Polycyclic Aromatic Hydrocarbons Adsorption onto Graphene: A DFT and AIMD Study


Abstract
1. Introduction
2. Computational Methods and Details
2.1. Density Functional Theory (DFT) Calculations
2.2. Ab-Initio Molecular Dynamics (AIMD) Simulations
3. Results
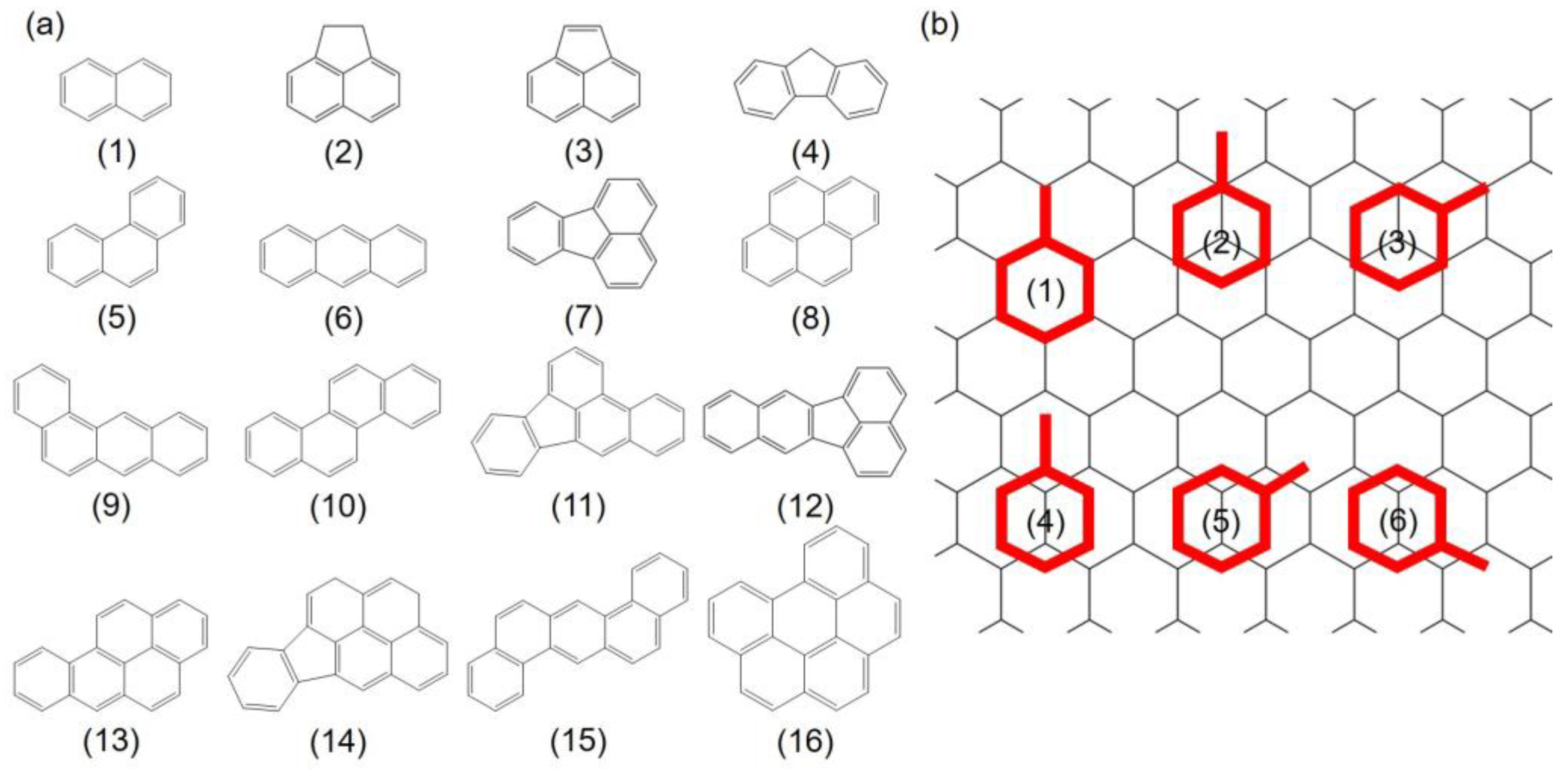
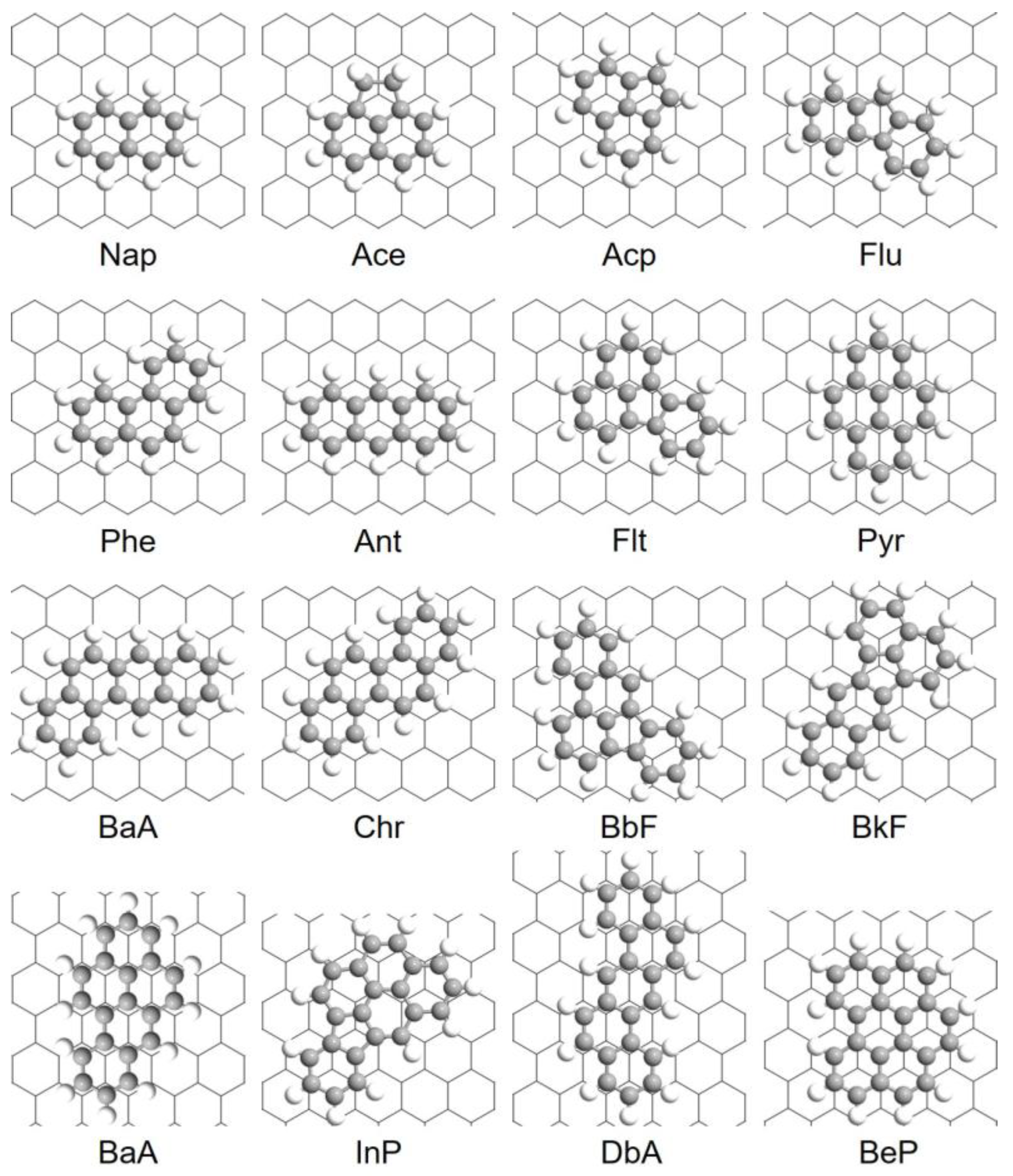
3.1. Adsorption Configurations
