Synthesis, Characterization, and Electrochemical Behavior of LiMnxFe(1−x)PO4 Composites Obtained from Phenylphosphonate-Based Organic-Inorganic Hybrids

 ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of Precursors
2.2. Synthesis of LiMn0.1Fe0.9PO4
2.3. Synthesis of LiMnxFe(1−x)PO4 (with x = 0.05, 0.1, 0.5, 0.9, and 0.95)
3. Results and Discussion
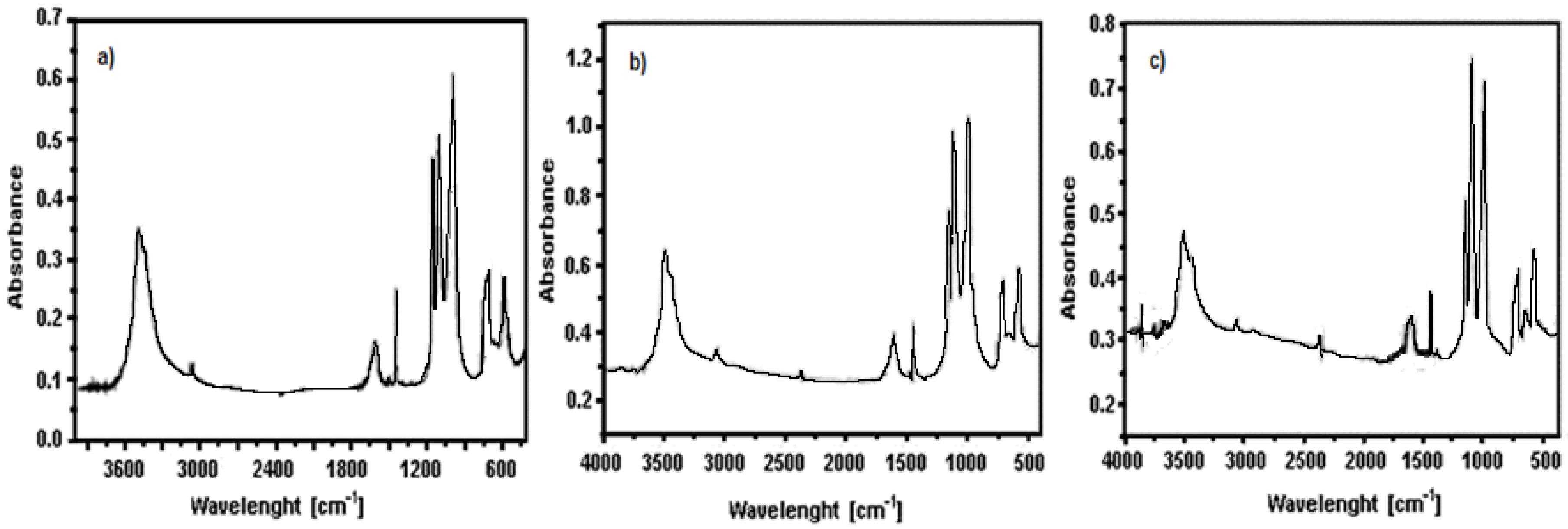
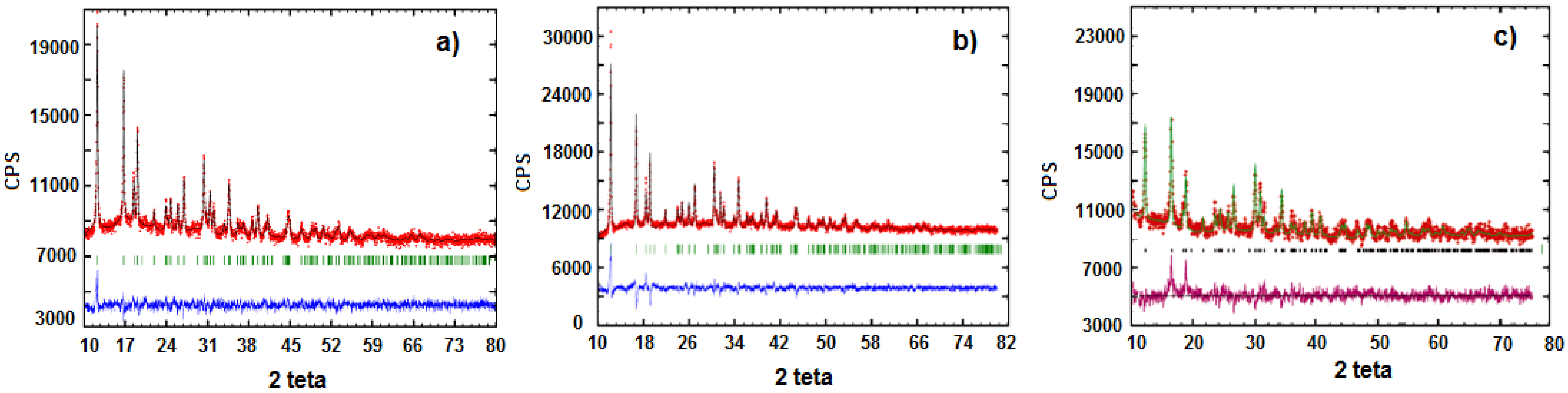
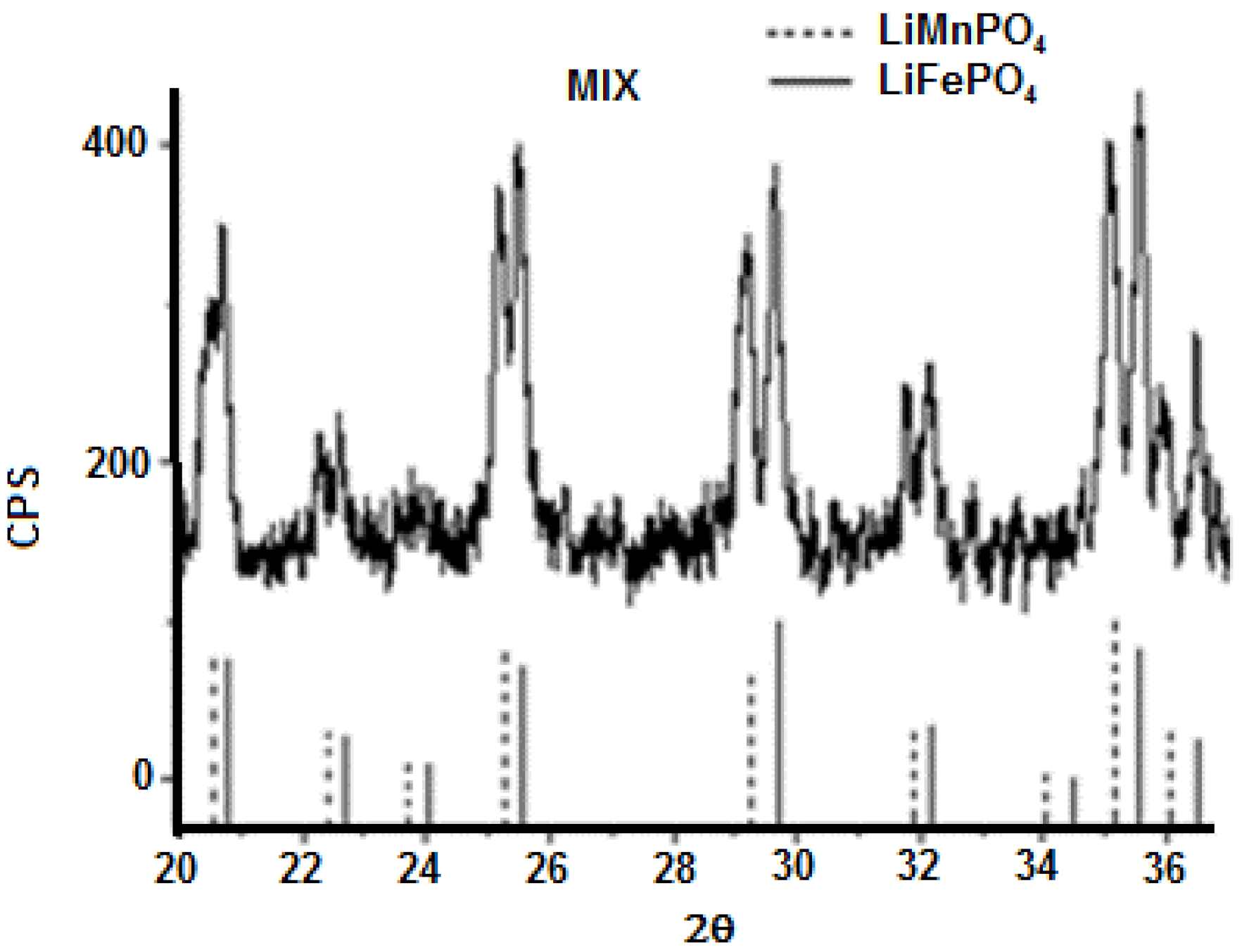
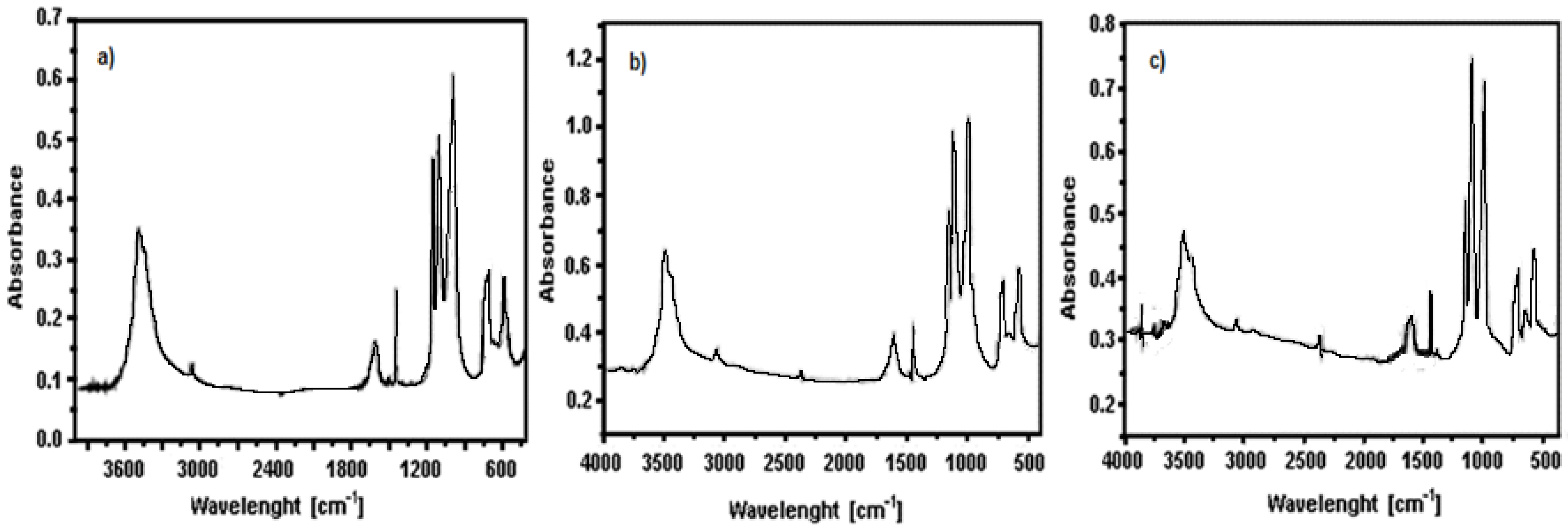
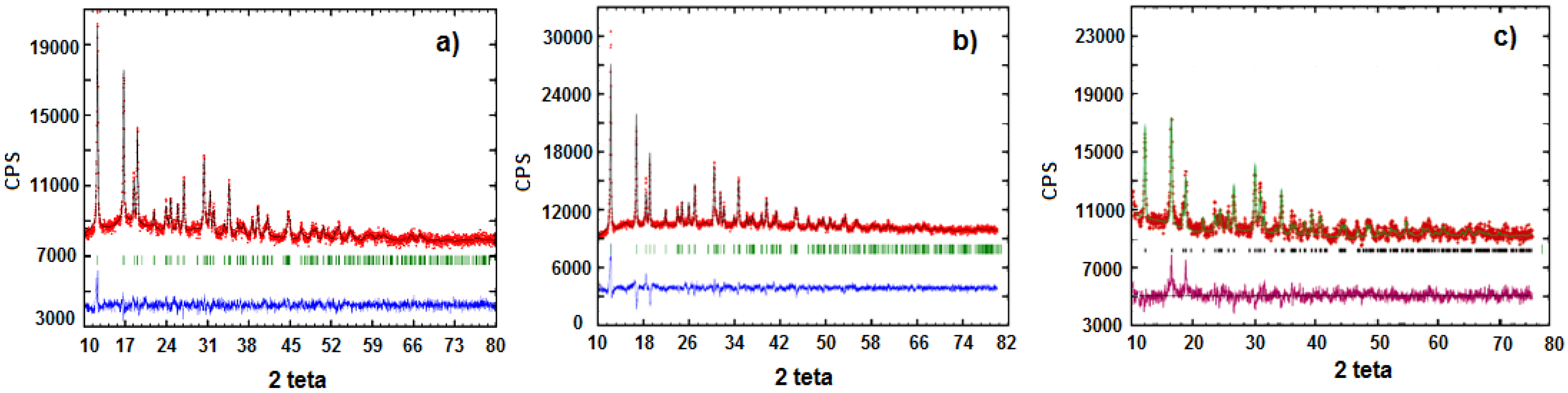
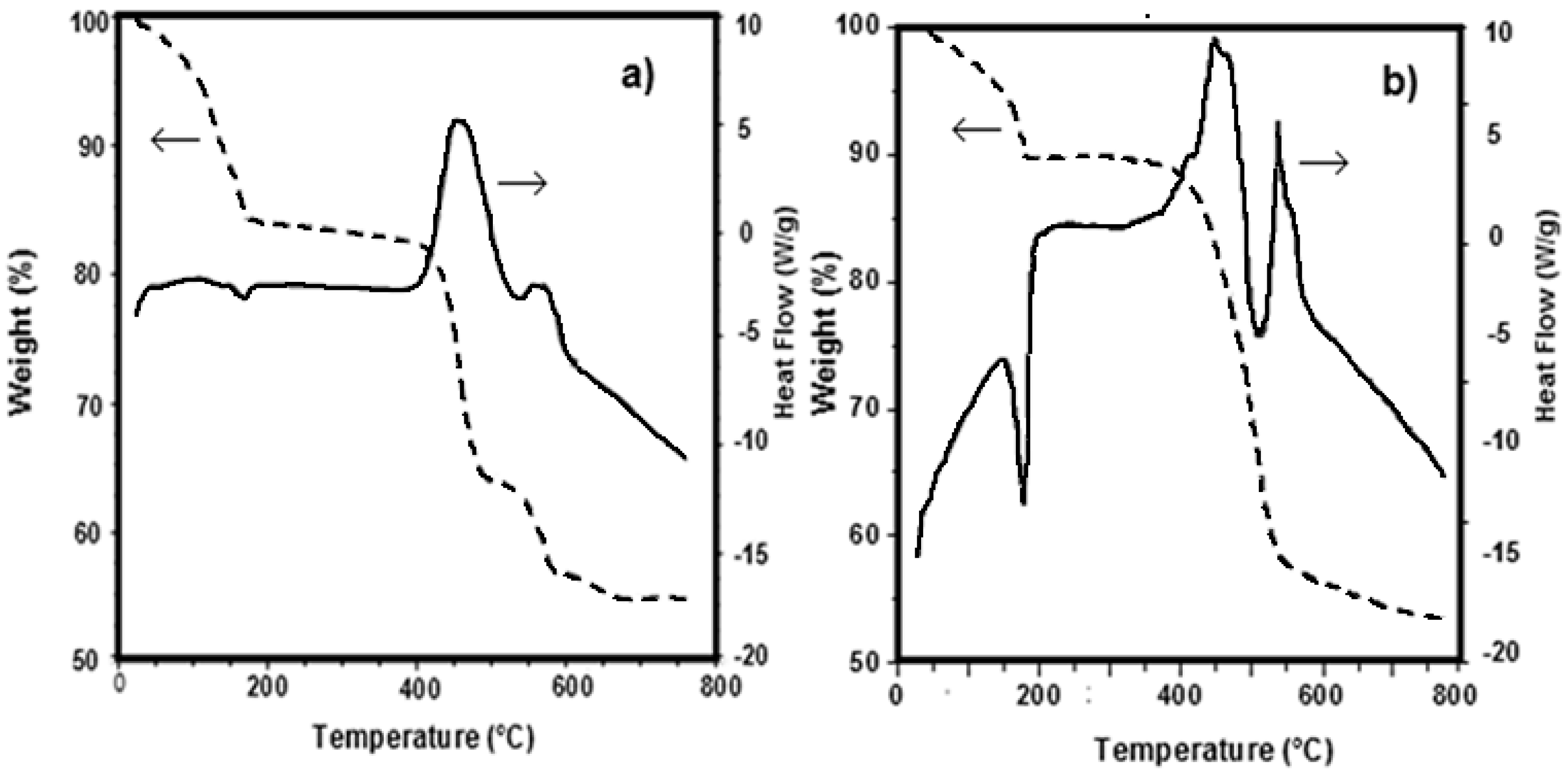
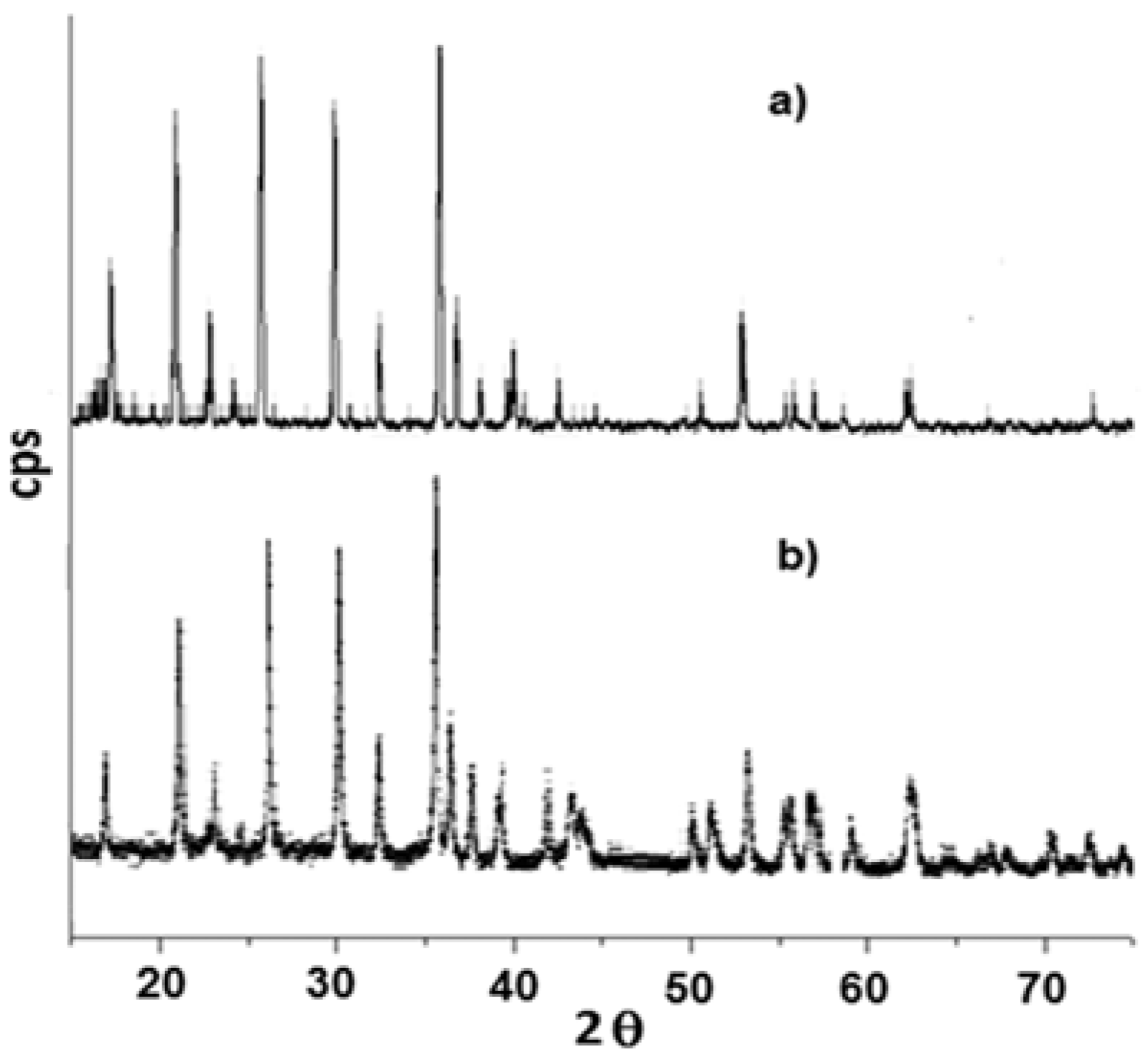
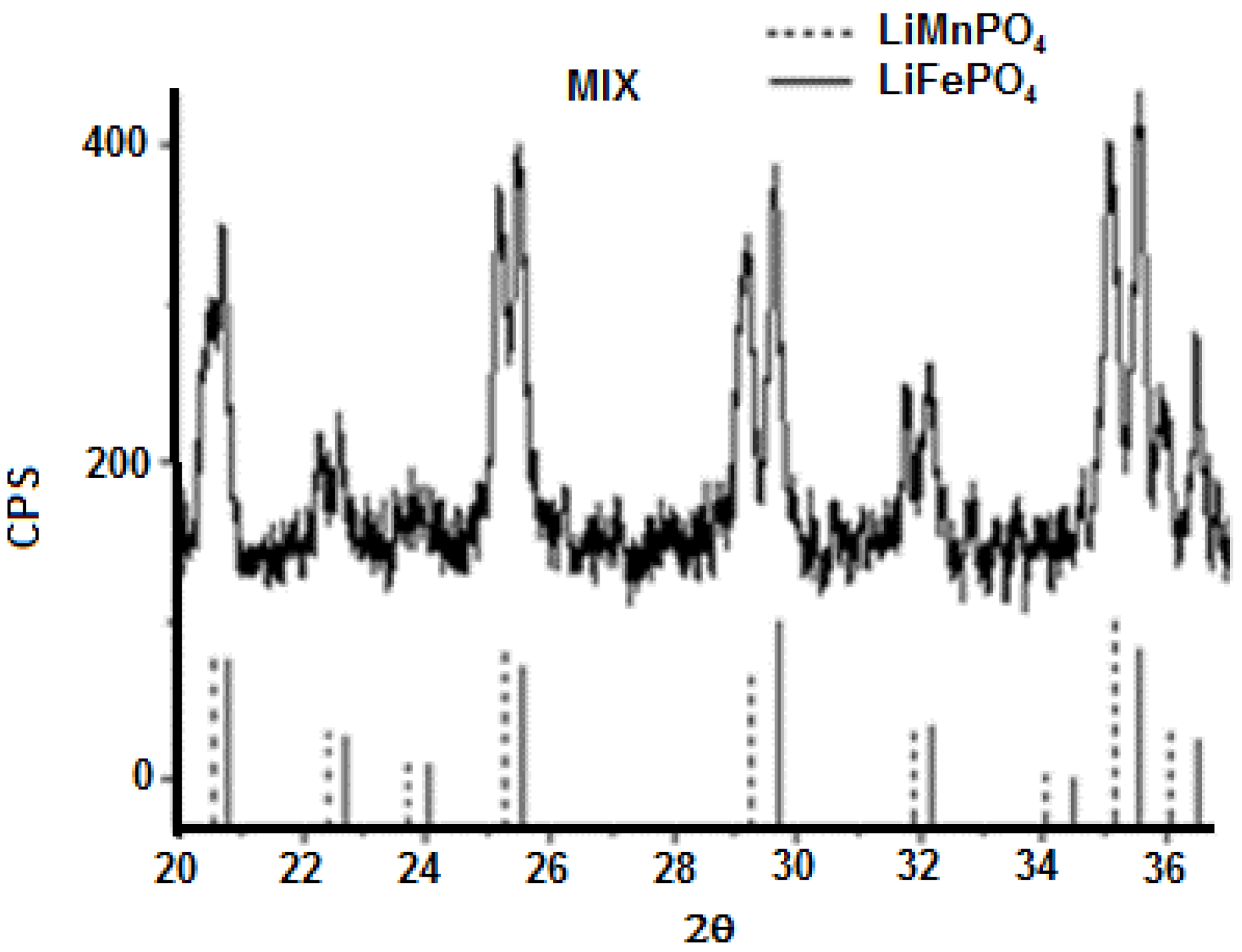
3.1. Precursors Characterization
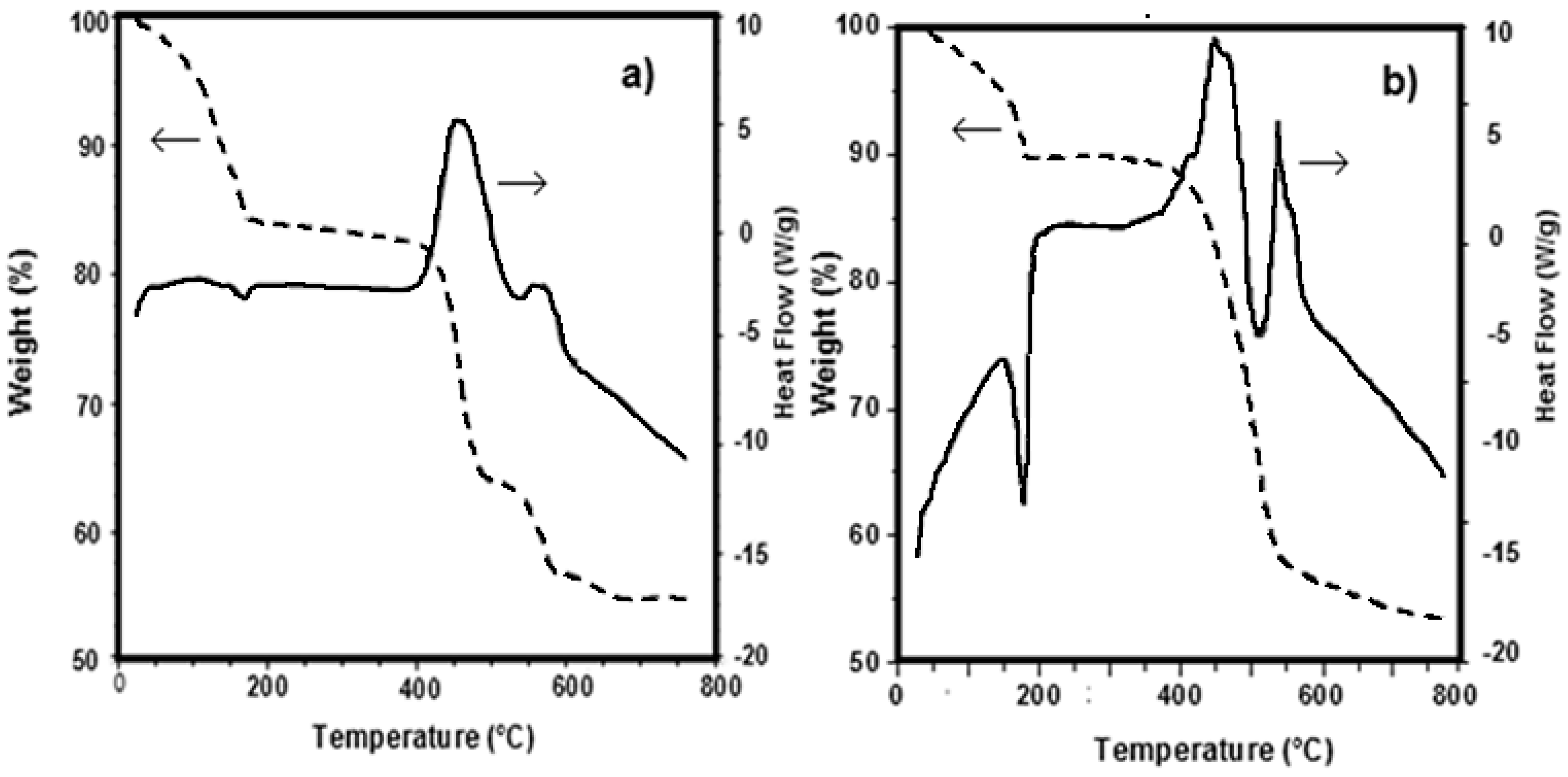
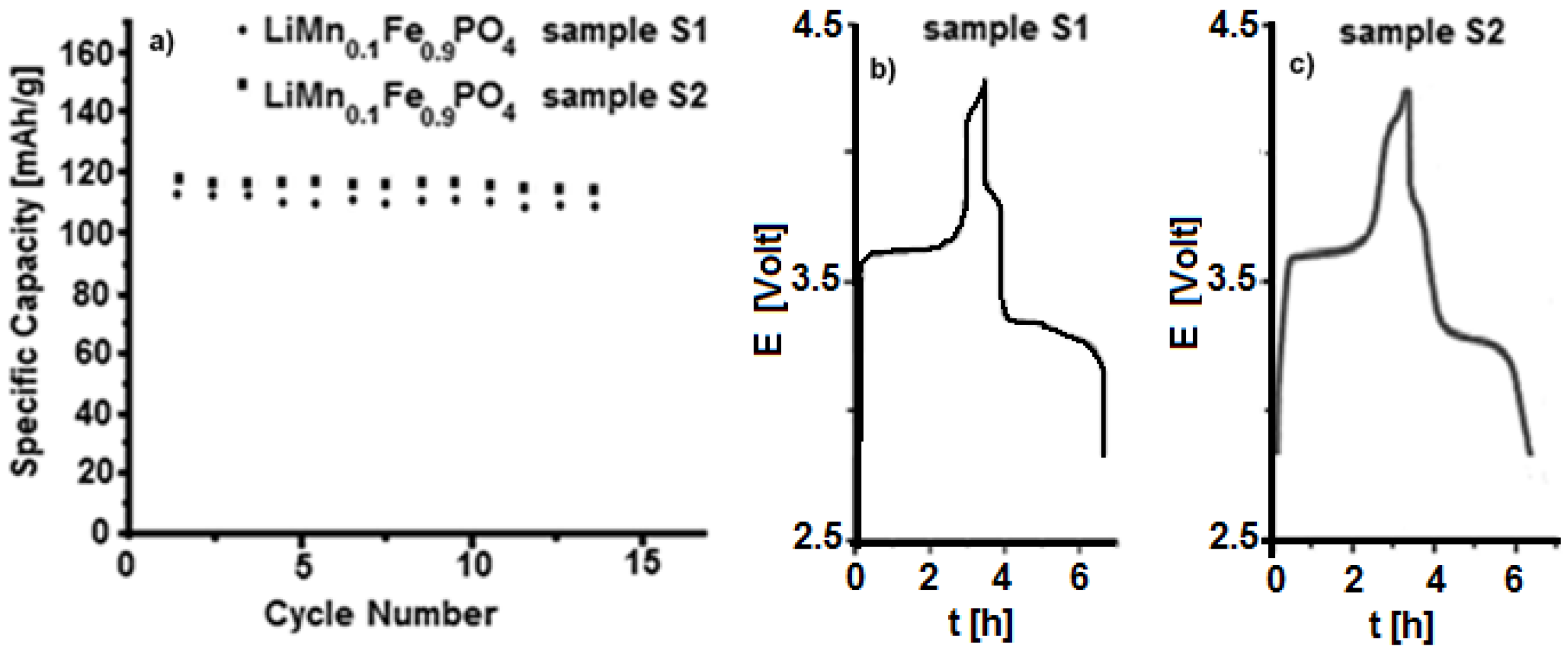
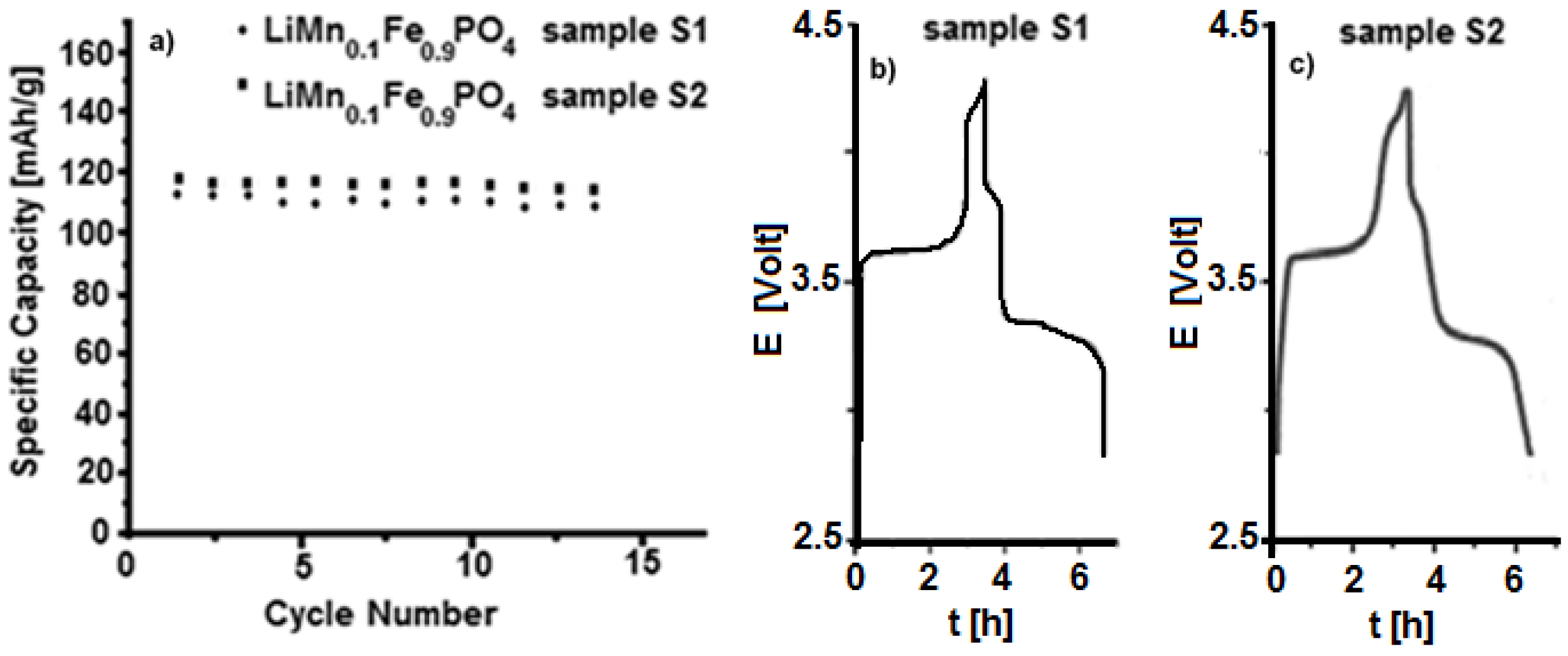
3.2. Characterization of LiFe0.9Mn0.1PO4
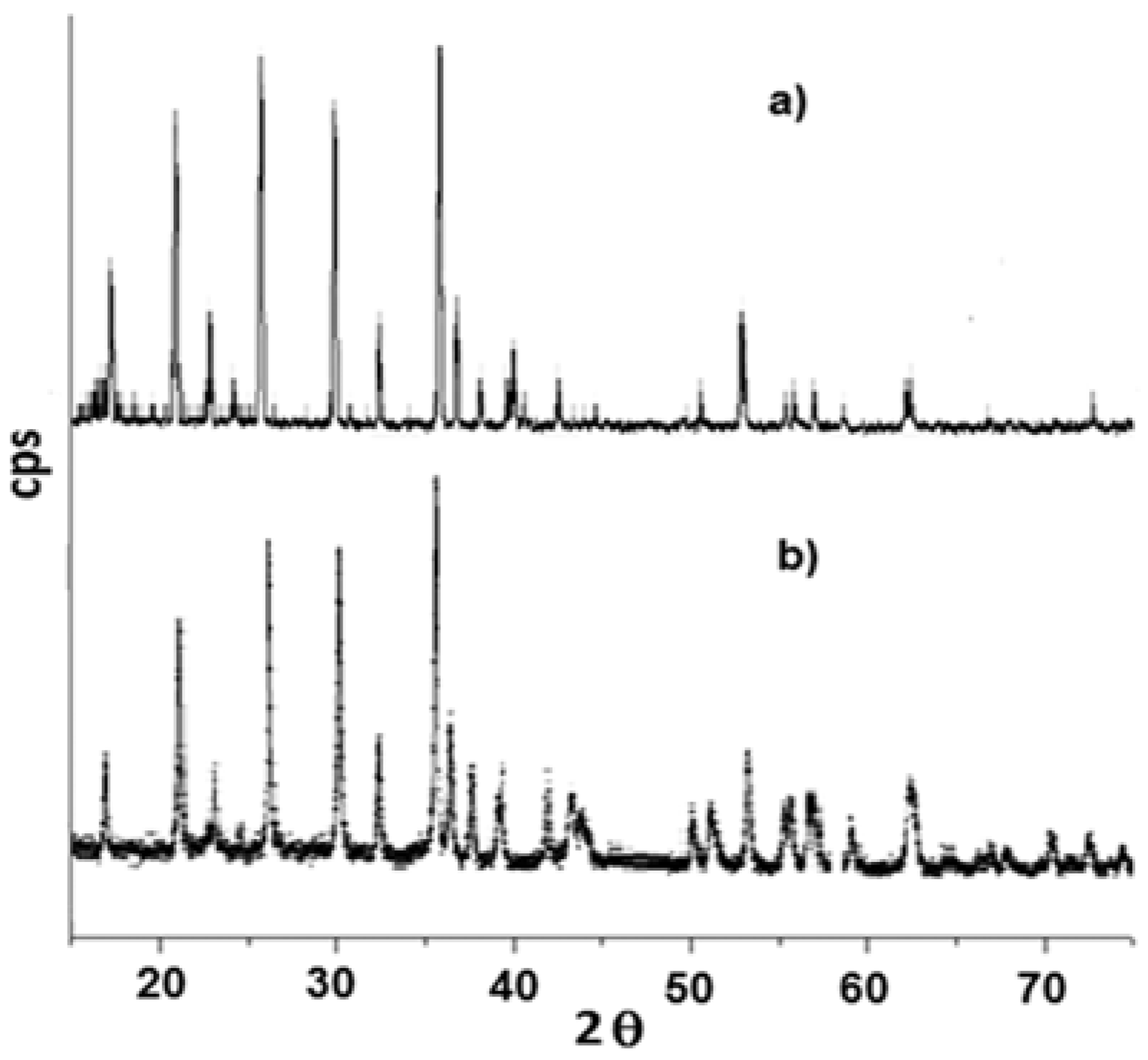
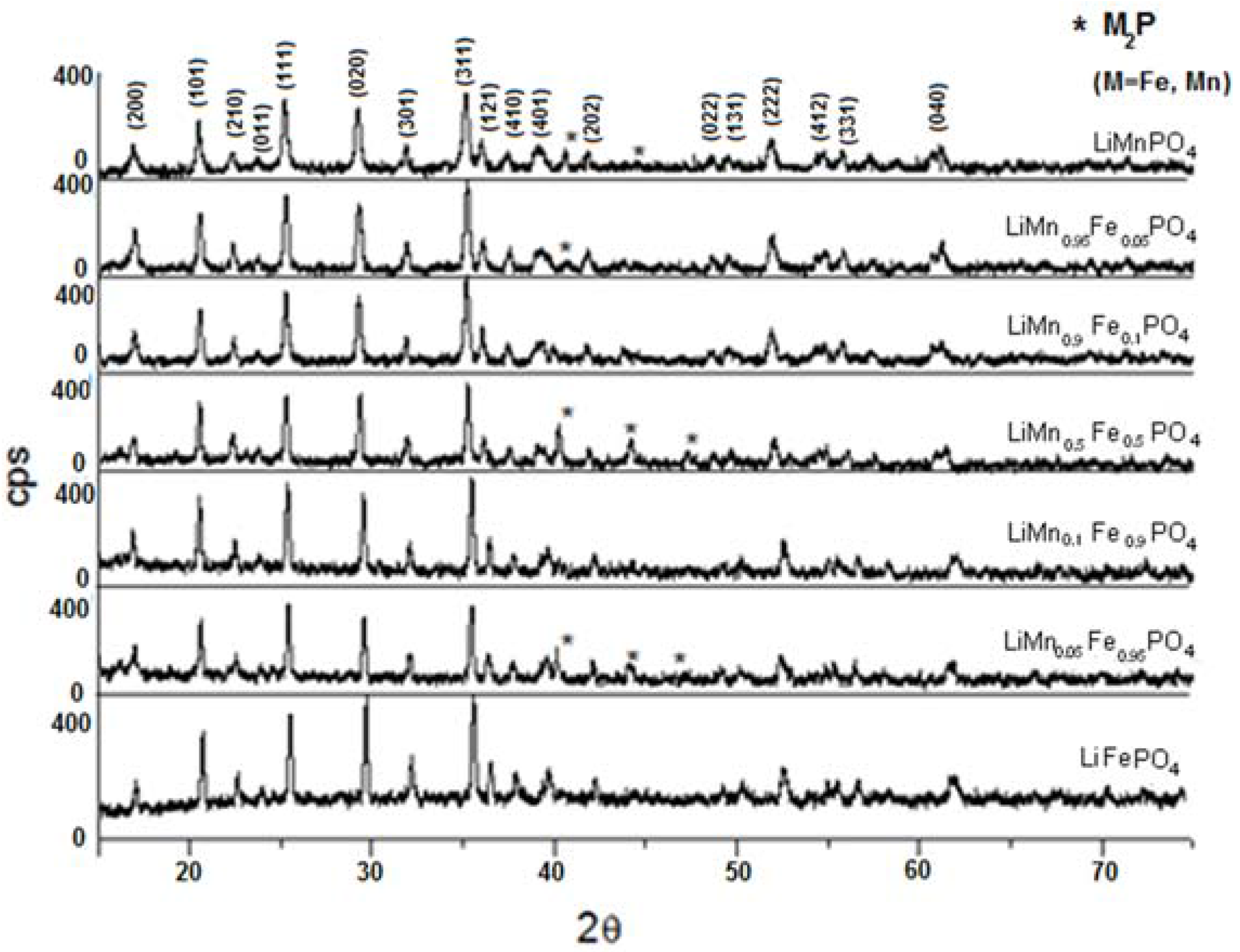
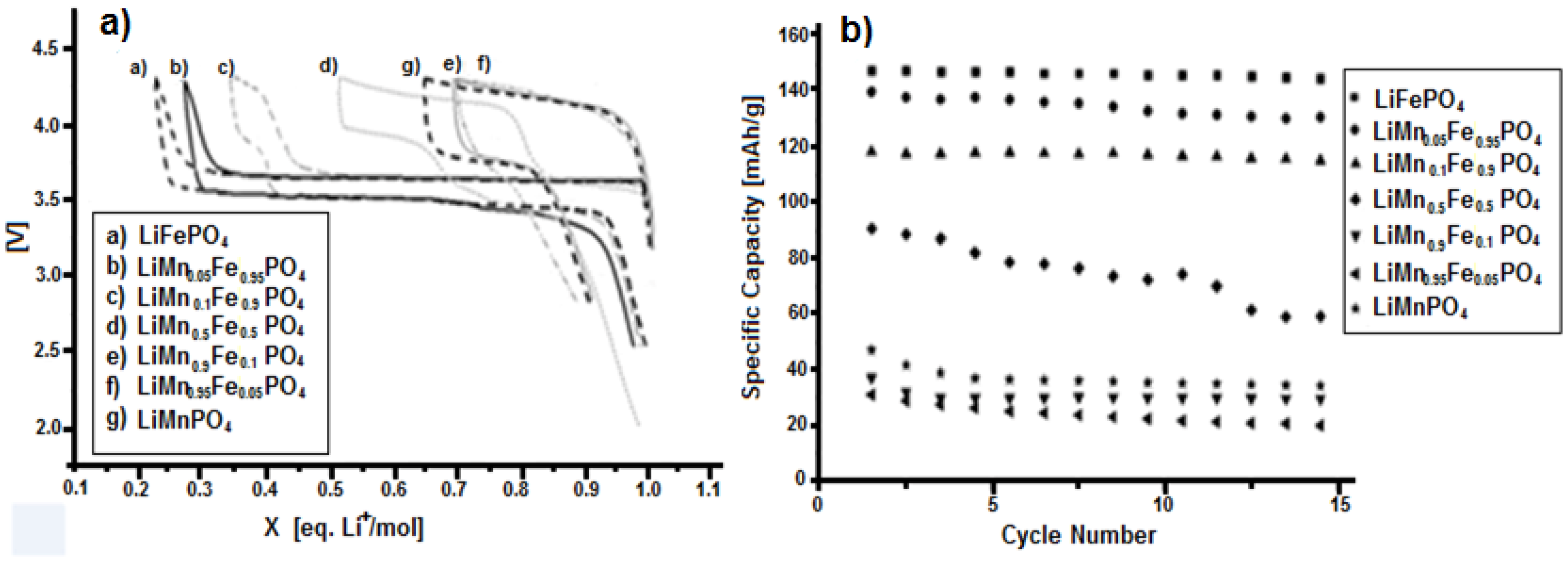
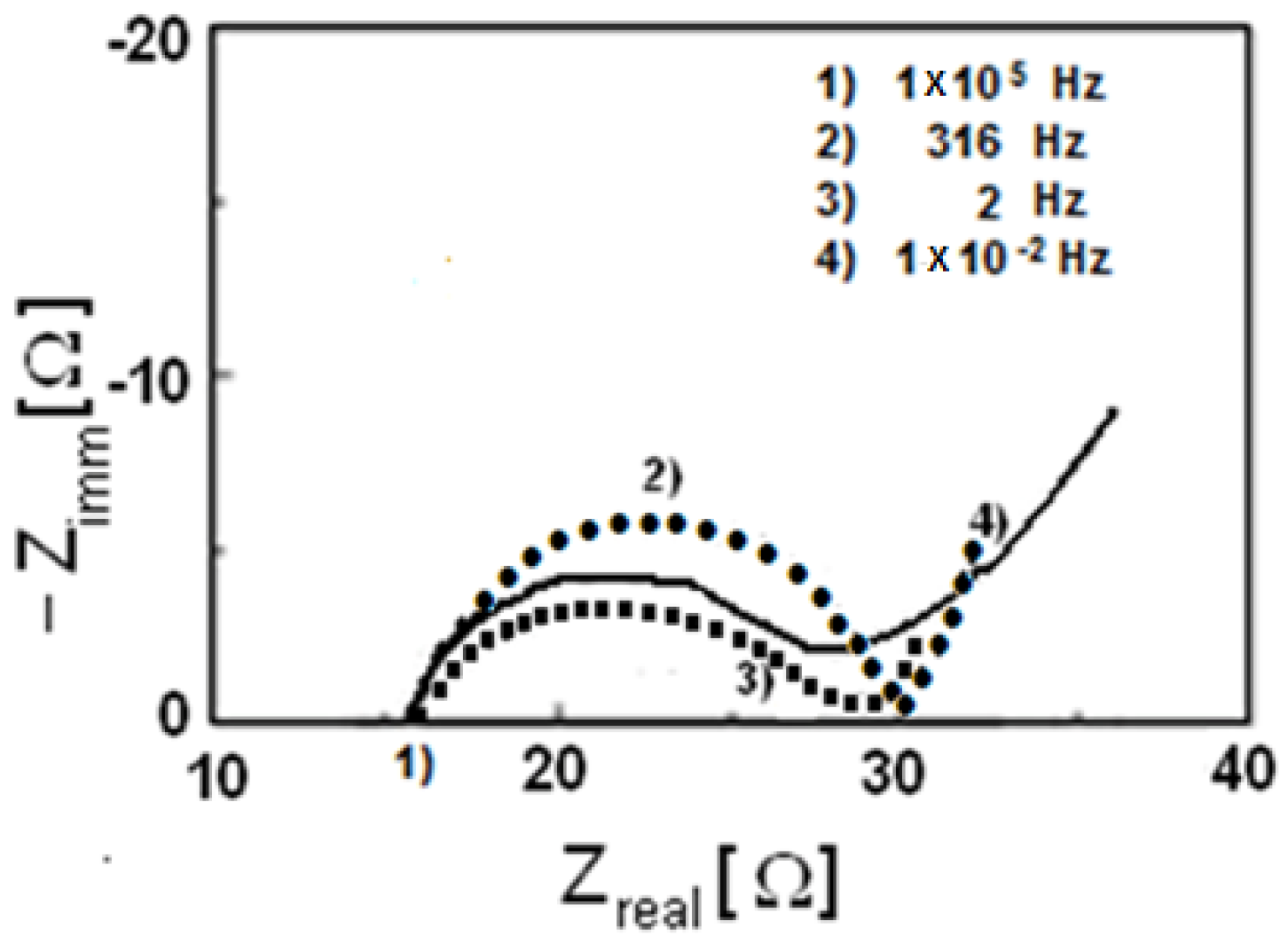
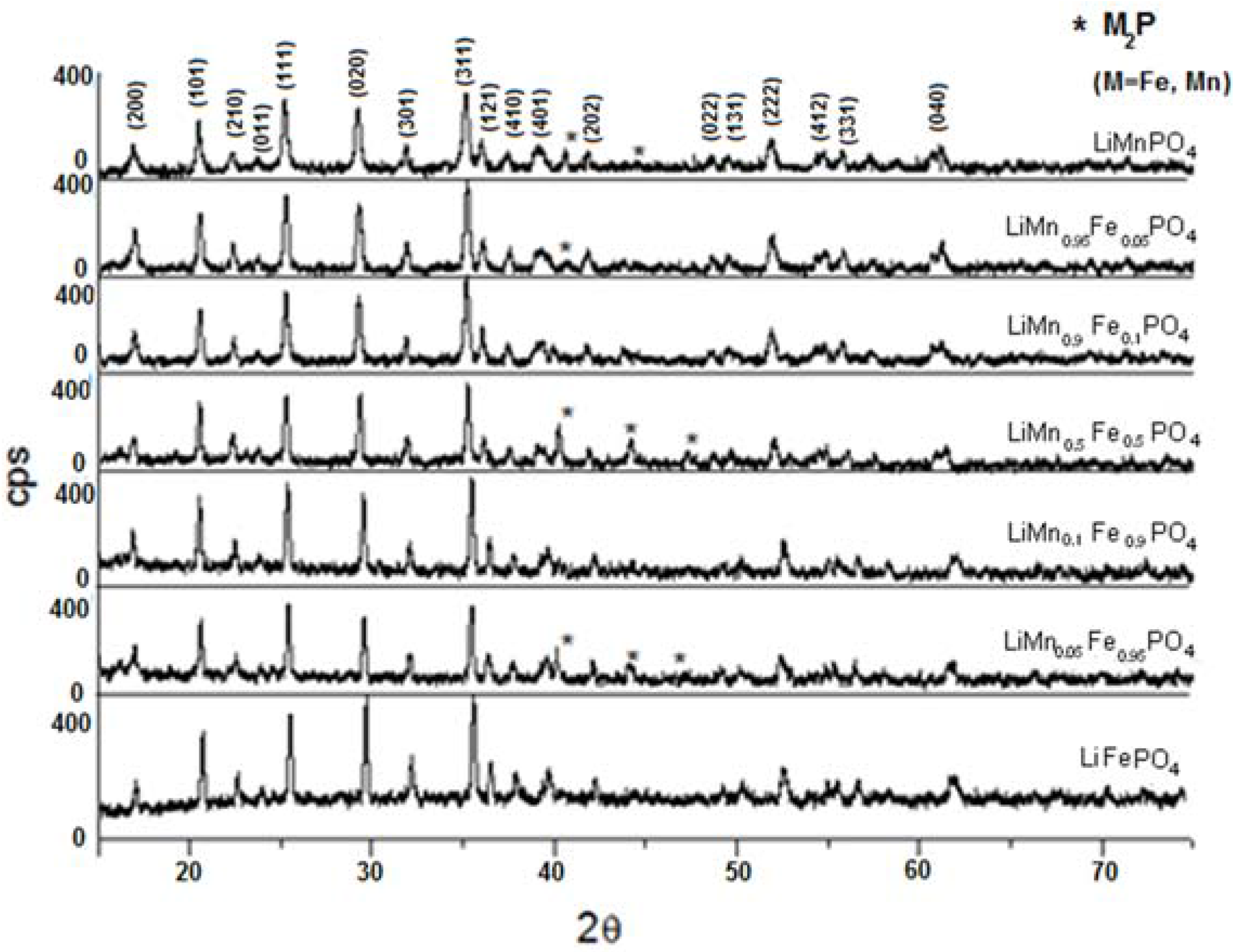
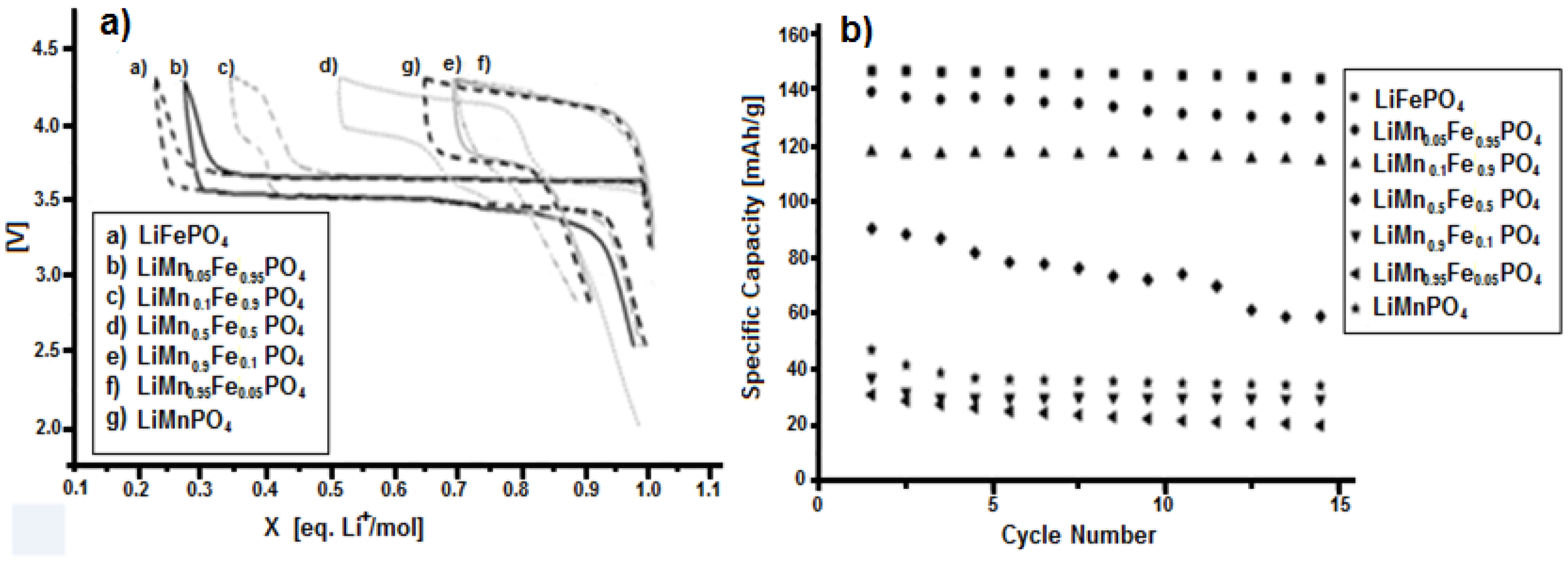
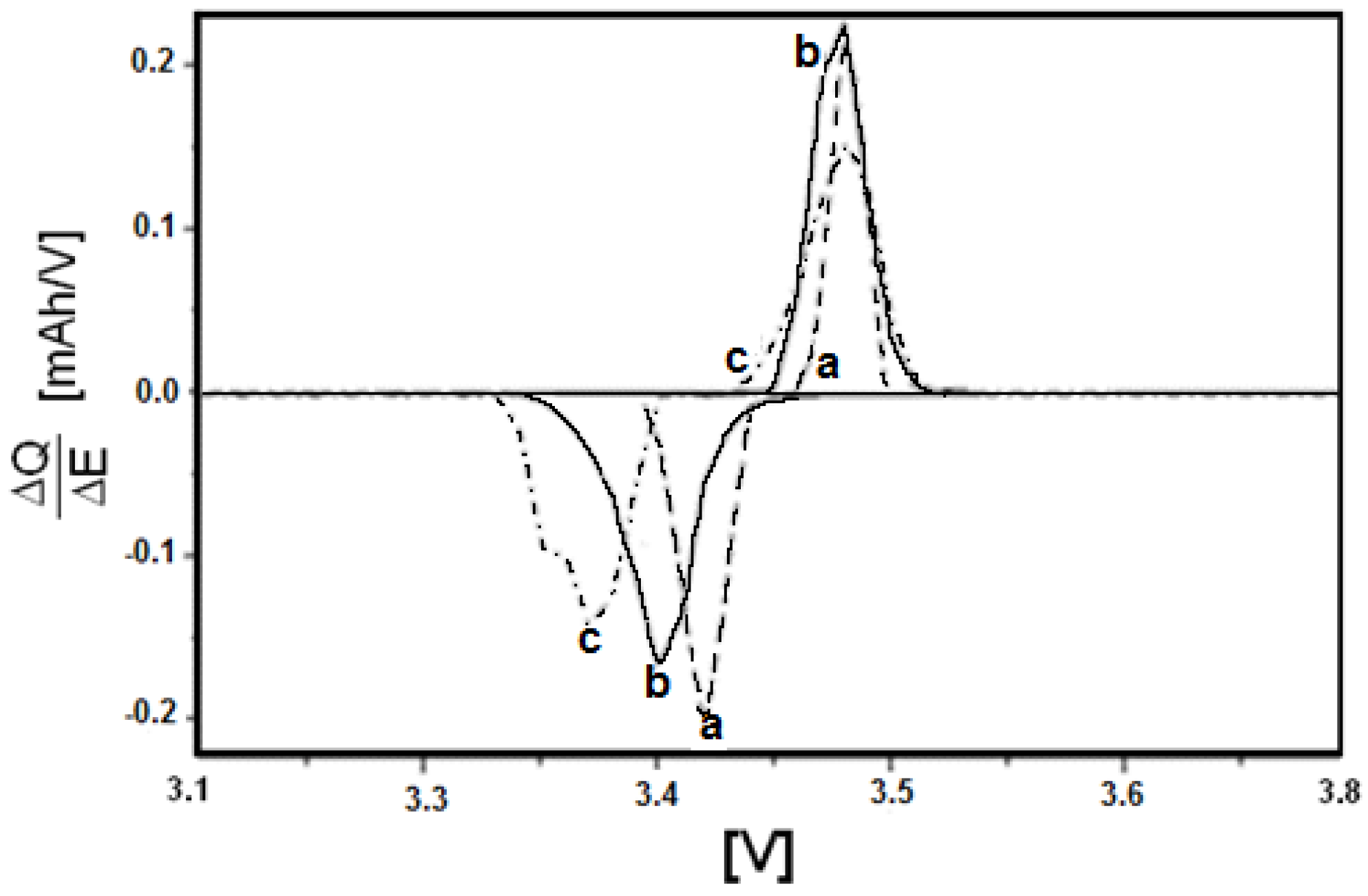
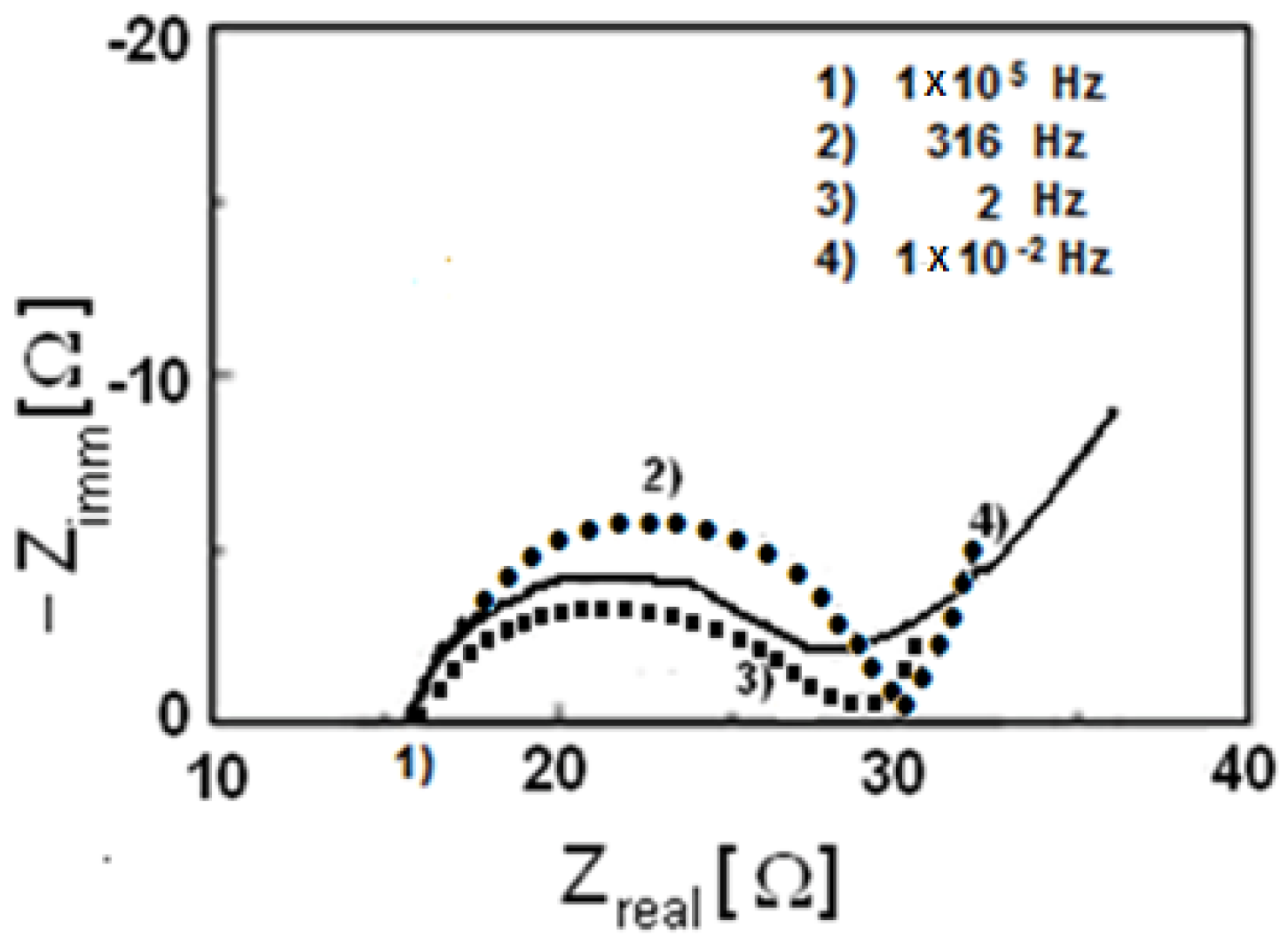
3.3. Characterization of LiMnxFe(1−x)PO4 (with x = 0, 0.05, 0.1, 0.5, 0.9, 0.95, and 1)
4. Experimental
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Lu, L.; Han, X.; Li, J.; Hua, J.; Ouyang, M. A review on the key issues for lithium-ion battery management in electric vehicles. J. Power Sources 2013, 226, 272–288. [Google Scholar] [CrossRef]
