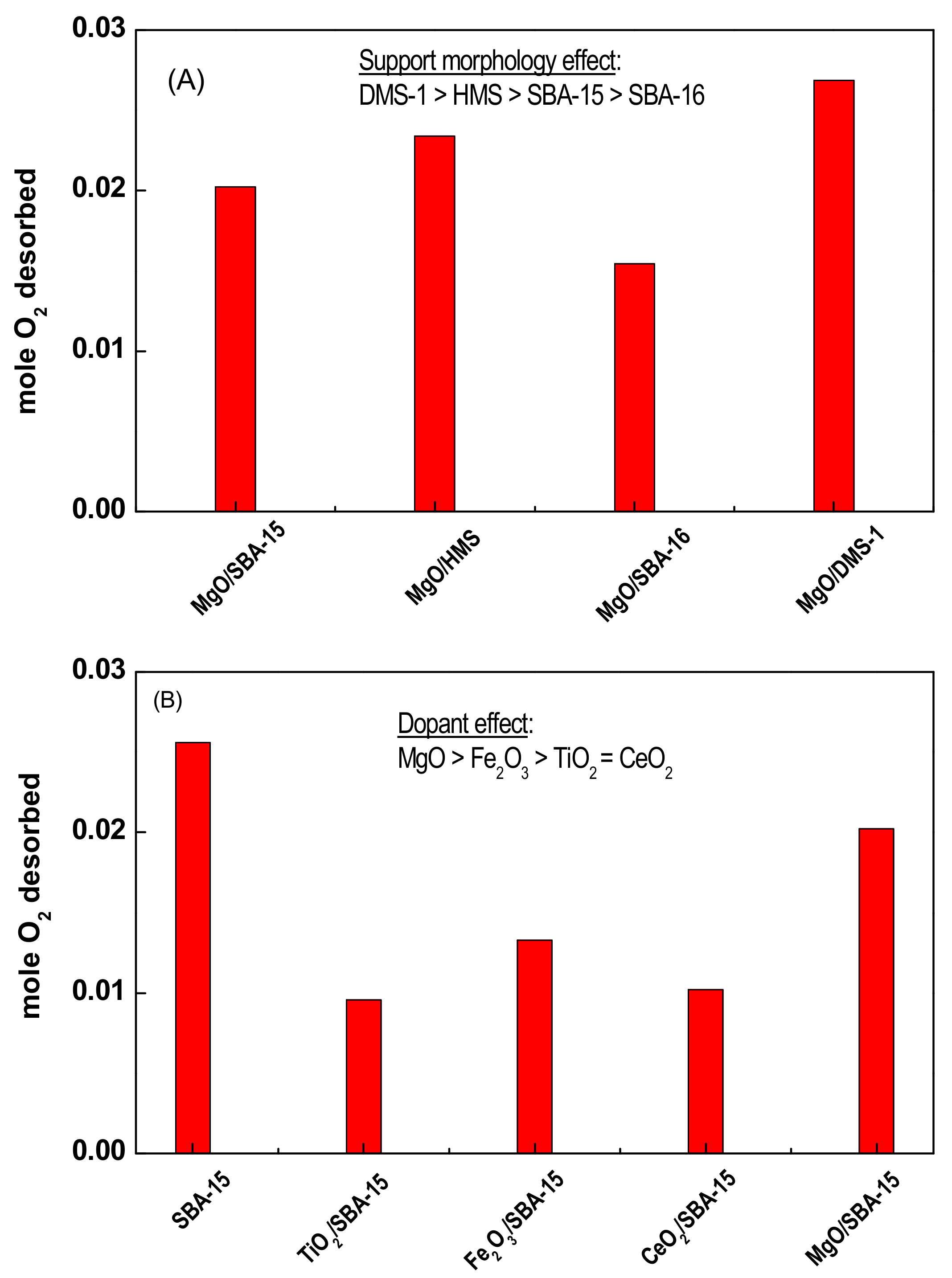
3.1.3. Oxygen Storage Capacity (OSC) of Naked Supports Determined by TPD-O2
3.1.4. Wide-Angle XRD Diffraction of Fresh Catalysts
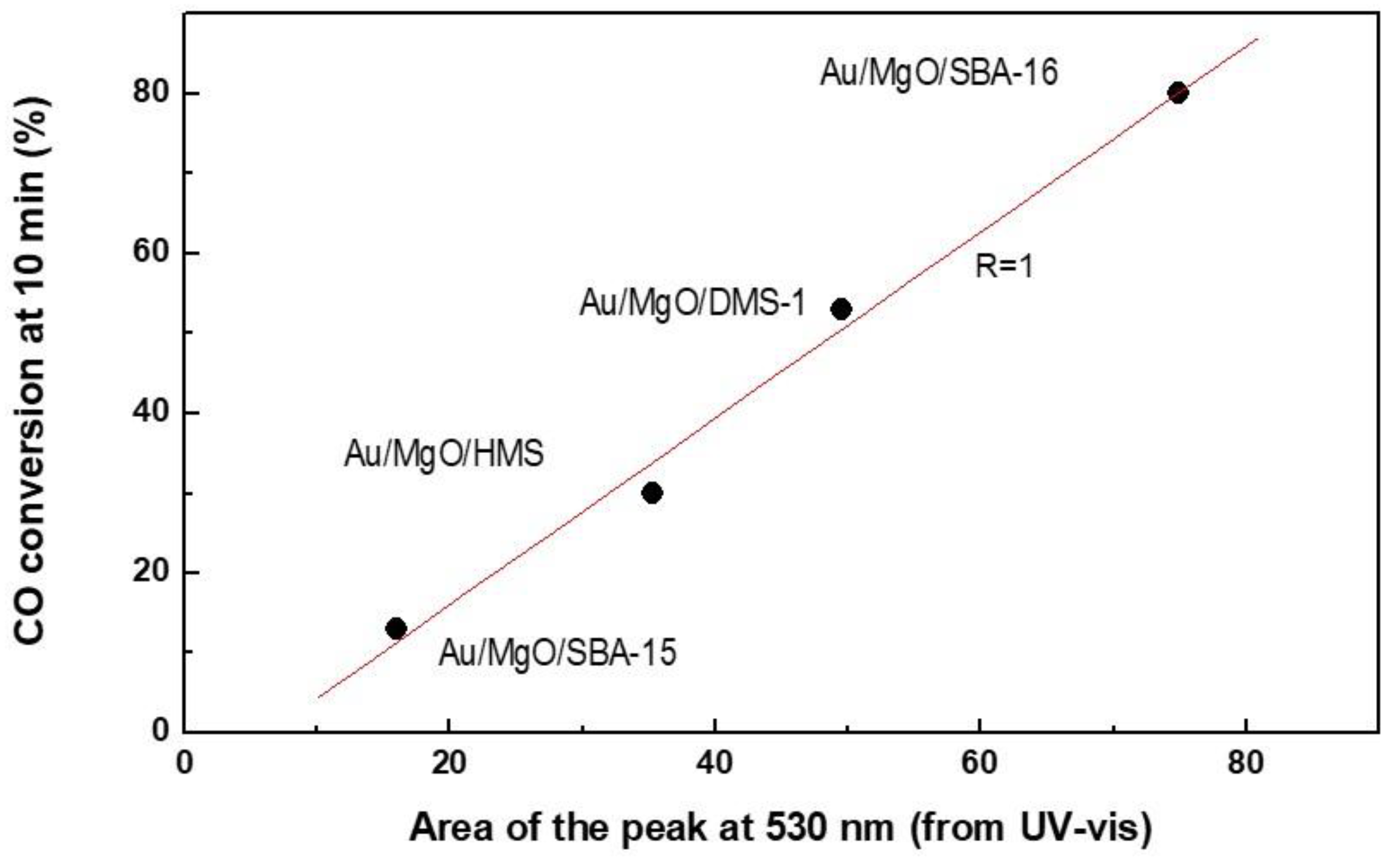
3.1.5. UV-vis Diffuse Reflectance Spectroscopy (UV-vis DRS)
3.1.6. DRIFTS Study of CO Adsorption
3.1.7. DRIFT Spectra of OH Region of Fresh Catalysts
3.1.8. X-ray Photoelectron Spectroscopy of Fresh Catalysts
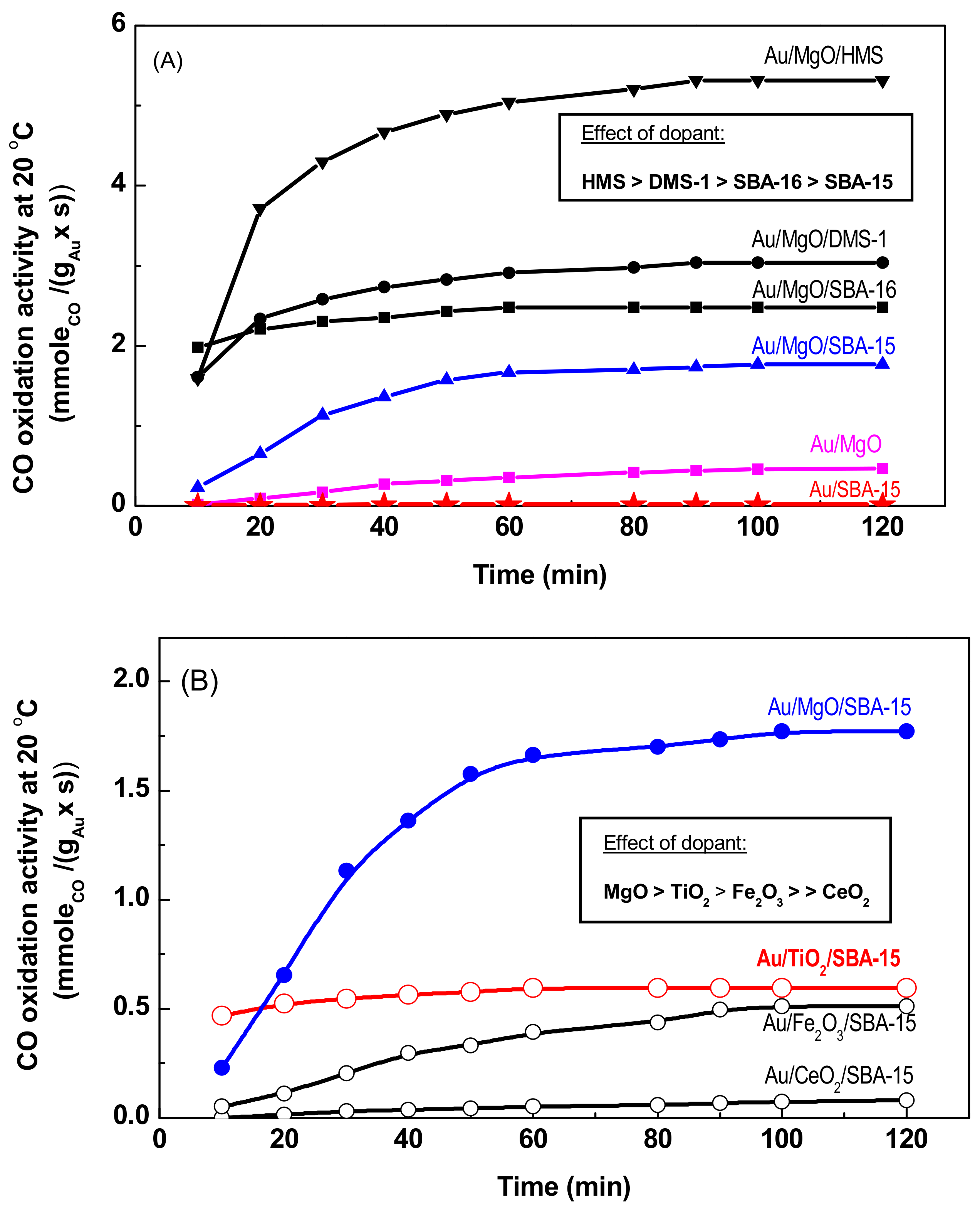
3.2. Catalytic Activity
3.3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M.J.; Delmon, B. Low-temperature oxidation of CO over gold supported on TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175–192. [Google Scholar] [CrossRef]
- Bond, G.C.; Thompson, D.T. Catalysis by gold. Catal. Rev.-Sci. Eng. 1999, 41, 319–388. [Google Scholar] [CrossRef]
- Palomino, R.M.; Gutiérrez, R.M.; Lui, Z.; Tenney, S.; Grinter, D.C.; Crumlin, E.; Waluyo, I.; Ramírez, P.J.; Rodriguez, J.A.; Senanayake, S.D. Inverse catalysts for CO oxidation: Enhanced oxide-metal interactions in MgO/Au(111), CeO2/Au(111), and TiO2/Au(111). ACS Sustsain. Chem. Eng. 2017, 5, 10783–10791. [Google Scholar] [CrossRef]
- Costello, C.K.; Kung, M.C.; Oh, S.-O.; Wang, Y.; Kung, H.H. Nature of the active site for CO oxidation on highly active Au/γ-Al2O3. Appl. Catal. A Gen. 2002, 232, 159–168. [Google Scholar] [CrossRef]
- Mavrikakis, M.; Stoltze, P.; Norskov, J.K. Making gold less noble. Catal. Lett. 2000, 64, 101–106. [Google Scholar] [CrossRef]
- Peza-Ledesma, C.L.; Escamilla-Perea, L.; Nava, R.; Pawelec, B.; Fierro, J.L.G. Supported gold catalysts in SBA-15 modified with TiO2 for oxidation of carbon monoxide. Appl. Catal. A Gen. 2010, 375, 37–48. [Google Scholar] [CrossRef]
- Escamilla-Perea, L.; Nava, R.; Pawelec, B.; Rosmaninho, M.G.; Peza-Ledesma, C.L.; Fierro, J.L.G. SBA-15-supported gold nanoparticles decorated by CeO2: Structural characteristics and CO oxidation activity. Appl. Catal. A Gen. 2010, 381, 42–53. [Google Scholar] [CrossRef]
- Escamilla-Perea, L.; Peza-Ledesma, C.L.; Nava, R.; Rivera-Muñoz, E.M.; Pawelec, B.; Fierro, J.L.G. CO oxidation at 20 °C over Au/SBA-15 catalysts decorated by Fe2O3 nanoparticles. Catal. Commun. 2011, 15, 108–112. [Google Scholar] [CrossRef]
- Kung, H.H.; Kung, M.C.; Costello, C.K. Supported Au catalysts for low temperature CO oxidation. J. Catal. 2003, 216, 425–432. [Google Scholar] [CrossRef]
- Liu, X.L.; Wang, A.; Zhang, T.; Mou, C.-Y. Catalysis by gold: New insights into the support effect. Nano Today 2013, 8, 403–416. [Google Scholar] [CrossRef]