3.2. Adsorption Engertics
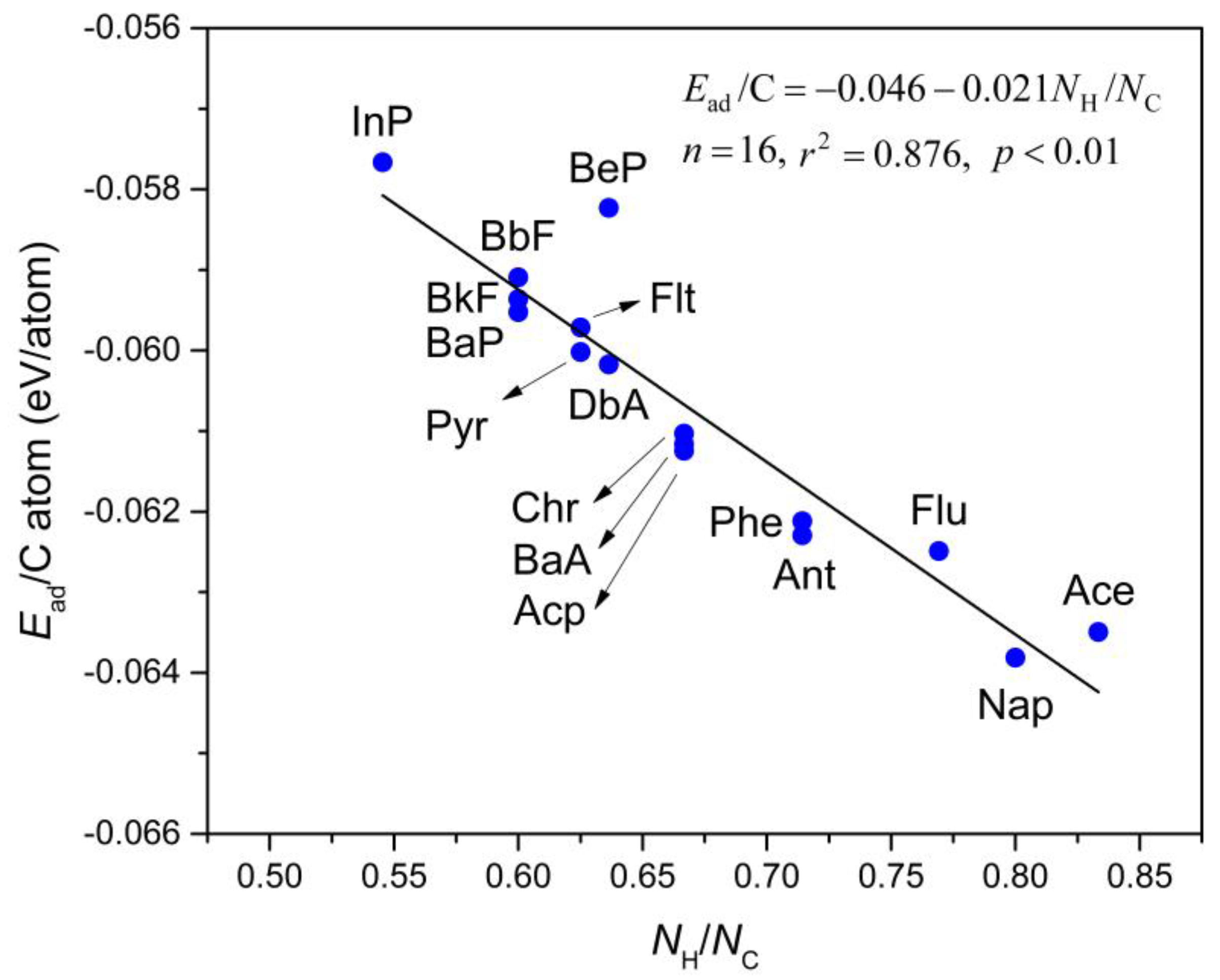
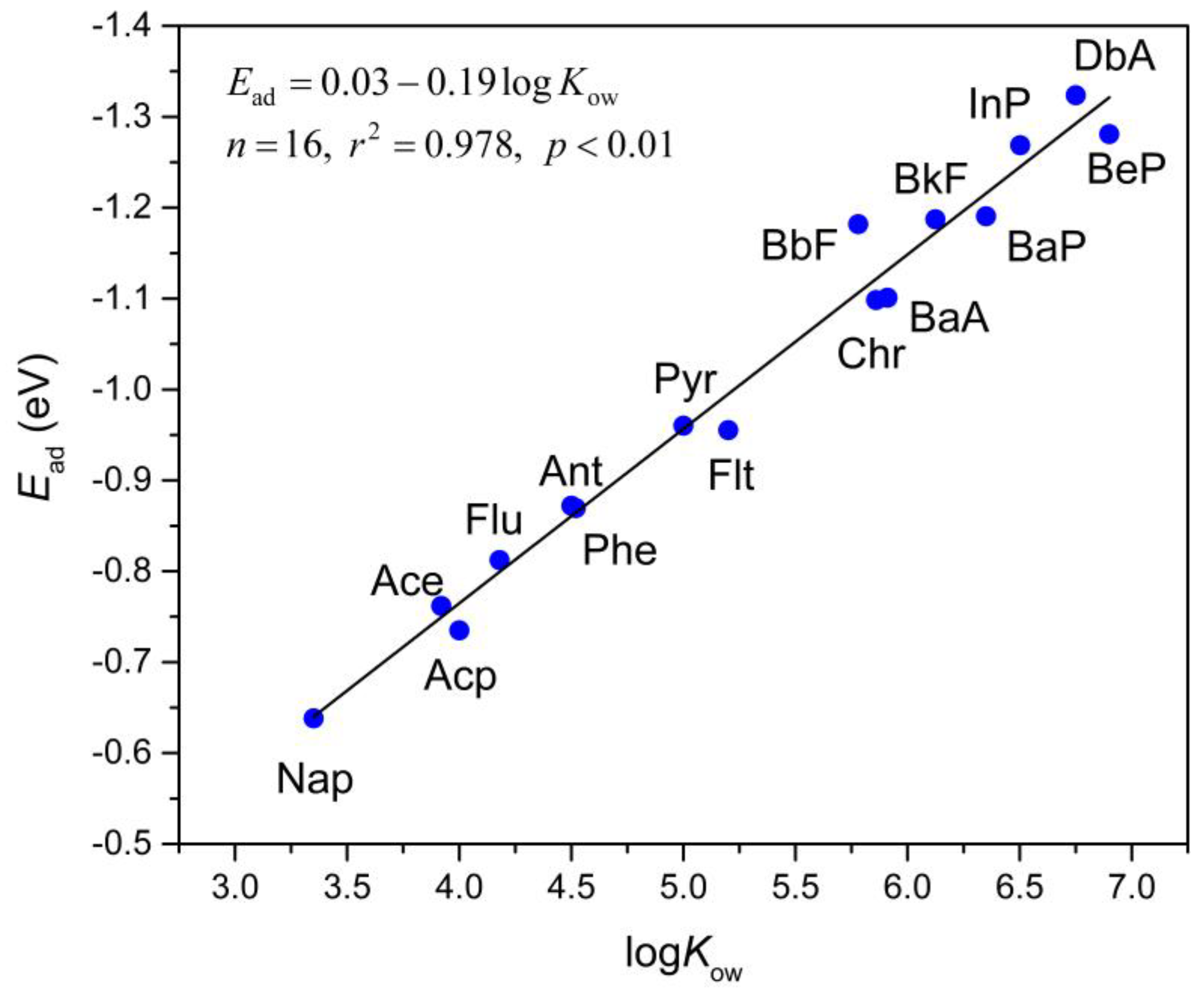
3.3. Correlation between Ead and logKow
n = 16, r2 = 0.978, p < 0.01.