- Mathew, M.; Kong, Q.H.; McGrory, J.; Fowler, M. Simulation of lithium ion battery replacement in a battery pack for application in electric vehicles. J. Power Sources 2017, 349, 94–104. [Google Scholar] [CrossRef]
- Pelletier, S.; Jabali, O.; Laporte, G.; Veneroni, M. Battery degradation and behaviour for electric vehicles: Review and numerical analyses of several models. Trans. Res. Part B Methodol. 2017, 103, 158–187. [Google Scholar] [CrossRef]
- Olivetti, E.A.; Ceder, G.; Gaustad, G.G.; Fu, X. Lithium-Ion Battery Supply Chain Considerations: Analysis of Potential Bottlenecks in Critical Metals. Joule 2017, 1, 229–243. [Google Scholar] [CrossRef]
- Santiangeli, A.; Fiori, C.; Zuccari, F.; Dell’Era, A.; Orecchini, F.; D’Orazio, A. Experimental analysis of the auxiliaries consumption in the energy balance of a pre-series plug-in hybrid-electric vehicle. Energy Procedia 2014, 45, 779–78868. [Google Scholar] [CrossRef]
- Orecchini, F.; Santiangeli, A.; Dell’Era, A. EVs and HEVs Using Lithium-Ion Batteries. In Lithium-Ion Batteries: Advances and Applications; Pistoia, G., Ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2014; pp. 205–248. ISBN 978-044459513-3. [Google Scholar] [CrossRef]
- Fabbri, G.; Mascioli, F.M.F.; Pasquali, M.; Mura, F.; Dell’Era, A. Automotive application of lithium-ion batteries: A new generation of electrode materials. In Proceedings of the IEEE 22nd International Symposium on Industrial Electronics (ISIE), Taipei, Taiwan, 28–31 May 2013. [Google Scholar] [CrossRef]
- Dell’Era, A.; Pasquali, M.; Fabbri, G.; Pasquali, L.; Tarquini, G.; Santini, E. Automotive application of lithium-ion batteries: Control of commercial batteries in laboratory test. In Proceedings of the IEEE 23rd International Symposium on Industrial Electronics (ISIE), Istanbul, Turkey, 1–4 June 2014; pp. 1616–1621. [Google Scholar] [CrossRef]