- Widmann, D.; Liu, Y.; Schüth, F.; Behm, R.J. Support effects in the Au-catalyzed CO oxidation—Correlation between activity, oxygen storage capacity, and support reducibility. J. Catal. 2010, 276, 292–305. [Google Scholar] [CrossRef]
- Schubert, M.M.; Hackenberg, S.; Veen, A.C. van; Muhler, M.; Plzak, V.; Behm, R.J. CO oxidation over supported gold catalysts-“inert” and “active” support materials and their role for the oxygen supply during reaction. J. Catal. 2001, 197, 113–122. [Google Scholar] [CrossRef]
- Hernandez, J.A.; Gómez, S.; Pawelec, B.; Zepeda, T.A. CO oxidation on Au nanoparticles supported on wormhole HMS material: Effect of support modification with CeO2. Appl. Catal. B Environ. 2009, 89, 128–136. [Google Scholar] [CrossRef]
- Liotta, L.F.; Pantaleo, G.; Venezia, A.M. Au/CeO2-SBA-15 catalysts for CO oxidation: Effect of ceria loading on physic-chemical properties and catalytic performance. Catal. Today 2012, 187, 10–19. [Google Scholar] [CrossRef]
- Zepeda, T.A.; Martinez-Hernández, A.; Guil-López, R.; Pawelec, B. Preferential CO oxidation in excess of hydrogen over Au/HMS catalysts modified by Ce, Fe and Ti oxides. Appl. Catal. B Environ. 2010, 100, 450–462. [Google Scholar] [CrossRef]
- Ramírez-Garza, R.E.; Pawelec, B.; Zepeda, T.A.; Martinez-Hernández, A. Total CO oxidation over Fe-containing Au/HMS catalysts: Effects of gold loading and catalyst pretreatment. Catal. Today. 2011, 172, 95–102. [Google Scholar] [CrossRef]
- Moreau, F.; Bond, G.C.; Linden, B.J. van der; Silberova, B.; Makkee, M. Gold supported on mixed oxides for the oxidation of carbon monoxide. Appl. Catal. B Environ. 2008, 347, 208–215. [Google Scholar] [CrossRef]
- Casaletto, M.P.; Longo, A.; Venezia, A.M.; Mortorana, A.; Prestianni, A. Metal-support and preparation influence on the structural and electronic properties of gold catalysts. Appl. Catal. A Gen. 2006, 302, 309–316. [Google Scholar] [CrossRef]
- Oh, H.S.; Yang, J.H.; Costello, C.K.; Wang, Y.M.; Bare, S.R.; Kung, H.H.; Kung, M.C. Selective catalytic oxidation of CO: Effect of chloride on supported Au catalysts. J. Catal. 2002, 210, 375–386. [Google Scholar] [CrossRef]
- Mizera, J.; Spiridis, N.; Socha, R.; Grabowski, R.; Samson, K.; Korecki, J.; Grzybowska, B.; Gurgul, J.; Kępiński, L.; Małecka, M.A. Au/FeOx catalysts of different degree of iron oxide reduction. Catal. Today 2012, 187, 20–29. [Google Scholar] [CrossRef]
- Socha, R.P.; Zackiewicz, E.; Spiridis, N.; Korecki, J. Au adsorption on defect-rich MgO(100) surfaces. Surf. Interface Anal. 2010, 42, 536–539. [Google Scholar] [CrossRef]
