Structure and Chemical Bonding of the Li-Doped Polar Intermetallic RE2In1−xLixGe2 (RE = La, Nd, Sm, Gd; x = 0.13, 0.28, 0.43, 0.53) System


Abstract
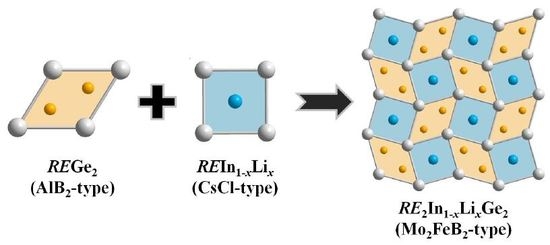
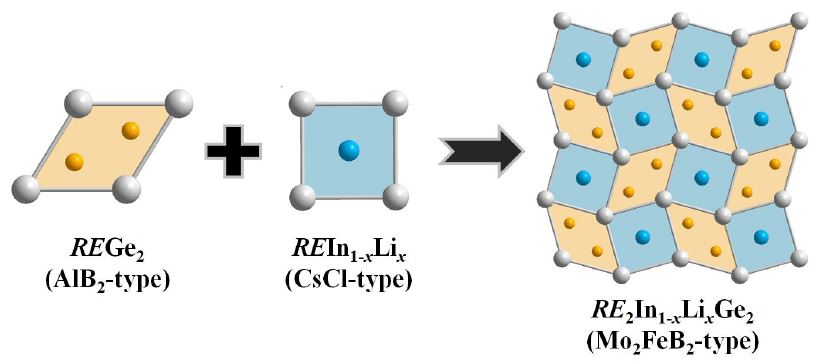
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. X-ray Diffraction Experiments
2.3. Electronic Structure Calculations
3. Results and Discussion
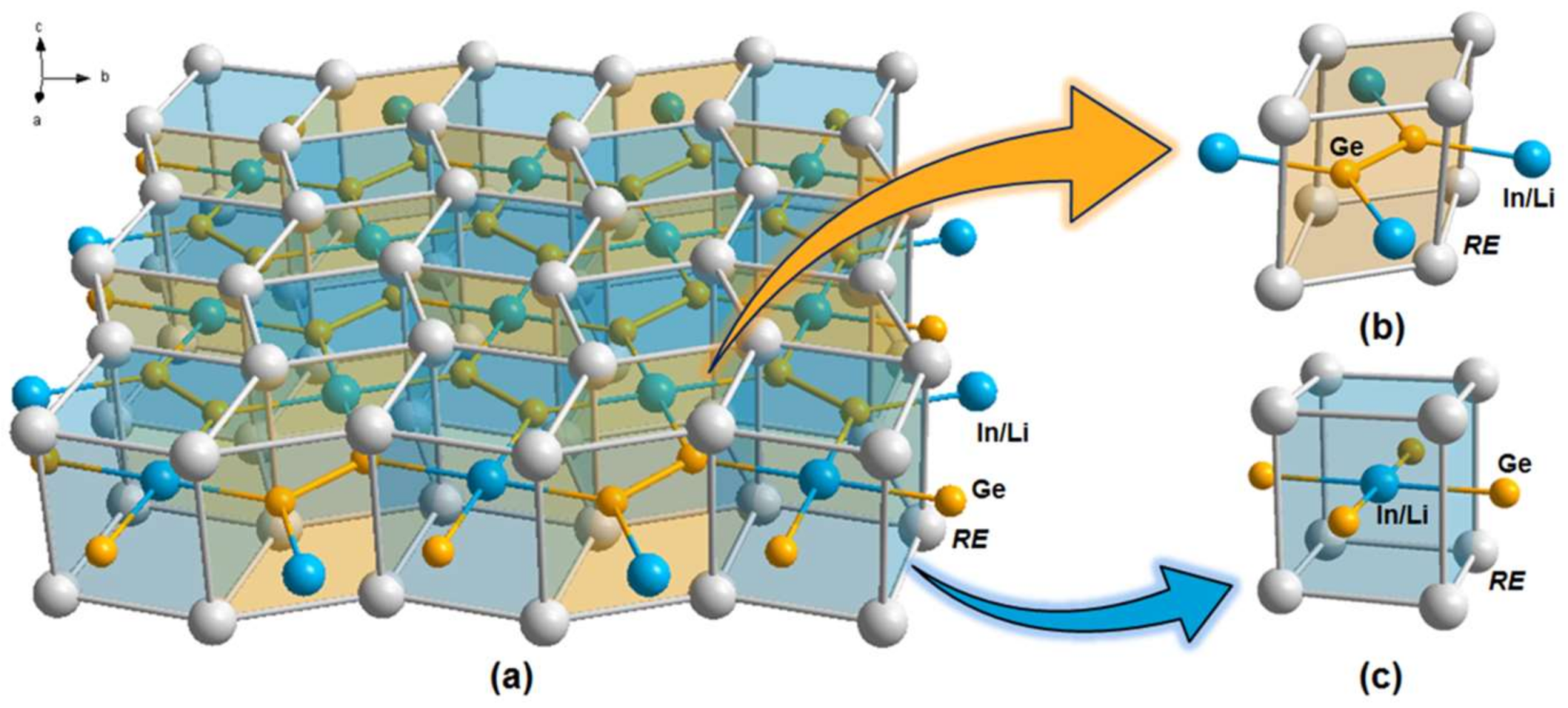
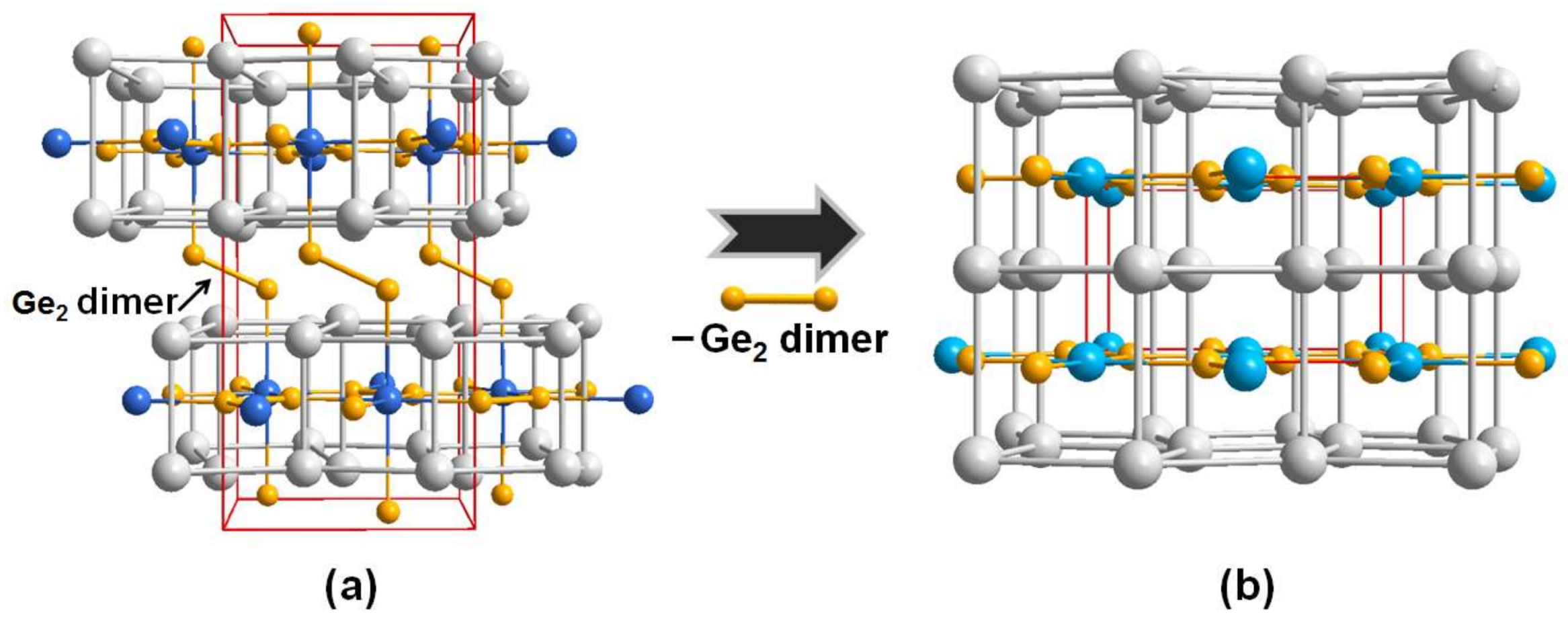
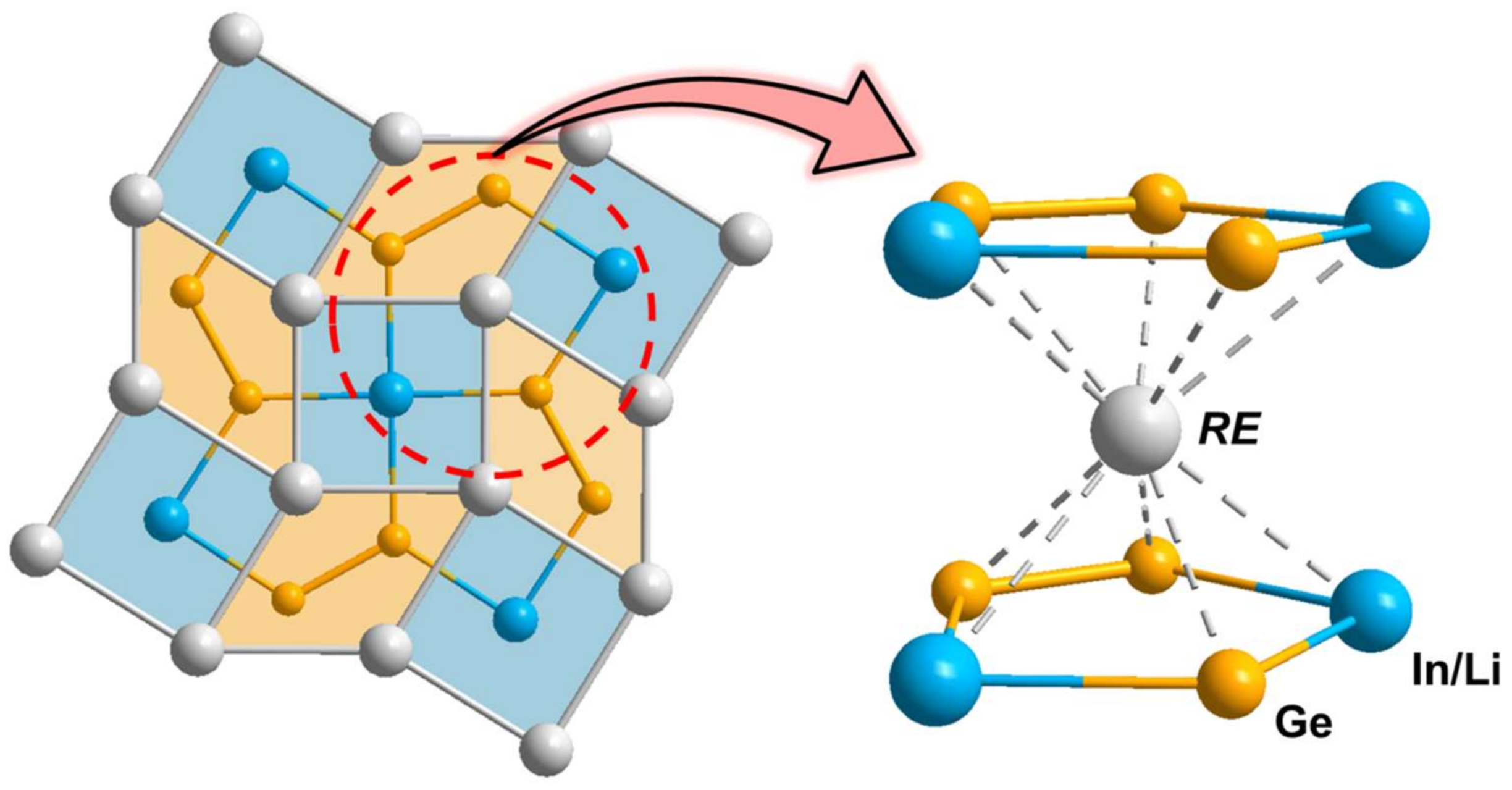
3.1. Crystal Structure Analysis
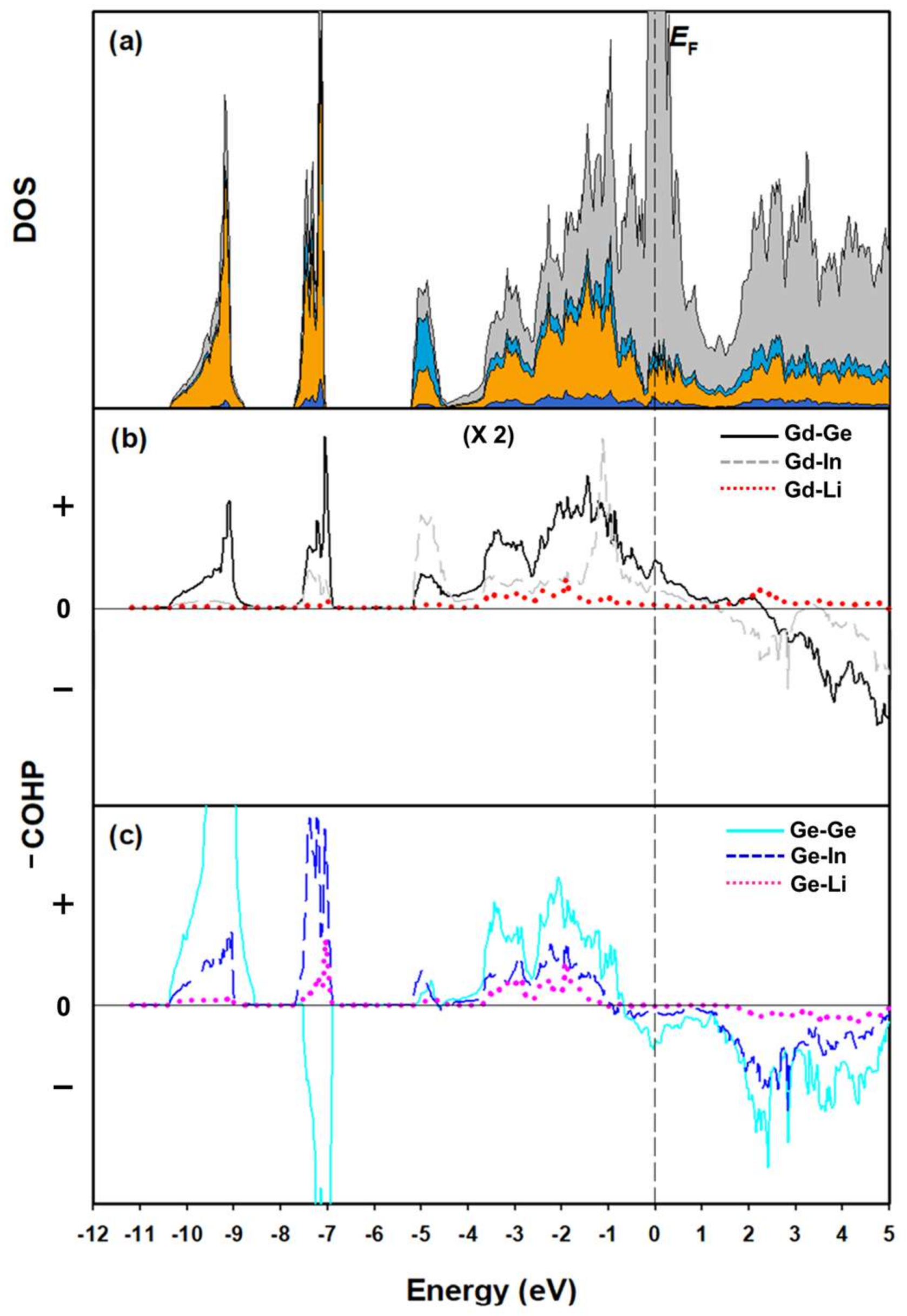
3.2. Electronic Structure and Chemical Bonding
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mozharivskyj, Y.; Pecharsky, A.O.; Pecharsky, V.K.; Miller, G.J. On the High-Temperature Phase Transition of Gd5Si2Ge2. J. Am. Chem. Soc. 2005, 127, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, F.; Jones, K.; Miller, G.J. Chemical Pressure and Rare-Earth Orbital Contributions in Mixed Rare-Earth Silicides La5–xYxSi4 (0 ≤ x ≤ 5). Inorg. Chem. 2011, 50, 12714–12723. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, L.-M.; Kim, S.-H.; Seo, D.-K. Electron-Precise/Deficient La5−xCaxGe4 (3.4 ≤ x ≤ 3.8) and Ce5−xCaxGe4 (3.0 ≤ x ≤ 3.3): Probing Low-Valence Electron Concentrations in Metal-Rich Gd5Si4-type Germanides. J. Am. Chem. Soc. 2005, 127, 15682–15683. [Google Scholar] [CrossRef] [PubMed]
- Morellon, L.; Blasco, J.; Algarabel, P.A.; Ibarra, M.R. Nature of the first-order antiferromagnetic-ferromagnetic transition in the Ge-rich magnetocaloric compounds Gd5(SixGe1−x)4. Phys. Rev. B 2000, 62, 1022–1026. [Google Scholar] [CrossRef]