- Fergus, J. Recent Developments in Cathode Materials for Lithium Ion Batteries. J. Power Sources 2010, 195, 939–954. [Google Scholar] [CrossRef]
- Scrosati, B.; Garche, J. Lithium Batteries: Status, Prospects and Future. J. Power Sources 2010, 195, 2419–2430. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, D.; Yang, F. Developments of Lithium Ion Batteries and Challenges of LiFePO4 as One Promising Cathode Material. J. Mater. Sci. 2009, 44, 2435–2443. [Google Scholar] [CrossRef]
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho_Olivines as Positive_Electrode Materials for Rechargeable Lithium Batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Bauer, E.M.; Bellitto, C.; Righini, G.; Pasquali, M.; Dell’Era, A.; Prosini, P.P. A versatile method of preparation of carbon-rich LiFePO4: A promising cathode material for Li-ion batteries. J. Power Sources 2005, 146, 544–549. [Google Scholar] [CrossRef]
- Pasquali, M.; Dell’Era, A.; Prosini, P.P. Fitting of the voltage-Li + insertion curve of LiFePO4. J. Solid State Electrochem. 2009, 13, 1859–1865. [Google Scholar] [CrossRef]
- Benoit, C.; Franger, S. Chemistry and Electrochemistry of Lithium Iron Phosphate. J. Solid State Electrochem. 2008, 12, 987–993. [Google Scholar] [CrossRef]
- Dell’era, A.; Pasquali, M. Comparison between different ways to determine diffusion coefficient and by solving Fick’s equation for spherical coordinates. J. Solid State Electrochem. 2009, 13, 849–859. [Google Scholar] [CrossRef]