- Ma, Z.; Dai, S. Development of novel gold catalysts for CO oxidation reaction: A materials perspective. Nano. Res. 2011, 4, 3–32. [Google Scholar] [CrossRef]
- Ma, G.; Binder, A.; Chi, M.; Liu, C.; Jin, R.; Jiang, D.; Fan, J.; Dai, S. Stabilizing gold clusters by heterostructured transition-metal oxide–mesoporous silica supports for enhanced catalytic activities for CO oxidation. Chem. Commun. 2012, 48, 11413–11415. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, L.-F.; Hamoudi, S.; Belkacemi, K. Synthesis of gold catalysts supported on mesoporous silica materials: Recent developments. Catalysts 2011, 1, 97–154. [Google Scholar] [CrossRef]
- Lopez, N.; Janssens, T.V.; Clausen, B.S.; Xu, Y.; Mavrikakis, M.; Bligaad, T.; Nørskov, J.K. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Bonelli, R.; Lucarelli, C.; Pasini, T.; Liotta, L.F.; Zacchini, S.; Albonetti, S. Total oxidation of valatile organic compounds on Au/FeOx catalysts supported on mesoporous SBA-15 silica. Appl. Catal. A Gen. 2011, 400, 54–60. [Google Scholar] [CrossRef]
- Stevens, W.J.J.; Lebeau, K.; Mertens, M.; van Tendeloo, G.; Cool, P.; Vansant, E.F. Investigation of the structural properties of the mesoporous SBA-16 and SBA-15 Materials. J. Phys. Chem. B 2006, 110, 9183–9187. [Google Scholar] [CrossRef] [PubMed]
- Grudzien, R.M.; Grabicka, B.E.; Jaroniec, M. Adsorption and structural properties of channel-like and cage-like organosilicas. Adsorption 2006, 12, 293–308. [Google Scholar] [CrossRef]
- Rivera-Muñoz, E.M.; Pawelec, B.; Nava, R.; Cortés, J.A.; Huirache-Acuña, R. SBA-16 Mesoporous Silica as Catalytic Support for Hydrodesulfurization Catalysts. In Comprehensive Guide for Mesoporous Materials; Aliofkazraei, M., Ed.; Nova Scientific Publisher: Hauppauge, NY, USA, 2015; Volume 4, Chapter 15; pp. 343–361. [Google Scholar]
- Bahaumik, A.; Samanta, S.; Kishor Mal, N. Highly active disordered extra large pore titanium silicate. Microporous Mesoporous Mater. 2004, 68, 29–35. [Google Scholar] [CrossRef]
- Somodi, F.; Borbáth, I.; Hegedus, M.; Tompos, A.; Sajó, I.E.; Szegedi, A.; Rojas, S.; Fierro, J.L.G.; Margifalvi, J.L. Modified preparation method for highly active Au/SiO2 catalysts used in CO oxidation. Appl. Catal. A Gen. 2008, 347, 216–222. [Google Scholar] [CrossRef]
- Szabó, E.G.; Hegedűs, M.; Szegedi, Á.; Sajó, I.; Margitfalvi, J.L. CO oxidation over Au/Al2O3 catalysts modified by MgO. React. Kinet. Catal. Lett. 2005, 86, 339–345. [Google Scholar] [CrossRef]