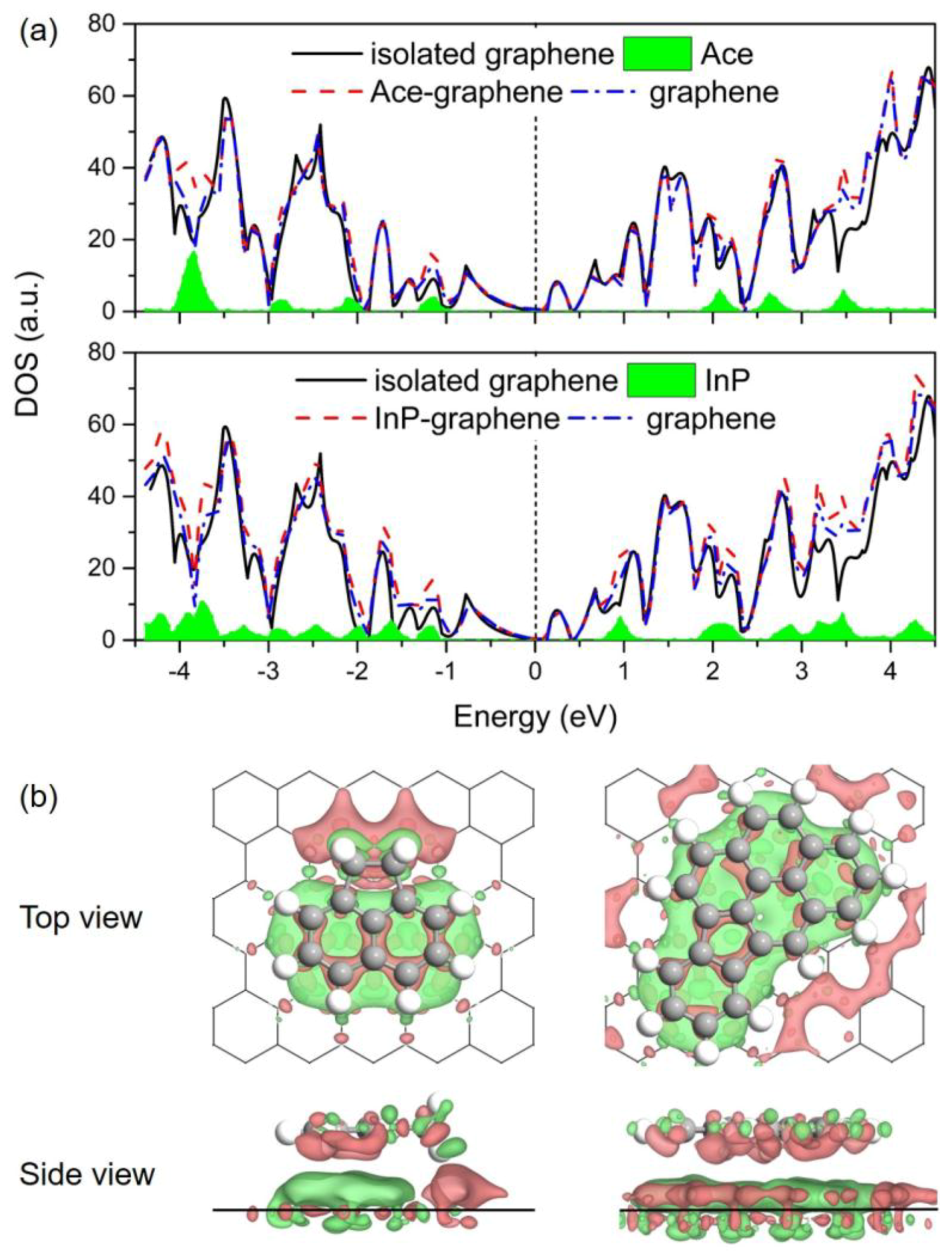
3.4. Electronic Properties
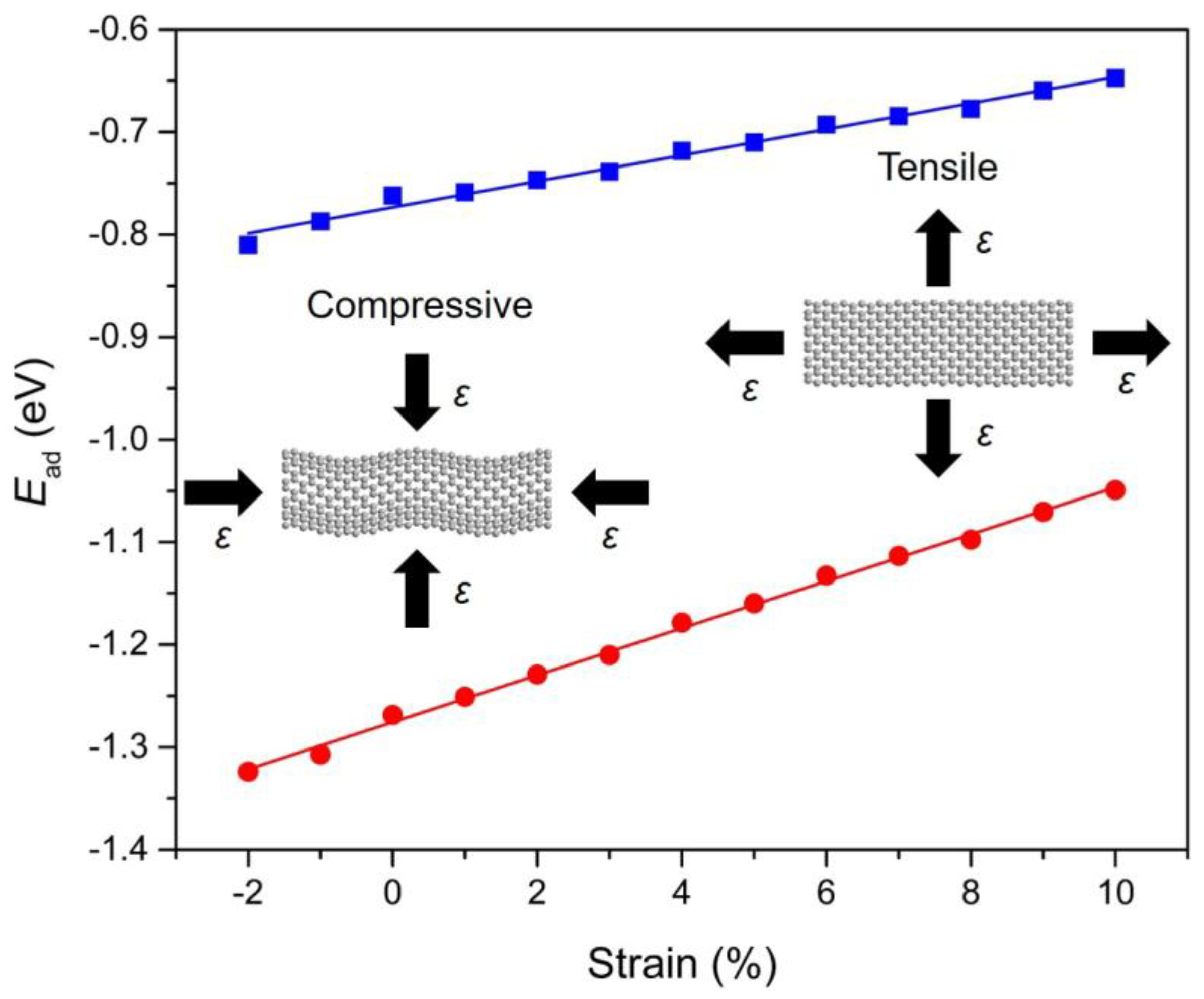
3.5. Strain-Dependent PAHs Adsorption
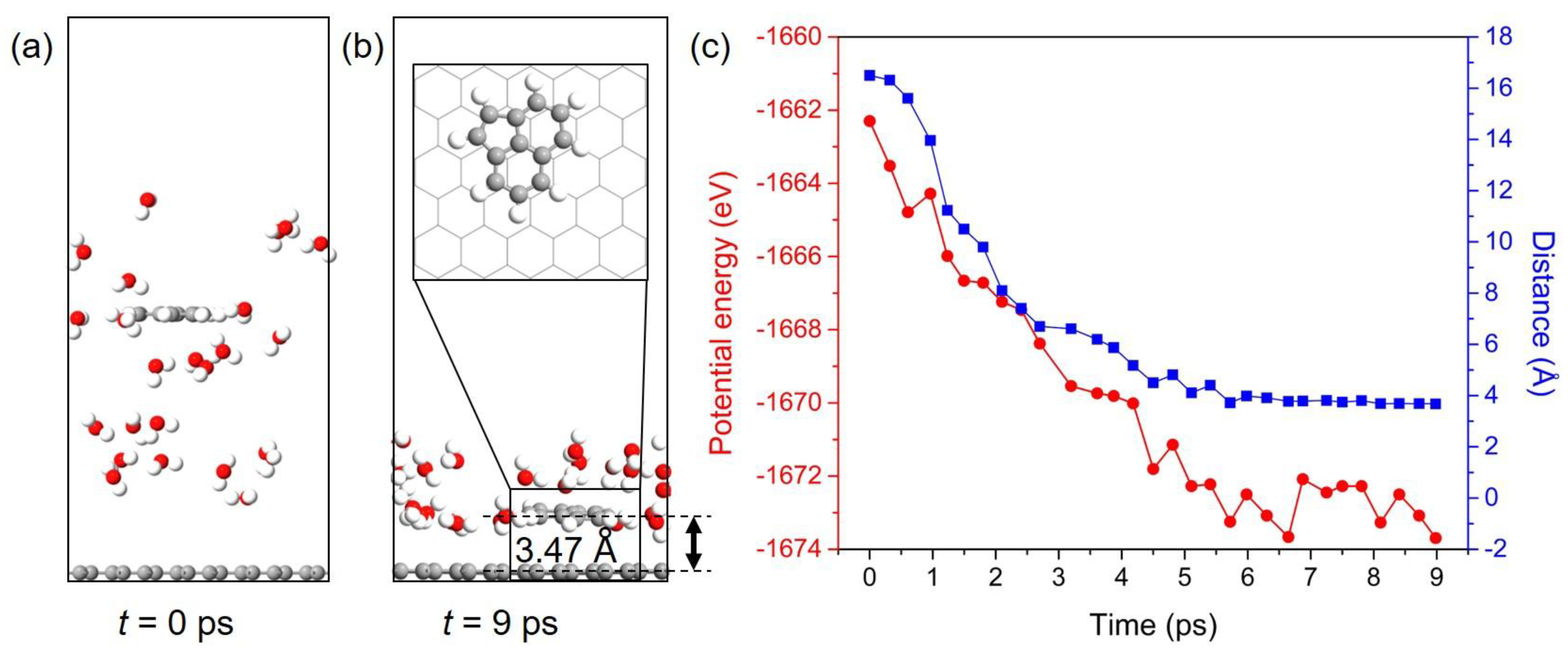
3.6. Dynamic Behavior of PAHs Adsorbed onto the Gr
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Gan, S.; Lau, E.; Ng, H. Remediation of soils contaminated with polycyclic aromatic hydrocarbons (PAHs). J. Hazard. Mater. 2009, 172, 532–549. [Google Scholar] [CrossRef] [PubMed]
- Achten, C.; Hofmann, T. Native polycyclic aromatic hydrocarbons (PAH) in coals—A hardly recognized source of environmental contamination. Sci. Total Environ. 2009, 407, 2461–2473. [Google Scholar] [CrossRef] [PubMed]
- Hale, S.E.; Lehmann, J.; Rutherford, D.; Zimmermanll, A.R.; Bachmann, R.T.; Shitumbanuma, V.; O’Toole, A.; Sundqvist, K.L.; Arp, H.P.H.; Cornelissen, G. Quantifying the total and bioavailable polycyclic aromatic hydrocarbons and dioxins in biochars. Environ. Sci. Technol. 2012, 46, 2830–2838. [Google Scholar] [CrossRef] [PubMed]
- Arfsten, D.P.; Schaeffer, D.J.; Mulveny, D.C. The effects of near ultraviolet radiation on the toxic effects of polycyclic aromatic hydrocarbons in animals and plants: A review. Ecotoxicol. Environ. Saf. 1996, 33, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Djomo, J.; Dauta, A.; Ferrier, V.; Narbonne, J.F.; Monkiedje, A.; Njine, T.; Garrigues, P. Toxic effects of some major polyaromatic hydrocarbons found in crude oil and aquatic sediments on Scenedesmus subspicatus. Water Res. 2004, 38, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhu, D.; Zhang, H.; Shi, X.; Sun, H.; Dang, F. Sorption of polar and nonpolar aromatic compounds to four surface soils of eastern China. Environ. Pollut. 2008, 156, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Zhang, H.; Tao, Q.; Xu, Z.; Zheng, S. Surface functionalized mesoporous silicas as adsorbents for aromatic contaminants in aqueous solution. Environ. Toxicol. Chem. 2009, 28, 1400–1408. [Google Scholar] [CrossRef] [PubMed]
- Walcarius, A.; Mercier, L. Mesoporous organosilica adsorbents: Nanoengineered materials for removal of organic and inorganic pollutants. J. Mater. Chem. 2010, 20, 4478–4511. [Google Scholar] [CrossRef]
- Kemer, B.; Ozdes, D.; Gundogdu, A.; Bulut, V.N.; Duran, C.; Soylak, M. Removal of fluoride ions from aqueous solution by waste mud. J. Hazard. Mater. 2009, 168, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Van Noort, P.C.; Jonker, M.T.; Koelmans, A.A. Modeling maximum adsorption capacities of soot and soot-like materials for PAHs and PCBs. Environ. Sci. Technol. 2004, 38, 3305–3309. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, R.; MacFarlane, J.; Gschwend, P. Importance of black carbon to sorption of native PAHs, PCBs, and PCDDs in Boston and New York harbor sediments. Environ. Sci. Technol. 2005, 39, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Chen, W.; Lin, D.; Yang, K. Influence of surface oxidation of multiwalled carbon nanotubes on the adsorption affinity and capacity of polar and nonpolar organic compounds in aqueous phase. Environ. Sci. Technol. 2012, 46, 5446–5454. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Arriagada, D. Adsorption of polycyclic aromatic hydrocarbons onto graphyne: Comparisons with graphene. Int. J. Quantum Chem. 2017, 117, e25346. [Google Scholar] [CrossRef]