- Levin, E.M.; Pecharsky, V.K.; Gschneidner, K.A., Jr. Spontaneous generation of voltage in Gd5(SixGe4−x) during a first-order phase transition induced by temperature or magnetic field. Phys. Rev. B 2001, 63, 174110–1474117. [Google Scholar] [CrossRef]
- Morellon, L.; Algarabel, P.A.; Ibara, M.R.; Blasco, J.; Garcia-Landa, B. Magnetic-field-induced structural phase transition in Gd5(Si1.8Ge2.2). Phys. Rev. B 1998, 58, R14721–R14724. [Google Scholar] [CrossRef]
- Morellon, L.; Stankiewicz, J.; Garcia-Landa, B.; Algarabel, P.A.; Ibarra, M.R. Giant magnetoresistance near the magnetostructural transition in Gd5(Si1.8Ge2.2). Appl. Phys. Lett. 1998, 73, 3462–3464. [Google Scholar] [CrossRef]
- Levin, E.M.; Pecharsky, V.K.; Gschneidner, K.A., Jr. Magnetic-field and temperature dependencies of the electrical resistance near the magnetic and crystallographic first-order phase transition of Gd5(Si2Ge2). Phys. Rev. B 1999, 60, 7993–7997. [Google Scholar] [CrossRef]
- Levin, E.M.; Pecharsky, V.K.; Gschneidner, K.A., Jr.; Tomlinson, P. Magnetic field and temperature-induced first-order transition in Gd5(Si1.5Ge2.5): A study of the electrical resistance behavior. J. Magn. Magn. Mater. 2000, 210, 181–188. [Google Scholar] [CrossRef]
- Percharsky, V.K.; Gschneidner, K.A., Jr. Giant Magnetocaloric Effect in Gd5(Si2Ge2). Phys. Rev. Lett. 1997, 78, 4494–4497. [Google Scholar] [CrossRef]