- Szabó, E.G.; Hegedűs, M.; Lónyi, F.; Szegedi, Á.; Datye, A.K.; Margitfalvi, J.L. Preparation, characterization and activity of Au/Al2O3 modified by MgO. Catal. Commun. 2009, 10, 889–893. [Google Scholar] [CrossRef]
- Walter, M.; Frondelius, P.; Honkala, K.; Hakkinen, H. Electronic Structure of MgO-supported Au clusters: Quantum dots probed by scanning tunneling microscopy. Phys. Chem. Rev. Lett. 2007, 99, 096102. [Google Scholar] [CrossRef] [PubMed]
- Landman, U.; Yoon, B.; Zhang, C.; Heiz, U.; Arenz, M. Factors in gold nanocatalysis: Oxidation of CO in the non-scalable size regime. Top. Catal. 2007, 44, 145–158. [Google Scholar] [CrossRef]
- Freund, H.-J. Metal-supported ultrathin oxide film systems as designable catalysts and catalyst supports. Surf. Sci. 2007, 601, 1438–1442. [Google Scholar] [CrossRef]
- Janssens, T.V.W.; Clausen, B.S.; Hyolbek, B.; Falsaig, H.; Christensen, C.H.; Bligaard, T.; Norskov, K. Insights into the reactivity of supported Au nanoparticles: Combining theory and experiments. Top. Catal. 2007, 44, 15–26. [Google Scholar] [CrossRef]
- Min, B.K.; Friend, C.M. Heterogeneous gold-based catalysts for green chemistry: Low temperature CO oxidation and propane oxidation. Chem. Rev. 2007, 107, 2709–2724. [Google Scholar] [CrossRef] [PubMed]
- Grisel, R.J.H.; Nieuwenhuys, B.E. Selective oxidation of CO over supported Au catalysts. J. Catal. 2001, 199, 48–59. [Google Scholar] [CrossRef]
- Grisel, R.J.H.; Nieuwenhuys, B.E. A comparative study of the oxidation of CO and CH4 over Au/MOx/Al2O3 catalysts. Catal. Today 2001, 64, 69–81. [Google Scholar] [CrossRef]
- Flodström, K.; Alfredsson, V. Influence of the block length of triblock copolymers on the formation of mesoporous silica. Microporous Mesoporous Mater. 2003, 59, 167–176. [Google Scholar]
- Zhao, D.Y.; Huo, Q.S.; Feng, J.L.; Chmelka, B.F.; Stucky, G.D. Nonionic Triblock and Star Diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J. Am. Chem. Soc. 1998, 120, 6024–6036. [Google Scholar] [CrossRef]
- Tanev, P.T.; Pinnavaia, T.J. Mesoporous silica sieves prepared by ionic and neutral surfactant templating: A comparison of physical properties. Chem. Mater. 1996, 8, 2068–2079. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Yuranov, I.; Moeckli, P.; Suvorova, E.; Buffat, P.; Kiwi-Minsker, L.; Renken, A. Pd/SiO2 catalysts: Synthesis of Pd nanoparticles with the controlled size in mesoporous silicas. J. Mol. Catal. A Chem. 2003, 192, 239–251. [Google Scholar] [CrossRef]