- Safronov, D.V.; PinusI, Y.; Profatilova, I.A.; Tarnopolskii, V.A.; Skundin, A.M.; Yaroslavtsev, A.B. Kinetics of Lithium Deintercalation from LiFePO4. Inorg. Mater. 2011, 47, 303–307. [Google Scholar] [CrossRef]
- Barker, J.; Saidi, M.Y.; Swoyer, J.L. Lithium Iron(II) Phospho-olivines Prepared by a Novel Carbothermal Reduction Method. Electrochem. Solid-State Lett. 2003, 6, A53–A55. [Google Scholar] [CrossRef]
- Wang, D.; Li, H.; Shi, S.; Huang, X.; Chen, L. Improving the Rate Performance of LiFePO4 by Fe_Site Doping. Electrochim. Acta 2005, 50, 2955–2958. [Google Scholar] [CrossRef]
- Roberts, M.R.; Vitins, G.; Owen, J.R. High Throughput Studies of Li1−x Mgx/2FePO4 and LiFe1−yMgyPO4 and the Effect of Carbon Coating. J. Power Sources 2008, 179, 754–762. [Google Scholar] [CrossRef]
- Wang, G.X.; Bewlay, S.L.; Konstantinov, K.; Liu, H.K.; Dou, S.X.; Ahn, J.H. Physical and electrochemical properties of doped lithium iron phosphate electrodes. Electrochim. Acta 2004, 50, 443–447. [Google Scholar] [CrossRef]
- Safronov, D.V.; Novikova, S.A.; Kulova, T.L.; Skundin, A.M.; Yaroslavtsev, A.B. Lithium Diffusion in Materials Based on LiFePO4 Doped with Cobalt and Magnesium. Inorg. Mater. 2012, 48, 513–519. [Google Scholar] [CrossRef]
- Nakamura, T.; Sakumoto, K.; Okamoto, M.; Seki, S.; Kobayashi, Y.; Takeuchi, T.; Tabuchi, M.; Yamada, Y. Electrochemical study on Mn2+substitution in LiFePO4 olivine compound. J. Power Sources 2007, 174, 435–441. [Google Scholar] [CrossRef]
- Nakamura, T.; Miwa, Y.; Tabuchi, M.; Yamada, Y. Structural and Surface Modifications of LiFePO4 Olivine Particles and Their Electrochemical Properties. J. Electrochem. Soc. 2006, 153, A1108–A1114. [Google Scholar] [CrossRef]