- Pecharsky, V.K.; Gschneidner, K.A., Jr. Tunable magnetic regenerator alloys with a giant magnetocaloric effect for magnetic refrigeration from ~20 to ~290 K. Appl. Phys. Lett. 1997, 70, 3299–3301. [Google Scholar] [CrossRef]
- Wang, H.; Misra, S.; Wang, F.; Miller, G.J. Structural and Magnetic Characteristics of Gd5GaxSi4−x. Inorg. Chem. 2010, 49, 4586–4593. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Poweleit, E.T.; Miller, G.J. On the Crystal Structure, Metal Atom Site Preferences and Magnetic Properties of Nd5–xErxTt4 (Tt = Si or Ge). Z. Anorg. Allg. Chem. 2009, 635, 889–897. [Google Scholar] [CrossRef]
- Miller, G.J. Complex rare-earth tetrelides, RE5(SixGe1−x)4: New materials for magnetic refrigeration and a superb playground for solid state chemistry. Chem. Soc. Rev. 2006, 35, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Nam, G.; Jeon, J.; Kim, Y.; Kang, S.; Ahn, K.; You, T.-S. Combined effect of chemical pressure and valence electron concentration through the electron-deficient Li substitution on the RE4LiGe4 (RE = La, Ce, Pr, and Sm) system. J. Solid State Chem. 2013, 205, 10–20. [Google Scholar] [CrossRef]
- Rieger, W.; Nowotny, H.; Benesovsky, F. Die Kristallstruktur von Mo2FeB2. Monatsh. Chem. 1964, 95, 1502–1503. [Google Scholar] [CrossRef]
- Choe, W.; Miller, G.J.; Levin, E.M. Crystal structure and magnetism of Gd2MgGe2. J. Alloy. Compd. 2001, 329, 121–130. [Google Scholar] [CrossRef]