- Kielbassa, S.; Häbich, A.; Schnaidt, J.; Bansmann, J.; Weigl, F.; Boyen, H.-G.; Ziemann, P.; Behm, R.J. On the morphology and stability of Au nanoparticles on TiO2(110) prepared from micelle-stabilized precursors. Langmuir 2006, 22, 7873–7880. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-C.; Huang, X.-S.; Liu, Q.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N.; Zhuang, J.-H. Gold nanoparticles deposited on manganese(III) oxide as novel efficient catalyst for low temperature CO oxidation. J. Catal. 2008, 259, 66–74. [Google Scholar] [CrossRef]
- Claus, P.; Brückner, A.; Mohr, C.; Hofmeister, H. Supported gold nanoparticles from quantum dot to mesoscopic size scale: Effect of electronic and structural properties on catalytic hydrogenation of conjugated functional groups. J. Am. Chem. Soc. 2000, 122, 11430–11439. [Google Scholar] [CrossRef]
- Zanella, R.; Giorgio, S.; Ho Shin, C.; Henry, C.R.; Louis, C. Characterization and reactivity in CO oxidation of gold nanoparticles supported on TiO2 prepared by deposition-precipitation with NaOH and urea. J. Catal. 2004, 222, 357–367. [Google Scholar] [CrossRef]
- Liu, H.; Lu, G.; Guo, Y.; Guo, Y.; Wang, J. Study on the synthesis and the catalytic properties of Fe-HMS materials in the hydroxylation of phenol. Microporous Mesoporous Mater. 2008, 108, 56–64. [Google Scholar] [CrossRef]
- Truel, A.; Hubert-Pfalzgraf, L.G. Nonometric monodispersed titanium oxide particles on mesoporous silica: Synthesis, characterization and catalytic activity in oxidation reactions in the liquid phase. J. Catal. 2003, 217, 343–353. [Google Scholar]
- Boccuzzi, F.; Chiorino, A.; Tsubota, S.; Haruta, M. FTIR Study of carbon monoxide oxidation and scrambling at room temperature over gold supported on ZnO and TiO2. J. Phys. Chem. 1996, 100, 3625–3631. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- Zaki, M.I.; Knözinger, H. Characterization of oxide surfaces by adsorption of carbon monoxide-a low temperature infrared spectroscopy study. Spectrochim. Acta Part A Mol. Spectrosc. 1987, 43, 1455–1459. [Google Scholar] [CrossRef]
- Luan, Z.; Fournier, J.A. In situ FRIR spectroscopic investigation of active sites and adsorbate interactions in mesoporous aluminosilicate SBA-15 molecular sieves. Microporous Mesoporous Mater. 2005, 79, 235–240. [Google Scholar] [CrossRef]
- Armaroli, T.; Bécue, T.; Gautier, S. Diffuse Reflection Infrared Spectroscopy (DRIFTS): Application to the in situ Analysis of Catalysts. Oil Gas Sci. Technol. Rev. IFP 2004, 59, 215–237. [Google Scholar] [CrossRef]
- Venezia, A.M.; La Parola, V.; Deganello, G.; Pawelec, B.; Fierro, J.L.G. Synergetic effect of gold in Au/Pd catalysts during hydrodesulfurization reactions of model compounds. J. Catal. 2003, 215, 317–325. [Google Scholar] [CrossRef]