- Lee, K.T.; Lee, K.S. Electrochemical properties of LiFe0.9Mn0.1PO4/Fe2P cathode material by mechanical alloying. J. Power Sources 2009, 189, 435–439. [Google Scholar] [CrossRef]
- Altomare, A.; Bellitto, C.; Ibrahim, S.A.; Mahmoud, M.R.; Rizzi, R. Iron(ii) phosphonates: A new series of molecule-based weak ferromagnets. J. Chem. Soc. Dalton Trans. 2000, 21, 3913–3919. [Google Scholar] [CrossRef]
- Bellitto, C.; Federici, F.; Colapietro, M.; Portalone, G.; Caschera, D. X-ray single-crystal structure and magnetic properties of Fe[CH3PO3)]·H2O: A layered weak ferromagnet. Inorg. Chem. 2002, 41, 709–714. [Google Scholar] [CrossRef]
- Bauer, E.M.; Bellitto, C.; Pasquali, M.; Prosini, P.P.; Righini, G. Versatile synthesis of carbon-Rich LiFePO4 enhancing its Electrochemical Properties. Electrochem. Solid-State Lett. 2004, 7, A85–A87. [Google Scholar] [CrossRef]
- Ho, C.; Raistrick, I.D.; Huggins, R.A. Application of A-C techniques to the study of lithium diffusion in tungsten trioxide thin films. J. Electrochem. Soc. 1980, 127, 343–350. [Google Scholar] [CrossRef]
- Levi, M.D.; Levi, E.A.; Aurbach, D. The mechanism of lithium intercalation in graphite film electrodes in aprotic media Part 2 potentiostatic intermittent titration and in situ XRD studies of the solid-state ionic diffusion. J. Electroanal. Chem. 1997, 421, 89–97. [Google Scholar] [CrossRef]
- Weppner, W.; Huggins, R.A. Determination of the kinetic parameter of mixed-conducting electrodes and application to the system Li3Sb. J. Electrochem. Soc. 1977, 1569–1578. [Google Scholar] [CrossRef]