- Zaremba, V.I.; Kaczorowski, D.; Nychyporuk, G.P.; Rodewald, U.C.; Pöttgen, R. Structure and physical properties of RE2Ge2In (RE = La, Ce, Pr, Nd). Solid State Sci. 2004, 6, 1301–1306. [Google Scholar] [CrossRef]
- Tobash, P.H.; Lins, D.; Bobev, S.; Lima, A.; Hundley, M.F.; Thompson, J.D.; Sarrao, J.L. Crystal Growth, Structural, and Property Studies on a Family of Ternary Rare-Earth Phases RE2InGe2 (RE = Sm, Gd, Tb, Dy, Ho, Yb). Chem. Mater. 2005, 17, 5567–5573. [Google Scholar] [CrossRef]
- Zaremba, V.I.; Tyvanchuk, Y.B.; Stepien-Damm, J. Crystal structure of diytterbium digermanium indide, Yb2Ge2In. Z. Kristallogr. New Cryst. Struct. 1997, 212, 291. [Google Scholar] [CrossRef]
- Zaremba, V.I.; Stepien-Damm, J.; Nichiporuk, G.P.; Tyvanchuk, Y.B.; Kalychak, Y.M. Structure of R2Ge2In crystals, where R is a rare-earth metal. Crystallogr. Rep. 1998, 43, 8–11. [Google Scholar]
- Steinberg, G.; Schuster, H.-U. Ternary Silieides of Lithium with Yttrium or Neodymium in a Modified U3Si2-Type Structure. Z. Naturforsch. 1979, 34, 1237–1239. [Google Scholar] [CrossRef]
- Dhar, S.K.; Manfrinetti, P.; Palenzona, A. Magnetic ordering in CeMg2Si2 and Ce2MgSi2. J. Alloys Compd. 1997, 252, 24–27. [Google Scholar] [CrossRef]
- Kranenberg, C.; Mewis, A. Darstellung und Kristallstrukturen von Ln2Al3Si2 and Ln2AlSi2 (Ln: Y, Tb–Lu). Z. Anorg. Allg. Chem. 2000, 626, 1448–1453. [Google Scholar] [CrossRef]
- Dhar, S.K.; Manfrinetti, P.; Palenzona, A.; Kimura, Y.; Kozaki, M.; Onuki, Y.; Takeuchi, T. Magnetic, transport and thermal behaviour of R3Si2, R2YSi2 (R = La and Ce), Ce2ScSi2 and Ce2Sc3Si4. Physics B 1999, 271, 150–157. [Google Scholar] [CrossRef]
