Structural and Electrochemical Characterization of Zn1−xFexO—Effect of Aliovalent Doping on the Li+ Storage Mechanism

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
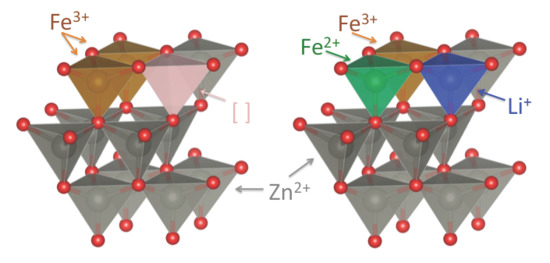
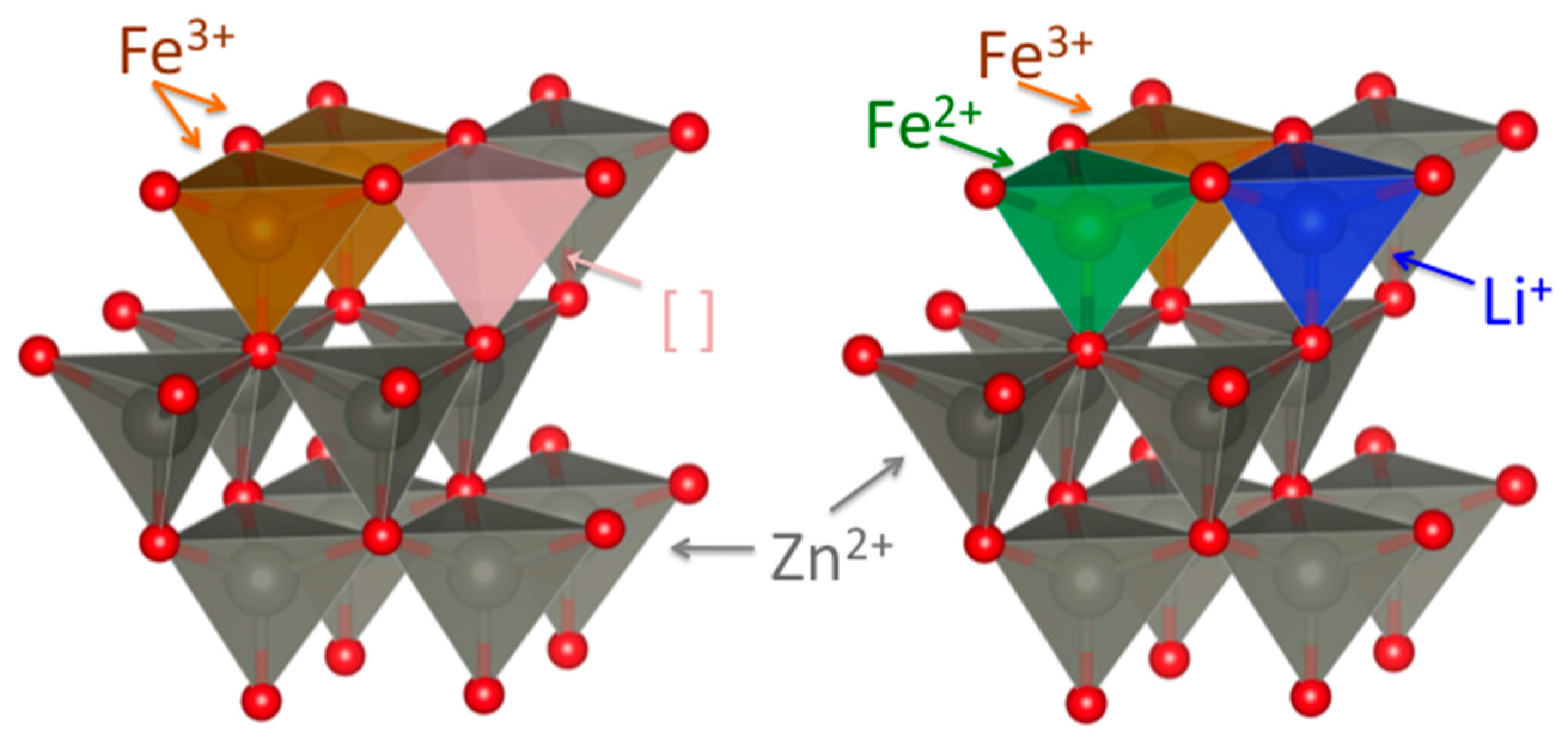
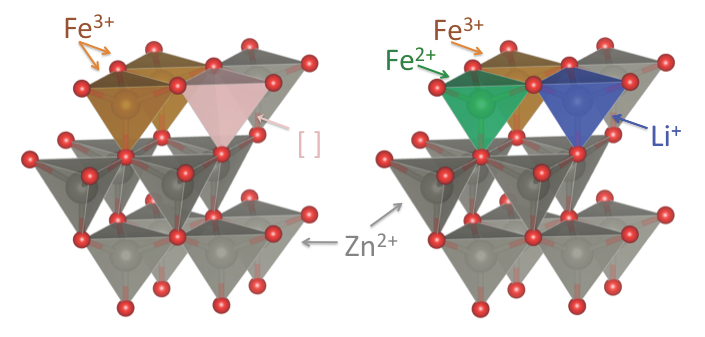
2.1. Synthesis and Basic Characterization
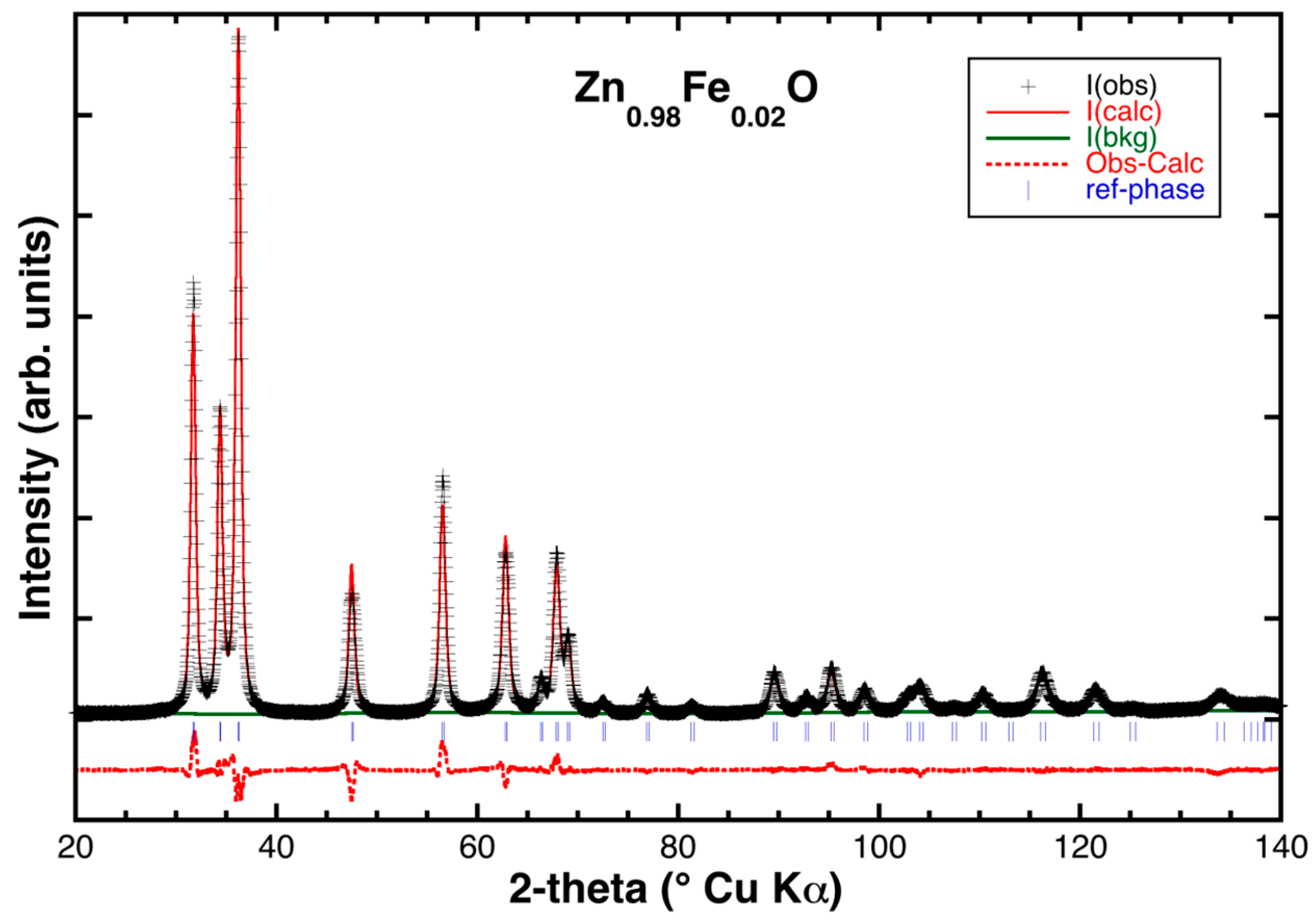
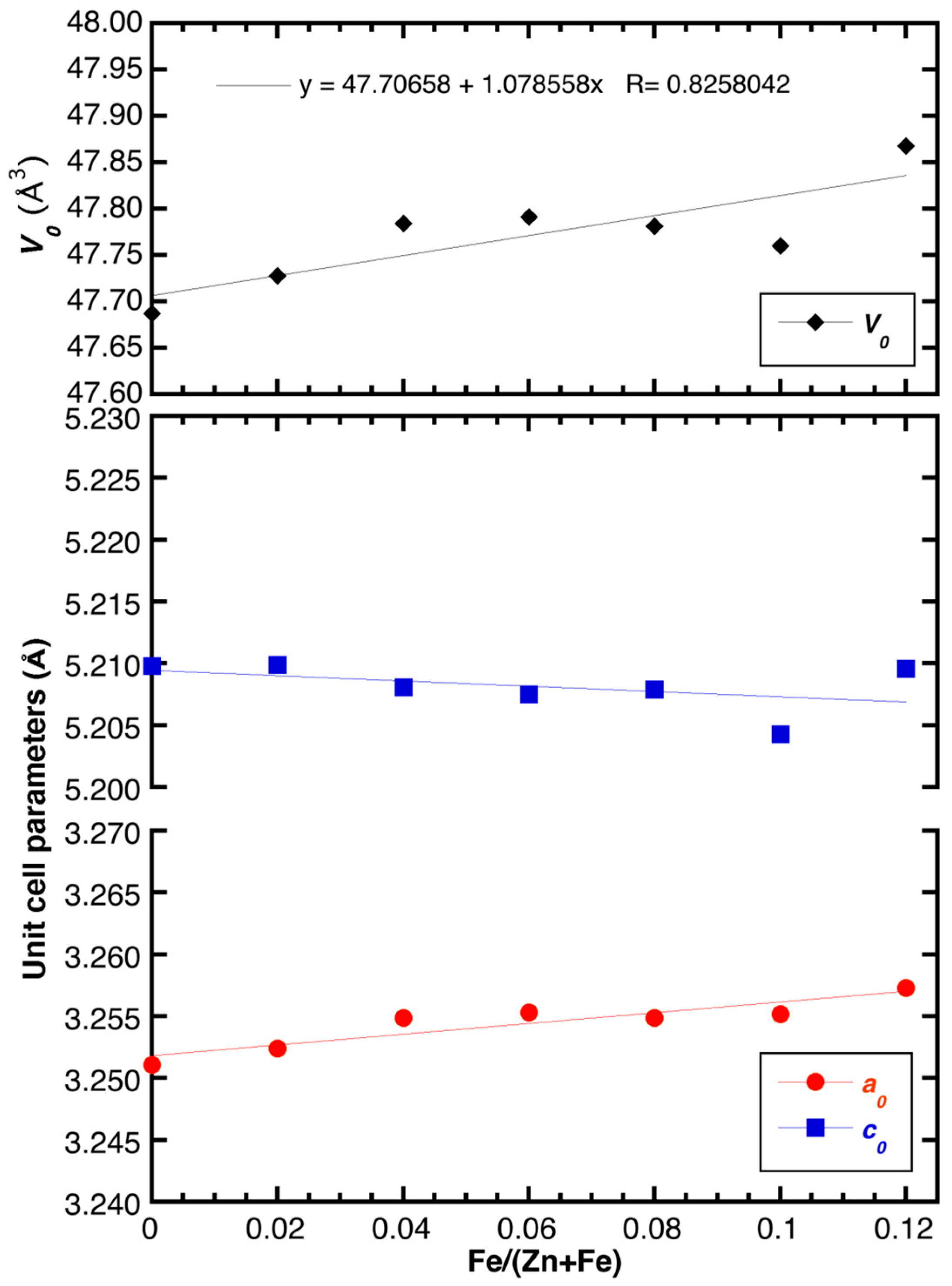
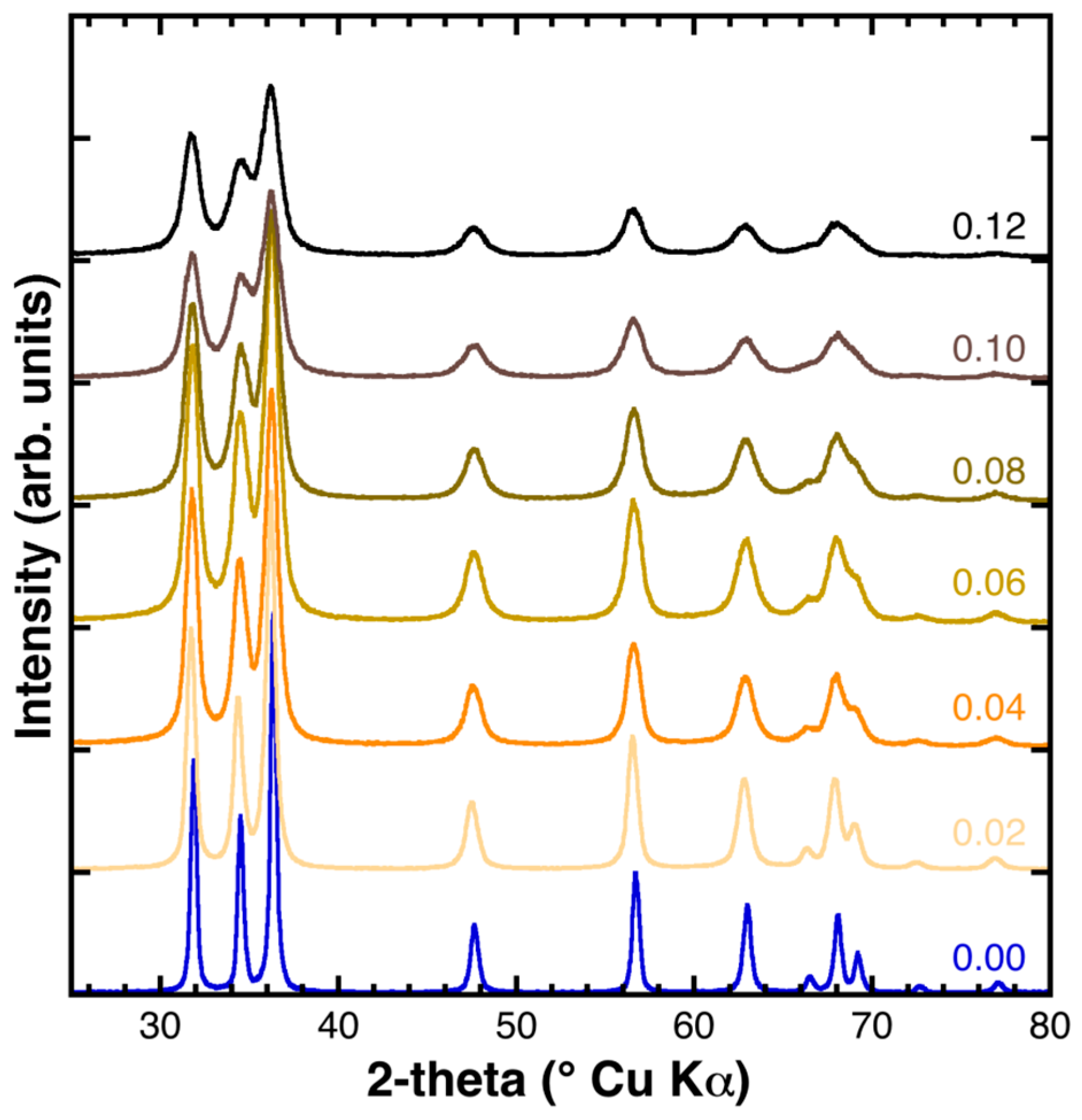
2.2. Powder XRD Characterization and Structural Refinement
2.3. Fe K-Edge XANES Spectroscopy
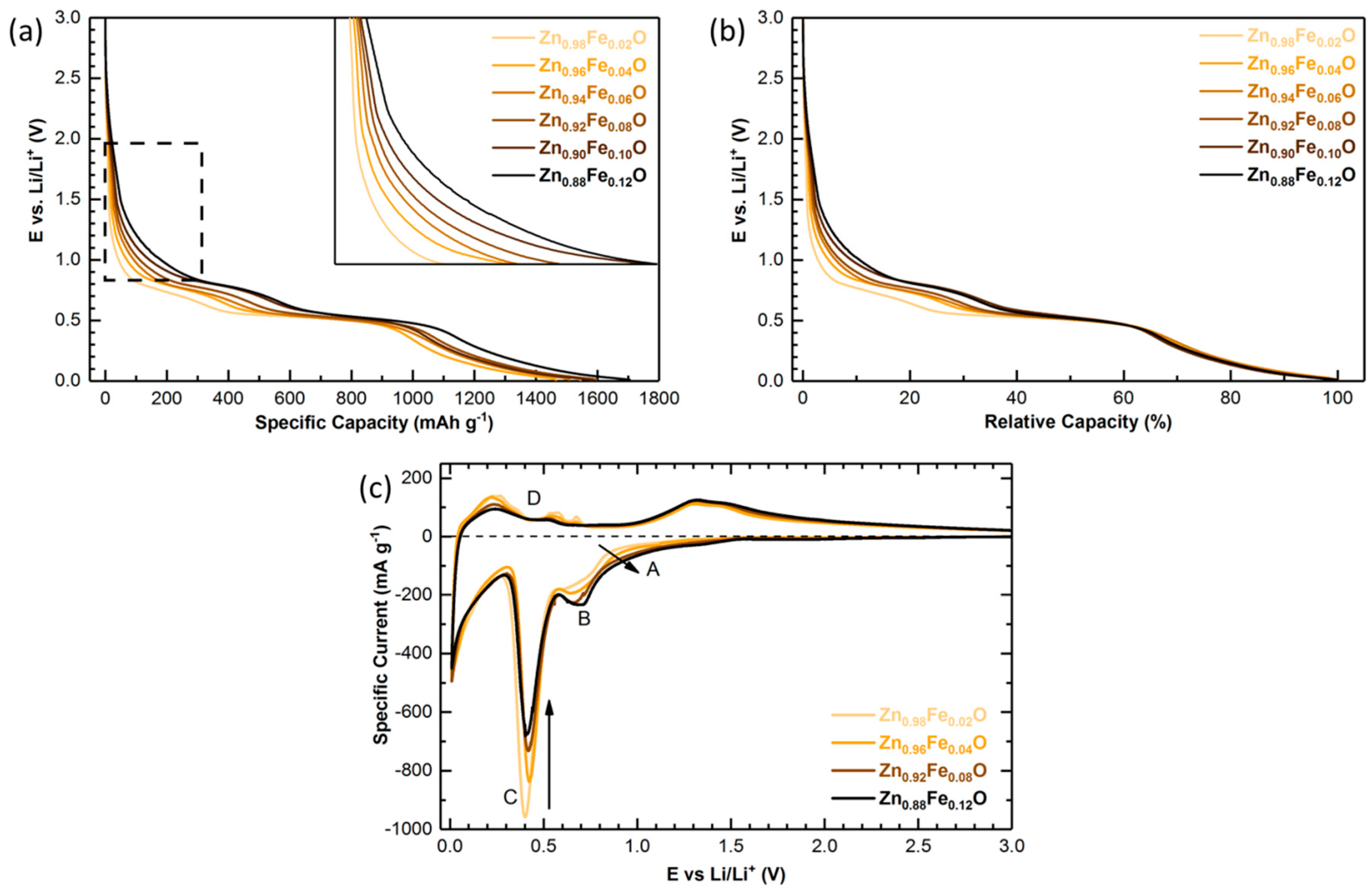
2.4. Electrochemical Characterization
3. Materials and Methods
3.1. Material Synthesis
3.2. Powder XRD Characterization and Structural Refinement
3.3. XAS Data Collection and Analysis
3.4. Electrochemical Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A


References
- Tarascon, J.-M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Armand, M.; Tarascon, J.-M. Building better batteries. Nature 2008, 451, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Scrosati, B.; Garche, J. Lithium batteries: Status, prospects and future. J. Power Sources 2010, 195, 2419–2430. [Google Scholar] [CrossRef]