- McKinnon, W.R.; Haering, R.R. Modern Aspect in Electrochemistry, 1st ed.; White, R.E., Bockris, J.O.M., Conway, B.E., Eds.; Plenum Press: New York, NY, USA; London, UK, 1983; ISBN 978-1-4615-7463-7. [Google Scholar] [CrossRef]
- Wang, C.; Hong, J. Ionic/Electronic Conducting Characteristics of LiFePO4 Cathode Materials. The Determining Factors for High Rate Performance. Electrochem. Solid-State Lett. 2007, 10, A65–A69. [Google Scholar] [CrossRef]
- Park, C.Y.; Park, S.B.; Oh, S.H.; Jang, H.; Cho, W.I.; Ion, L. Diffusivity and Improved Electrochemical Performances of the Carbon Coated LiFePO4. Bull. Korean Chem. Soc. 2011, 32, 836–840. [Google Scholar] [CrossRef]
- Saravanan, K.; Vitta, J.J.; Reddy, M.V.; Chowdari, B.V.R.; Balaya, P. Storage performance of LiFe1−xMnxPO4 nanoplates (x = 0, 0.5, and 1). J. Solid State Electrochem. 2010, 14, 1755–1760. [Google Scholar] [CrossRef]
- Yoncheva, M.; Koleva, V.; Mladenov, M.; Sendova-Vassileva, M.; Nikolaeva-Dimitrova, M.; Stoyanova, R.; Zhecheva, E. Carbon-coated nano-sized LiFe1−xMnxPO4 solid solutions (0 ≤ x ≤ 1) obtained from phosphate–formate precursors. J. Mater. Sci. 2011, 46, 7082–7089. [Google Scholar] [CrossRef]
- Xu, J.; Chen, G.; Li, H.J.; Lv, Z.S. Direct-hydrothermal synthesis of LiFe1−xMnxPO4 cathode materials. J. Appl. Electrochem. 2010, 40, 575–580. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, X.; Li, H.; Huang, X. Electrochemical performances of LiFe1−xMnxPO4 with high Mn content. J. Power Sources 2011, 196, 6992–6996. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Yu, X.; Cai, R.; Shao, Z.; Liao, X.Z.; Ma, Z.F. Mechanoactivation-assisted synthesis and electrochemical characterization of manganese lightly doped LiFePO4. J. Alloy. Compd. 2010, 492, 675–680. [Google Scholar] [CrossRef]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Formula |