- Kraft, R.; Pöttgen, R. Syntheses and Crystal Structure of the Ternary Silicides RE2Si2Mg (RE = Y, La–Nd, Sm, Gd–Lu) and Structure Refinement of Dy5Si3. Monatsh. Chem. 2005, 136, 1707–1713. [Google Scholar] [CrossRef]
- Kraft, R.; Pöttgen, R. Ternary Germanides RE2Ge2Mg (RE = Y, La–Nd, Sm, Gd, Tb). Monatsh. Chem. 2004, 135, 1327–1334. [Google Scholar] [CrossRef]
- Dinges, T.; Rodewald, U.C.; Matar, S.F.; Eckert, H.; Pöttgen, R. New Ternary Silicide LiRh2Si2—Structure and Bonding Peculiarities. Z. Anorg. Allog. Chem. 2009, 635, 1894–1903. [Google Scholar] [CrossRef]
- Bruker. APEX2; Bruker AXS Inc.: Madison, WI, USA, 2007. [Google Scholar]
- Bruker. SAINT; Bruker AXS Inc.: Madison, WI, USA, 2002. [Google Scholar]
- Sheldrick, G.M. SADABS; University of Göttingen: Göttingen, Germany, 2003. [Google Scholar]
- Gelato, L.M.; Parthé, E. STRUCTURE TIDY—A computer program to standardize crystal structure data. J. Appl. Crystallogr. 1987, 20, 139–143. [Google Scholar] [CrossRef]
- Andersen, O.K. Linear methods in band theory. Phys. Rev. B. 1975, 12, 3060–3083. [Google Scholar] [CrossRef]
- Andersen, O.K.; Jepsen, O. Explicit, first-principles tight-binding theory. Phys. Rev. Lett. 1984, 53, 2571–2574. [Google Scholar] [CrossRef]
- Andersen, O.K. Minimal basis sets in the linear muffin-tin orbital method: Application to the diamond-structure crystals C, Si, and Ge. Phys. Rev. B. 1986, 34, 2439–2449. [Google Scholar] [CrossRef]