- Yang, K.; Zhu, L.; Xing, B. Adsorption of polycyclic aromatic hydrocarbons by carbon nanomaterials. Environ. Sci. Technol. 2006, 40, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Al-Degs, Y.S.; El-Barghouthi, M.I.; El-Sheikh, A.H.; Walker, G.M. Effect of solution pH, ionic strength, and temperature on adsorption behavior of reactive dyes on activated carbon. Dyes Pigments 2008, 77, 16–23. [Google Scholar] [CrossRef]
- Bi, H.; Xie, X.; Yin, K.; Zhou, Y.; Wan, S.; He, L.; Ruoff, R.S. Spongy graphene as a highly efficient and recyclable sorbent for oils and organic solvents. Adv. Funct. Mater. 2012, 22, 4421–4425. [Google Scholar] [CrossRef]
- Ncibi, M.C.; Sillanpää, M. Optimized removal of antibiotic drugs from aqueous solutions using single, double and multi-walled carbon nanotubes. J. Hazard. Mater. 2015, 298, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Radian, A.; Mishael, Y. Effect of humic acid on pyrene removal from water by polycation-clay mineral composites and activated carbon. Environ. Sci. Technol. 2012, 46, 6228–6235. [Google Scholar] [CrossRef] [PubMed]
- Apul, O.G.; Wang, Q.; Zhou, Y.; Karanfil, T. Adsorption of aromatic organic contaminants by graphene nanosheets: Comparison with carbon nanotubes and activated carbon. Water Res. 2013, 47, 1648–1654. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yang, S.; Zhao, G.; Wang, Q.; Wang, X. Adsorption of polycyclic aromatic hydrocarbons on graphene oxides and reduced graphene oxides. Chem. Asian J. 2013, 8, 2755–2761. [Google Scholar] [CrossRef] [PubMed]
- Perreault, F.; de Faria, A.F.; Elimelech, M. Environmental applications of graphene-based nanomaterials. Chem. Soc. Rev. 2015, 44, 5861–5896. [Google Scholar] [CrossRef] [PubMed]
- Du Jiayuan, W.Y.; Feifei, L.; Dai Yanhui, Z.J.; Zhenyu, W. Adsorption behavior and mechanism of environmental pollutants on graphene oxide. Adv. Earth Sci. 2016, 31, 1125–1136. [Google Scholar]
- Zhao, G.; Jiang, L.; He, Y.; Li, J.; Dong, H.; Wang, X.; Hu, W. Sulfonated graphene for persistent aromatic pollutant management. Adv. Mater. 2011, 23, 3959–3963. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Chen, B. Sulfonated graphene nanosheets as a superb adsorbent for various environmental pollutants in water. Environ. Sci. Technol. 2015, 49, 7364–7372. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, Z.; Chen, B. Adsorption of polycyclic aromatic hydrocarbons by graphene and graphene oxide nanosheets. Environ. Sci. Technol. 2014, 48, 4817–4825. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, Y.; Zhang, L.; Huang, H.; Hu, J.; Shah, S.M.; Su, X. Adsorption and removal of tetracycline antibiotics from aqueous solution by graphene oxide. J. Colloid Interface Sci. 2012, 368, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Ramesha, G.K.; Kumara, A.V.; Muralidhara, H.B.; Sampath, S. Graphene and graphene oxide as effective adsorbents toward anionic and cationic dyes. J. Colloid Interface Sci. 2011, 361, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, J.N.; Mahesh, K.; Le, N.H.; Kemp, K.C.; Timilsina, R.; Tiwari, R.N.; Kim, K.S. Reduced graphene oxide-based hydrogels for the efficient capture of dye pollutants from aqueous solutions. Carbon 2013, 56, 173–182. [Google Scholar] [CrossRef]