- Dunn, B.; Kamath, H.; Tarascon, J.-M. Electrical Energy Storage for the Grid: A Battery of Choices. Science 2011, 334, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, M.M.; Wolverton, C.; Isaacs, E.D. Electrical energy storage for transportation—Approaching the limits of, and going beyond, lithium-ion batteries. Energy Environ. Sci. 2012, 5, 7854–7863. [Google Scholar] [CrossRef]
- Aravindan, V.; Lee, Y.S.; Madhavi, S. Research Progress on Negative Electrodes for Practical Li-Ion Batteries: Beyond Carbonaceous Anodes. Adv. Energy Mater. 2015, 5. [Google Scholar] [CrossRef]
- Yamada, Y.; Iriyama, Y.; Abe, T.; Ogumi, Z. Kinetics of Lithium Ion Transfer at the Interface between Graphite and Liquid Electrolytes: Effects of Solvent and Surface Film. Langmuir 2009, 25, 12766–12770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J. A review of the electrochemical performance of alloy anodes for lithium-ion batteries. J. Power Sources 2011, 196, 13–24. [Google Scholar] [CrossRef]
- Cabana, J.; Monconduit, L.; Larcher, D.; Palacín, M.R. Beyond Intercalation-Based Li-Ion Batteries: The State of the Art and Challenges of Electrode Materials Reacting Through Conversion Reactions. Adv. Mater. 2010, 22, 170–192. [Google Scholar] [CrossRef] [PubMed]
- Obrovac, M.N.; Chevrier, V.L. Alloy Negative Electrodes for Li-Ion Batteries. Chem. Rev. 2014, 114, 11444–11502. [Google Scholar] [CrossRef] [PubMed]
- Özgür, Ü.; Alivov, Y.I.; Liu, C.; Teke, A.; Reshchikov, M.A.; Doǧan, S.; Avrutin, V.; Cho, S.J.; Morko̧, H. A comprehensive review of ZnO materials and devices. J. Appl. Phys. 2005, 98. [Google Scholar] [CrossRef]
- Bresser, D.; Mueller, F.; Fiedler, M.; Krueger, S.; Kloepsch, R.; Baither, D.; Winter, M.; Paillard, E.; Passerini, S. Transition-Metal-Doped Zinc Oxide Nanoparticles as a new Lithium-Ion Anode Material. Chem. Mater. 2013, 25, 4977–4985. [Google Scholar] [CrossRef]