- Karadas, F.; Ertas, G.; Ozkaraoglu, E.; Suzer, S. X-ray-induced production of gold nanoparticles on a SiO2/Si system and in a poly(methyl methacrylate) matrix. Langmuir 2005, 21, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Carley, A.F.; Chalker, P.R.; Riviere, J.C.; Wyn Roberts, M. The identification and characterisation of mixed oxidation states at oxidised titanium surfaces by analysis of X-ray photoelectron spectra. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 1987, 83, 351–370. [Google Scholar] [CrossRef]
- Nava, R.; Ortega, R.A.; Alonso, G.; Ornelas, C.; Pawelec, B.; Fierro, J.L.G. CoMo/Ti-SBA-15 catalysts for dibenzothiophene desulfurization. Catal. Today 2007, 127, 70–84. [Google Scholar] [CrossRef]
- Herranz, T.; Rojas, S.; Pérez-Alonso, F.J.; Ojeda, M.; Terreros, P.; Fierro, J.L.G. Carbon oxide hydrogenation over silica-supported iron-based catalysts: Influence of the preparation route. Appl. Catal. A Gen. 2006, 308, 19–30. [Google Scholar] [CrossRef]
- Parks, G.A. The isoelectric points of solid oxides, solid hydroxides, and aqueous hydroxo complex systems. Chem. Rev. 1965, 65, 177–198. [Google Scholar] [CrossRef]
- Xu, X.; Goodman, D.W. The preparation and characterization of ultra-thin silicon dioxide films on Mo(110) surface. Surf. Sci. 1993, 282, 323–333. [Google Scholar] [CrossRef]
- Rainer, D.R.; Goodman, D.W. Metal clusters on ultrathin oxide films: Model catalysts for surface science studies. J. Mol. Catal. A Chem. 1998, 131, 259–283. [Google Scholar] [CrossRef]
- Grunwaldt, J.-D.; Maciejewski, M.; Becker, O.S.; Fabrizioli, P.; Baiker, A. Comparative study of Au/TiO2 and Au/ZrO2 catalysts for low-temperature CO oxidation. J. Catal. 1999, 186, 458–469. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Malwadkar, S.; Chilukuri, S. Cu-Ce mixed oxides on Al-pillared clay: Effect of method of preparation on catalytic activity in the preferential oxidation of carbon monoxide. Appl. Catal. B Environ. 2008, 84, 21–29. [Google Scholar] [CrossRef]
- Bond, G.C.; Rawle, A.F. Catalytic hydrogenation in the liquid phase. Part 1. Hydrogenation of isoprene catalysed by palladium, palladium-gold and palladium-silver catalysts. J. Mol Catal. A Chem. 1996, 199, 261–271. [Google Scholar] [CrossRef]
- Li, Z.; Ciobanu, C.V.; Hu, J.; Palomares-Báez, J.P.; Rodriguez-López, J.L.; Richards, R. Experimental and DFT studies of gold nanoparticles supported on MgO(111) nano-sheets and their catalytic activity. Phys. Chem. Chem. Phys. 2011, 13, 2582–2589. [Google Scholar] [CrossRef] [PubMed]