|---|---|
| P1 | Fe0.9Mn0.1(C6H5PO3)(H2O) |
| P2 | Fe(C6H5PO3)(H2O) |
| P3 | Mn(C6H5PO3)(H2O) |
| Material (Symbol) | a | b | c |
|---|---|---|---|
| Fe0.9Mn0.1(C6H5PO3)·H2O (P1) | 5.680 | 14.410 | 4.900 |
| Fe(C6H5PO3)·H2O (P2) | 5.652 | 14.404 | 4.882 |
| Mn(C6H5PO3)·H2O (P3) | 5.751 | 14.401 | 4.953 |
| Compound | a (Å) | b (Å) | c (Å) | Crystallite Size T (Å) |
|---|---|---|---|---|
| LiFePO4 | 10.330 | 6.010 | 4.690 | 393 |
| LiFe0.95Mn0.05PO4 | 10.335 | 6.011 | 4.693 | 370 |
| LiFe0.9Mn0.1PO4 | 10.347 | 6.020 | 4.699 | 368 |
| LiFe0.5Mn0.5PO4 | 10.372 | 6.054 | 4.706 | 365 |
| LiFe0.1Mn0.9PO4 | 10.448 | 6.103 | 4.743 | 282 |
| LiFe0.05Mn0.95PO4 | 10.448 | 6.104 | 4.743 | 280 |
| LiMnPO4 | 10.450 | 6.108 | 4.732 | 265 |
| Material (Symbol) | Specific Surface Area (m2·g−1) | Carbon Content (%) |
|---|---|---|
| LiFePO4 | 105 | 10.0 |
| LiMn0.05Fe0.95PO4 | 100 | 10.2 |
| LiMn0.1Fe0.9PO4 | 130 | 12.5 |
| LiMn0.5Fe0.5PO4 | 115 | 10.3 |
| LiMn0.9Fe0.1PO4 | 110 | 11.3 |
| LiMn0.95Fe0.05PO4 | 151 | 13.7 |
| LiMnPO4 | 105 | 11.5 |
| Material (Symbol) | D (cm2·s−1) |
|---|---|
| LiFePO4 | 2.0 × 10−14 |
| LiMn0.05Fe0.95PO4 | 5.7 × 10−14 |
| LiMn0.1Fe0.9PO4 | 7.7 × 10−14 |
| Compound | Method | C-Rate | Capacity (mAh/g) | Reference |
|---|---|---|---|---|
| LiMnxFe(1−x)PO4/C (x = 0; 0.5; 1) | Solvothermal process | C/5 | 150; 65; 50 | [35] |
| LiMnxFe(1−x)PO4/C (x = 0; 0.5; 1) | freeze-dry process | C/20 | 140; 120; 95 | [36] |