- Mueller, F.; Geiger, D.; Kaiser, U.; Passerini, S.; Bresser, D. Elucidating the Impact of Cobalt Doping on the Lithium Storage Mechanism in Conversion/Alloying-Type Zinc Oxide Anodes. ChemElectroChem 2016, 3, 1311–1319. [Google Scholar] [CrossRef]
- Bresser, D.; Passerini, S.; Scrosati, B. Leveraging valuable synergies by combining alloying and conversion for lithium-ion anodes. Energy Environ. Sci. 2016, 9, 3348–3367. [Google Scholar] [CrossRef]
- Mueller, F.; Gutsche, A.; Nirschl, H.; Geiger, D.; Kaiser, U.; Bresser, D.; Passerini, S. Iron-Doped ZnO for Lithium-Ion Anodes: Impact of the Dopant Ratio and Carbon Coating Content. J. Electrochem. Soc. 2017, 164, A6123–A6130. [Google Scholar] [CrossRef]
- Mote, V.; Purushotham, Y.; Dole, B. Williamson-Hall analysis in estimation of lattice strain in nanometer-sized ZnO particles. J. Theor. Appl. Phys. 2012, 6, 6–13. [Google Scholar] [CrossRef]
- Giuli, G.; Trapananti, A.; Mueller, F.; Bresser, D.; Dácapito, F.; Passerini, S. Insights into the Effect of Iron and Cobalt Doping on the Structure of Nanosized ZnO. Inorg. Chem. 2015, 54, 9393–9400. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.D. Revised Effective Ionic Eadii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Kumar, S.; Mukherjee, S.; Singh, R.K.; Chatterjee, S.; Ghosh, A.K. Structural and optical properties of sol-gel derived nanocrystalline Fe-doped ZnO. J. Appl. Phys. 2011, 110, 103508–103516. [Google Scholar] [CrossRef]
- Reddy, A.J.; Kokila, M.K.; Nagabhushana, H.; Sharma, S.C.; Rao, J.L.; Shivakumara, C.; Nagabhushana, B.M.; Chakradhar, R.P.S. Structural, EPR, photo and thermoluminescence properties of ZnO:Fe nanoparticles. Mater. Chem. Phys. 2012, 133, 876–883. [Google Scholar] [CrossRef]
- Hazen, R.M.; Jeanloz, R. Wüstite (Fe1−xO): A review of its defect structure and physical properties. Rev. Geophys. 1984, 22, 37–46. [Google Scholar] [CrossRef]
- Wilke, M.; Farges, F.; Petit, P.E.; Brown, G.E.; Martin, F. Oxidation state and coordination of Fe in minerals: An Fe K-XANES spectroscopic study. Am. Mineral. 2001, 86, 714–730. [Google Scholar] [CrossRef]
- Giuli, G.; Pratesi, G.; Cipriani, C.; Paris, E. Iron local structure in tektites and impact glasses by extended X-ray absorption fine structure and high-resolution X-ray absorption near-edge structure spectroscopy. Geochim. Cosmochim. Acta 2002, 66, 4347–4353. [Google Scholar] [CrossRef]
- Giuli, G.; Pratesi, G.; Eeckhout, S.G.; Koeberl, C.; Paris, E. Iron reduction in silicate glass produced during the 1945 nuclear test at the Trinity site (Alamogordo, New Mexico, USA). In Large Meteorite Impacts and Planetary Evolution IV; Gibson, R.L., Reimold, W.U., Eds.; Geological Society of America: Boulder, CO, USA, 2010; ISBN 9780813724652. [Google Scholar]
- Larson, A.C.; Von Dreele, R.B. General Structure Analysis System (GSAS); Los Alamos National Laboratory Report Laur 86–748; The Regents of the University of California: Oakland, CA, USA, 2004. [Google Scholar]
- Xu, Y.-N.; Ching, W.Y. Electronic, optical, and structural properties of some wurtzite crystals. Phys. Rev. B 1993, 48, 4335–4351. [Google Scholar] [CrossRef]