- Molina, L.M.; Hammer, B. Some recent theoretical advances in the understanding of the catalytic activity of Au. Appl. Catal. A Gen. 2005, 291, 21–31. [Google Scholar] [CrossRef]
- Comotti, M.; Li, W.-C.; Spliethoff, B.; Schüth, F. Support Effect in High Activity Gold Catalysts for CO Oxidation. J. Am. Chem. Soc. 2006, 128, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.; Häkkinen, H.; Landman, U.; Wörz, A.S.; Antonietti, J.M.; Abbet, S.; Judai, K.; Heiz, U. Charging effects on bonding and catalyzed oxidation of CO on Au8 clusters on MgO. Science 2005, 307, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Costello, C.K.; Guzman, J.; Yang, J.H.; Wang, Y.M.; Kung, M.C.; Gates, B.C.; Kung, H.H. Activation of Au/γ-Al2O3 catalysts for CO oxidation: Characterization by X-ray absorption near edge structure and temperature programmed reduction. J. Phys. Chem. B 2004, 108, 12529–12536. [Google Scholar] [CrossRef]
- Valden, M.; Lai, X.; Goodman, D.W. Onset of Catalytic Activity of Gold Clusters on Titania with the Appearance of Nonmetallic Properties. Science 1998, 281, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Hodge, N.A.; Kiely, C.J.; Whyman, R.; Siddiqui, M.R.H.; Hutchings, G.J.; Bond, G.C.; Thompson, D.T. Gold-catalysed oxidation of carbon monoxide. Gold Bull. 2000, 33, 41–50. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Support | M (wt %) | SBET (m² g−1) | NSBET | Vtotal (m3 g−1) | d (nm) |
|---|---|---|---|---|---|
| HMS | - | 976 | - | n.c. | 5.6 |
| MgO/HMS | 5.2 | 573 | 0.6 | n.c. | 5.7 |
| DMS-1 | - | 772 | - | 0.46 | 2.4 |
| MgO/DMS-1 | 5.6 | 165 | 0.2 | 0.11 | 2.8 |
| SBA-16 | 275 | - | 0.18 | 2.6 | |
| MgO/SBA-16 | 5.6 | 262 | 1.0 | 0.18 | 2.8 |
| SBA-15 | - | 819 | - | 0.96 | 4.7 |
| MgO/SBA-15 | 5.3 | 302 | 0.4 | 0.43 | 5.7 |
| Fe2O3/SBA-15 | 5.3 | 634 | 0.8 | 0.84 | 5.7 |
| TiO2/SBA-15 | 7.3 | 555 | 0.7 | 0.69 | 5.0 |
| CeO2/SBA-15 | 5.6 | 584 | 0.8 | 0.69 | 4.8 |
| Catalyst | Au a (wt %) | dAu c (nm) | d MOx c (nm) | SBET (m² g−1) | NSBET d | Vtotal (m3 g−1) | dpores (nm) |
|---|---|---|---|---|---|---|---|
| Au/MgO/HMS | 0.9 | 6.3 | n.d. | 574 | 1.0 | n.c. | 7.2 |
| Au/MgO/DMS-1 | 0.5 | 7.4 | n.d. | 268 | 1.6 | 0.27 | 4.1 |
| Au/MgO/SBA-16 | 0.6 | 7.9 | n.d. | 306 | 1.2 | 0.38 | 4.9 |
| Au/MgO/SBA-15 | 0.8 | 7.3 (5.6) | n.d. | 398 | 1.3 | 0.70 | 7.0 |
| Au/SBA-15 | 1.0 | n.d. (15.0) | n.d | 487 | 0.6 | 0.72 | 5.9 |
| Au/Fe2O3/SBA-15 | 2.9 | 5.9 (7.5) | 16.7 | 467 | 0.7 | 0.68 | 6.3 |