- Jepsen, O.; Burkhardt, A.; Andersen, O.K. The TB-LMTO-ASA Program, Version 4.7; Max-Plank-Institut fur Festkorperforschung: Shuttgart, Germany, 1999. [Google Scholar]
- Andersen, O.K.; Jepsen, O.; Glötzel, D. Highlights of Condensed Matter Theory; Bassani, F., Fumi, F., Tosi, M., Eds.; Elsevier Science: North Holland, NY, USA, 1985. [Google Scholar]
- Jepsen, O.; Andersen, O.K. Calculated electronic structure of the sandwich d1 metals LaI2 and CeI2: Application of new LMTO techniques. Z. Phys. B Condens. Matter 1995, 97, 35–47. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Zachariasen, W.H. Crystal chemical studies of the 5f-series of elements. VIII. Crystal structure studies of uranium silicides and of CeSi2, NpSi2, and PuSi2. Acta Crysallogr. 1949, 2, 94–99. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Dronskowski, R.; Blöchl, P.E. Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Refined Composition | La2In0.72(1)Li0.28Ge2 | Nd2In0.87(1)Li0.13Ge2 | Sm2In0.57(1)Li0.43Ge2 | Gd2In0.47(1)Li0.53Ge2 | |
|---|---|---|---|---|---|
| Formula weight (g/mol) | 923.06 | 917.41 | 940.93 | 989.65 | |
| Space group; Z | P4/mbm; 2 | ||||
| Lattice parameters (Å) | a | 7.6140(3) | 7.4368(2) | 7.3834(3) | 7.2885(3) |
| c | 4.4496(2) | 4.3263(10) | 4.2704(2) | 4.2416(2) | |
| Volume (Å3) | 257.957 | 239.121 | 232.799 | 225.323 | |
| dcalcd (g/cm3) | 11.88 | 12.74 | 13.42 | 14.59 | |
| Independent reflections | 594 (Rint = 0.0406) | 251 (Rint = 0.0460) | 120 (Rint = 0.0355) | 153 (Rint = 0.0529) | |