- Maliyekkal, S.M.; Sreeprasad, T.S.; Krishnan, D.; Kouser, S.; Mishra, A.K.; Waghmare, U.V.; Pradeep, T. Graphene: A reusable substrate for unprecedented adsorption of pesticides. Small 2013, 9, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.M.; Viñes, F.; Görling, A. On the interaction of polycyclic aromatic compounds with graphene. Carbon 2012, 50, 2482–2492. [Google Scholar] [CrossRef]
- Chen, D.-M.; Shenai, P.M.; Zhao, Y. Tight binding description on the band gap opening of pyrene-dispersed graphene. Phys. Chem. Chem. Phys. 2011, 13, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.M.; Viñes, F.; Görling, A. Bandgap engineering of graphene by physisorbed adsorbates. Adv. Mater. 2011, 23, 2638–2643. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Ceperley, D.M.; Alder, B. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 1980, 45, 566. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Rajesh, C.; Majumder, C.; Mizuseki, H.; Kawazoe, Y. A theoretical study on the interaction of aromatic amino acids with graphene and single walled carbon nanotube. J. Chem. Phys. 2009, 130, 124911. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Bostwick, A.; Seyller, T.; Horn, K.; Rotenberg, E. Controlling the electronic structure of bilayer graphene. Science 2006, 313, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Björk, J.; Hanke, F.; Palma, C.-A.; Samori, P.; Cecchini, M.; Persson, M. Adsorption of aromatic and anti-aromatic systems on graphene through π−π stacking. J. Phys. Chem. Lett. 2010, 1, 3407–3412. [Google Scholar] [CrossRef]
- Hsun Su, Y.; Kai Wu, Y.; Tu, S.L.; Chang, S.-J. Electrostatic studies of π–π interaction for benzene stacking on a graphene layer. Appl. Phys. Lett. 2011, 99, 163102. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Z.; Moe, Y.N. On the performance of local density approximation in describing the adsorption of electron donating/accepting molecules on graphene. Chem. Phys. 2012, 406, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Y.; Wang, Y.-B. Noncovalent π⋅⋅⋅π interaction between graphene and aromatic molecule: Structure, energy, and nature. J. Chem. Phys. 2014, 140, 094302. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, B.; Sarkar, U.; Seriani, N. Electronic properties of homo-and heterobilayer graphyne: The idea of a nanocapacitor. J. Phys. Chem. C 2016, 120, 26579–26587. [Google Scholar] [CrossRef]
- Mackay, D.; Shiu, W.-Y.; Ma, K.-C.; Lee, S.C. Handbook of Physical-Chemical Properties and Environmental Fate for Organic Chemicals; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Sabljić, A.; Güsten, H.; Verhaar, H.; Hermens, J. QSAR modelling of soil sorption. Improvements and systematics of log Koc vs. log Kow correlations. Chemosphere 1995, 31, 4489–4514. [Google Scholar] [CrossRef]