| LiMnxFe(1−x)PO4/C (x = 0.7; 0.8; 0.9) | Solid state reaction | C/10 | 110; 120; 130 | [37] |
| LiMnxFe(1−x)PO4 (x = 0; 0.05; 0.1; 0.2; 0.4) | Hydrothermal process | C/10 | 140; 110; 95; 90; 78 | [38] |
| LiMnxFe(1−x)PO4 (x = 0; 0.1; 0.2; 0.3) | Mechano-activation synthesis | C/10 | 135; 108; 125; 80 | [39] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dell’Era, A.; Pasquali, M.; Bauer, E.M.; Vecchio Ciprioti, S.; Scaramuzzo, F.A.; Lupi, C. Synthesis, Characterization, and Electrochemical Behavior of LiMnxFe(1−x)PO4 Composites Obtained from Phenylphosphonate-Based Organic-Inorganic Hybrids. Materials 2018, 11, 56. https://doi.org/10.3390/ma11010056
Dell’Era A, Pasquali M, Bauer EM, Vecchio Ciprioti S, Scaramuzzo FA, Lupi C. Synthesis, Characterization, and Electrochemical Behavior of LiMnxFe(1−x)PO4 Composites Obtained from Phenylphosphonate-Based Organic-Inorganic Hybrids. Materials. 2018; 11(1):56. https://doi.org/10.3390/ma11010056
Chicago/Turabian StyleDell’Era, Alessandro, Mauro Pasquali, Elvira Maria Bauer, Stefano Vecchio Ciprioti, Francesca A. Scaramuzzo, and Carla Lupi. 2018. "Synthesis, Characterization, and Electrochemical Behavior of LiMnxFe(1−x)PO4 Composites Obtained from Phenylphosphonate-Based Organic-Inorganic Hybrids" Materials 11, no. 1: 56. https://doi.org/10.3390/ma11010056
APA StyleDell’Era, A., Pasquali, M., Bauer, E. M., Vecchio Ciprioti, S., Scaramuzzo, F. A., & Lupi, C. (2018). Synthesis, Characterization, and Electrochemical Behavior of LiMnxFe(1−x)PO4 Composites Obtained from Phenylphosphonate-Based Organic-Inorganic Hybrids. Materials, 11(1), 56. https://doi.org/10.3390/ma11010056