| Data/restraints/parameters | 594/0/13 | 251/0/13 | 120/0/13 | 153/0/13 | |
| Ra indices (I > 2σI) | R1 | 0.0201 | 0.0146 | 0.0161 | 0.0188 |
| wR2 | 0.0375 | 0.0309 | 0.0349 | 0.0397 | |
| GOF on F2 | 1.096 | 1.098 | 1.249 | 1.161 | |
| Largest differences of peak/hole (e/Å3) | 1.373/−1.276 | 2.320/−2.693 | 0.768/−0.705 | 2.436/−0.846 | |
| Atom | Wyckoff Site | Occupation | x | y | z | Ueq (Å2) |
|---|---|---|---|---|---|---|
| La2In0.72(1)Li0.28Ge2 | ||||||
| La | 4h | 1 | 0.1796(1) | 0.6796(1) | 1/2 | 0.009(1) |
| Ge | 4g | 1 | 0.6179(1) | 0.1179(1) | 0 | 0.011(1) |
| In/Li | 2a | 0.72(1)/0.28 | 0 | 0 | 0 | 0.018(1) |
| Nd2In0.87(1)Li0.13Ge2 | ||||||
| Nd | 4h | 1 | 0.1791(1) | 0.6791(1) | 1/2 | 0.007(1) |
| Ge | 4g | 1 | 0.6197(1) | 0.1197(1) | 0 | 0.008(1) |
| In/Li | 2a | 0.87(1)/0.13 | 0 | 0 | 0 | 0.014(1) |
| Sm2In0.57(1)Li0.43Ge2 | ||||||
| Sm | 4h | 1 | 0.1785(1) | 0.6785(1) | 1/2 | 0.008(1) |
| Ge | 4g | 1 | 0.6209(1) | 0.1209(1) | 0 | 0.009(1) |
| In/Li | 2a | 0.57(1)/0.43 | 0 | 0 | 0 | 0.015(1) |
| Gd2In0.47(1)Li0.53Ge2 | ||||||
| Gd | 4h | 1 | 0.1792(1) | 0.6792(1) | 1/2 | 0.008(1) |
| Ge | 4g | 1 | 0.6213(1) | 0.1213(1) | 0 | 0.010(1) |
| In/Li | 2a | 0.47(1)/0.53 | 0 | 0 | 0 | 0.010(1) |
| Atomic Pair | Bond Distance (Å) | |||
|---|---|---|---|---|
| La2In0.72(1)Li0.28Ge2 | Nd2In0.87(1)Li0.13Ge2 | Sm2In0.57(1)Li0.43Ge2 | Gd2In0.47(1)Li0.53Ge2 | |
| RE-RE (shorter) | 3.8670(2) | 3.7681(3) | 3.7273(6) | 3.6942(5) |
| RE-RE (longer) | 3.9552(2) | 3.8649(3) | 3.8398(6) | 3.7876(5) |