| Au/TiO2/SBA-15 | 2.5 | 7.6 (5.5) | n.d. | 616 | 1.1 | 0.81 | 5.3 |
| Au/CeO2/SBA-15 | 2.7 | 6.1 (6.2) | 4.8 | 462 | 0.8 | 0.67 | 5.8 |
| Catalyst | Au 4f7/2 | Fe 2p3/2 | Mg 2p | Ti 2p3/2 | Ce 3d5/2 | |
|---|---|---|---|---|---|---|
| Au0 | Au1+ | |||||
| Au/MgO/HMS | 83.8 (62) | 85.1 (38) | - | 50.8 | - | - |
| Au/MgO/DMS-1 | 83.9 (33) | 85.1 (67) | - | 50.7 | - | - |
| Au/MgO/SBA-16 | 83.8 (29) | 85.0 (71) | - | 50.8 | - | - |
| Au/MgO/SBA-15 | 83.8 (33) | 85.0 (67) | - | 50.7 | - | - |
| Au/Fe2O3/SBA-15 | 83.9 (82) | 85.1 (18) | 710.7 | - | - | - |
| Au/TiO2/SBA-15 | 84.0 (100) | - | - | - | 459.3 | - |
| Au/CeO2/SBA-15 | 83.8 (80) | 85.0 (20) | - | - | - | 882.8 |
| Au/SBA-15 | 83.8 (70) | 85.0 (30) | - | - | - | - |
| Catalyst | M/Si a | Autotal/(Si + M) b × 103 | Au0/(Si + M) × 103 | Au1+/(Si + M) × 103 | Au1+/Au0 |
|---|---|---|---|---|---|
| Au/MgO/HMS | 0.037 | 1.0 (2.6) | 0.6 | 0.4 | 0.6 |
| Au/MgO/DMS-1 | 0.033 | 0.6 (1.4) | 0.2 | 0.4 | 2.0 |
| Au/MgO/SBA-16 | 0.045 | 0.8 (1.7) | 0.2 | 0.6 | 2.4 |
| Au/MgO/SBA-15 | 0.049 | 0.9 (2.3) | 0.3 | 0.6 | 2.0 |
| Au/Fe2O3/SBA-15 | 0.025 | 1.4 (8.4) | 1.1 | 0.3 | 0.3 |
| Au/TiO2/SBA-15 | 0.059 | 1.7 (7.1) | 1.7 | - | - |
| Au/CeO2/SBA-15 | 0.024 | 5.0 (5.7) | 4.0 | 1.0 | 0.3 |
| Au/SBA-15 | - | 10.1 (3.1) | 7.1 | 3.0 | 0.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Martell, A.; Pawelec, B.; Nava, R.; Mota, N.; Escamilla-Perea, L.; Navarro, R.M.; Fierro, J.L.G. CO Oxidation at 20 °C on Au Catalysts Supported on Mesoporous Silica: Effects of Support Structural Properties and Modifiers. Materials 2018, 11, 948. https://doi.org/10.3390/ma11060948
Moreno-Martell A, Pawelec B, Nava R, Mota N, Escamilla-Perea L, Navarro RM, Fierro JLG. CO Oxidation at 20 °C on Au Catalysts Supported on Mesoporous Silica: Effects of Support Structural Properties and Modifiers. Materials. 2018; 11(6):948. https://doi.org/10.3390/ma11060948
Chicago/Turabian StyleMoreno-Martell, Abigail, Barbara Pawelec, Rufino Nava, Noelia Mota, Luis Escamilla-Perea, Rufino M. Navarro, and Jose L.G. Fierro. 2018. "CO Oxidation at 20 °C on Au Catalysts Supported on Mesoporous Silica: Effects of Support Structural Properties and Modifiers" Materials 11, no. 6: 948. https://doi.org/10.3390/ma11060948
APA StyleMoreno-Martell, A., Pawelec, B., Nava, R., Mota, N., Escamilla-Perea, L., Navarro, R. M., & Fierro, J. L. G. (2018). CO Oxidation at 20 °C on Au Catalysts Supported on Mesoporous Silica: Effects of Support Structural Properties and Modifiers. Materials, 11(6), 948. https://doi.org/10.3390/ma11060948