- Chu, W.; Chan, K.-H. The prediction of partitioning coefficients for chemicals causing environmental concern. Sci. Total Environ. 2000, 248, 1–10. [Google Scholar] [CrossRef]
- Toropov, A.; Toropova, A.; Raska, I., Jr. QSPR modeling of octanol/water partition coefficient for vitamins by optimal descriptors calculated with SMILES. Eur. J. Med. Chem. 2008, 43, 714–740. [Google Scholar] [CrossRef] [PubMed]
- Sangster, J. Octanol-water partition coefficients of simple organic compounds. J. Phys. Chem. 1989, 18, 1111–1229. [Google Scholar] [CrossRef]
- Zacharia, R.; Ulbricht, H.; Hertel, T. Interlayer cohesive energy of graphite from thermal desorption of polyaromatic hydrocarbons. Phys. Rev. B 2004, 69, 155406. [Google Scholar] [CrossRef]
- Girifalco, L.; Lad, R. Energy of cohesion, compressibility, and the potential energy functions of the graphite system. J. Chem. Phys. 1956, 25, 693–697. [Google Scholar] [CrossRef]
- Brandenburg, J.G.; Alessio, M.; Civalleri, B.; Peintinger, M.F.; Bredow, T.; Grimme, S. Geometrical correction for the inter-and intramolecular basis set superposition error in periodic density functional theory calculations. J. Phys. Chem. A 2013, 117, 9282–9292. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.; Visontai, D.; Lambert, C.J.; Bryce, M.R.; Frampton, H.; Chappell, D. A study of planar anchor groups for graphene-based single-molecule electronics. J. Chem. Phys. 2014, 140, 054708. [Google Scholar] [CrossRef] [PubMed]
- Hamada, I.; Otani, M. Comparative van der Waals density-functional study of graphene on metal surfaces. Phys. Rev. B 2010, 82, 153412. [Google Scholar] [CrossRef]
- Ma, Y.; Dai, Y.; Guo, M.; Huang, B. Graphene-diamond interface: Gap opening and electronic spin injection. Phys. Rev. B 2012, 85, 235448. [Google Scholar] [CrossRef]
- Lee, J.-H.; Choi, Y.-K.; Kim, H.-J.; Scheicher, R.H.; Cho, J.-H. Physisorption of DNA nucleobases on h-BN and graphene: vdW-corrected DFT calculations. J. Phys. Chem. C 2013, 117, 13435–13441. [Google Scholar] [CrossRef]
- Ershova, O.V.; Lillestolen, T.C.; Bichoutskaia, E. Study of polycyclic aromatic hydrocarbons adsorbed on graphene using density functional theory with empirical dispersion correction. Phys. Chem. Chem. Phys. 2010, 12, 6483–6491. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xie, S.; Cai, H.; Bai, X.; Xue, Z. Quantitative structure–property relationships for octanol–water partition coefficients of polybrominated diphenyl ethers. Chemosphere 2008, 72, 1602–1606. [Google Scholar] [CrossRef] [PubMed]
- Kah, M.; Zhang, X.; Jonker, M.T.; Hofmann, T. Measuring and modeling adsorption of PAHs to carbon nanotubes over a six order of magnitude wide concentration range. Environ. Sci. Technol. 2011, 45, 6011–6017. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Zhang, J.; Chen, J.; Li, X. Simulating adsorption of organic pollutants on finite (8, 0) single-walled carbon nanotubes in water. Environ. Sci. Technol. 2012, 46, 8887–8894. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Nabi, D.; Gros, J.; Dimitriou-Christidis, P.; Arey, J.S. Mapping environmental partitioning properties of nonpolar complex mixtures by use of GC × GC. Environ. Sci. Technol. 2014, 48, 6814–6826. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Name | Abbreviation | CAS Number | Formula |