| RE-Ge (shorter) | 3.1157(3) | 3.0246(4) | 2.9911(7) | 2.9543(7) |
| RE-Ge (longer) | 3.2092(2) | 3.1319(4) | 3.1026(8) | 3.0776(7) |
| RE-In/Li | 3.5737(1) | 3.4845(2) | 3.4541(4) | 3.4362(3) |
| Ge-Ge | 2.5384(8) | 2.5185(7) | 2.5252(14) | 2.4998(12) |
| Ge-In/Li | 3.0448(3) | 2.9649(5) | 2.9378(10) | 2.8985(9) |
| Atomic Pair | Gd-Ge | Gd-In | Gd-Li | Ge-Ge | Ge-In | Ge-Li |
|---|---|---|---|---|---|---|
| ICOHP | 0.088 | 0.06 | 0.009 | 0.180 | 0.098 | 0.027 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Jeon, J.; You, T.-S. Structure and Chemical Bonding of the Li-Doped Polar Intermetallic RE2In1−xLixGe2 (RE = La, Nd, Sm, Gd; x = 0.13, 0.28, 0.43, 0.53) System. Materials 2018, 11, 495. https://doi.org/10.3390/ma11040495
Lee J, Jeon J, You T-S. Structure and Chemical Bonding of the Li-Doped Polar Intermetallic RE2In1−xLixGe2 (RE = La, Nd, Sm, Gd; x = 0.13, 0.28, 0.43, 0.53) System. Materials. 2018; 11(4):495. https://doi.org/10.3390/ma11040495
Chicago/Turabian StyleLee, Junsu, Jieun Jeon, and Tae-Soo You. 2018. "Structure and Chemical Bonding of the Li-Doped Polar Intermetallic RE2In1−xLixGe2 (RE = La, Nd, Sm, Gd; x = 0.13, 0.28, 0.43, 0.53) System" Materials 11, no. 4: 495. https://doi.org/10.3390/ma11040495
APA StyleLee, J., Jeon, J., & You, T.-S. (2018). Structure and Chemical Bonding of the Li-Doped Polar Intermetallic RE2In1−xLixGe2 (RE = La, Nd, Sm, Gd; x = 0.13, 0.28, 0.43, 0.53) System. Materials, 11(4), 495. https://doi.org/10.3390/ma11040495