|---|---|---|---|---|
| 1 | Naphthalene | Nap | 91-20-3 | C10H8 |
| 2 | Acenaphthene | Ace | 83-32-9 | C12H10 |
| 3 | Acenaphthylene | Acp | 208-96-8 | C12H8 |
| 4 | Fluorene | Flu | 86-73-7 | C13H10 |
| 5 | Phenanthrene | Phe | 85-01-8 | C14H10 |
| 6 | Anthracene | Ant | 120-12-7 | C14H10 |
| 7 | Fluoranthene | Flt | 206-44-0 | C16H10 |
| 8 | Pyrene | Pyr | 129-00-0 | C16H10 |
| 9 | Benzo[a]anthracene | BaA | 56-55-3 | C18H12 |
| 10 | Chrysene | Chr | 218-01-9 | C18H12 |
| 11 | Benzo[b]fluoranthene | BbF | 205-99-2 | C20H12 |
| 12 | Benzo[k]fluoranthene | BkF | 207-08-9 | C20H12 |
| 13 | Benzo[a]pyrene | BaP | 50-32-8 | C20H12 |
| 14 | Indeno[1,2,3-c,d]pyrene | InP | 193-39-5 | C22H12 |
| 15 | Dibenz[a,h]anthracene | DbA | 53-70-3 | C22H14 |
| 16 | Benzo[g,h,i]perylene | BeP | 191-24-2 | C22H12 |
| No. | PAHs | Configurations | Ead (eV) | dinter (Å) | logKow | QPAH (e) | |
|---|---|---|---|---|---|---|---|
| PBE-D3 | LDA | ||||||
| 1 | Nap | Top | −0.638 | −0.353 | 3.45 | 3.37 [52] | 0.051 |
| 2 | Ace | Top | −0.762 | −0.458 | 3.43 | 3.92 [52] | 0.054 |
| 3 | Acp | Top | −0.735 | −0.427 | 3.44 | 4.00 [52] | 0.052 |
| 4 | Flu | Bridge | −0.812 | −0.471 | 3.38 | 4.18 [52] | 0.064 |
| 5 | Phe | Top | −0.870 | −0.491 | 3.44 | 4.57 [52] | 0.061 |
| 6 | Ant | Top | −0.872 | −0.500 | 3.44 | 4.54 [52] | 0.063 |
| 7 | Flt | Bridge | −0.955 | −0.537 | 3.45 | 5.22 [52] | 0.064 |
| 8 | Pyr | Bridge | −0.961 | −0.540 | 3.50 | 4.88 [53,54] | 0.062 |
| 9 | BaA | Top | −1.101 | −0.636 | 3.51 | 5.91 [52] | 0.073 |
| 10 | Chr | Top | −1.099 | −0.628 | 3.45 | 5.86 [52] | 0.072 |
| 11 | BbF | Bridge | −1.182 | −0.672 | 3.42 | 6.06 [54] | 0.077 |
| 12 | BkF | Top | −1.187 | −0.673 | 3.42 | 6.12 [55] | 0.077 |
| 13 | BaP | Bridge | −1.190 | −0.677 | 3.46 | 6.04 [52] | 0.072 |
| 14 | InP | Top | −1.269 | −0.716 | 3.42 | 6.50 [52] | 0.079 |
| 15 | DbA | Bridge | −1.324 | −0.757 | 3.43 | 6.75 [56] | 0.080 |
| 16 | BeP | Top | −1.281 | −0.734 | 3.44 | 6.50 [52] | 0.077 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Ou, P.; Wei, Y.; Zhang, X.; Song, J. Polycyclic Aromatic Hydrocarbons Adsorption onto Graphene: A DFT and AIMD Study. Materials 2018, 11, 726. https://doi.org/10.3390/ma11050726
Li B, Ou P, Wei Y, Zhang X, Song J. Polycyclic Aromatic Hydrocarbons Adsorption onto Graphene: A DFT and AIMD Study. Materials. 2018; 11(5):726. https://doi.org/10.3390/ma11050726
Chicago/Turabian StyleLi, Bing, Pengfei Ou, Yulan Wei, Xu Zhang, and Jun Song. 2018. "Polycyclic Aromatic Hydrocarbons Adsorption onto Graphene: A DFT and AIMD Study" Materials 11, no. 5: 726. https://doi.org/10.3390/ma11050726
APA StyleLi, B., Ou, P., Wei, Y., Zhang, X., & Song, J. (2018). Polycyclic Aromatic Hydrocarbons Adsorption onto Graphene: A DFT and AIMD Study. Materials, 11(5), 726. https://doi.org/10.3390/ma11050726