- D‘Acapito, F.; Trapananti, A.; Torrengo, S.; Mobilio, S. X-ray Absorption Spectroscopy: The Italian Beamline GILDA of the ESRF. Not. Neutroni Luce Sincrotrone 2014, 19, 14–23. [Google Scholar]
- Giuli, G.; Cicconi, M.R.; Paris, E. The [4]Fe3+–O distance in synthetic kimzeyite garnet, Ca3Zr2 [Fe2SiO12]. Eur. J. Mineral. 2012, 24, 783–790. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZnO | Zn0.98Fe0.02O | Zn0.96Fe0.04O | Zn0.94Fe0.06O | Zn0.92Fe0.08O | Zn0.9Fe0.1O | Zn0.88Fe0.12O | |
|---|---|---|---|---|---|---|---|
| a0 (Å) | 3.2511 (3) | 3.2524 (1) | 3.2549 (1) | 3.2553 (1) | 3.2549 (2) | 3.2552 (2) | 3.2573 (2) |
| c0 (Å) | 5.2098 (1) | 5.2099 (1) | 5.2081 (3) | 5.2075 (3) | 5.2079 (5) | 5.2043 (4) | 5.2096 (4) |
| V0 (Å3) | 47.687 (1) | 47.728 (1) | 47.784 (3) | 47.791 (3) | 47.781 (5) | 47.760 (5) | 47.868 (4) |
| <T-O> | 1.9795 (50) | 1.9800 (50) | 1.9802 (50) | 1.9815 (50) | 1.9818 (50) | 1.9815 (50) | 1.9830 (50) |
| wRp | 8.36 | 7.11 | 7.44 | 7.88 | 7.88 | 7.60 | 8.7.44 |
| Rp | 6.60 | 5.64 | 5.79 | 6.26 | 6.07 | 5.87 | 5.92 |
| RF2 | 3.49 | 3.43 | 4.51 | 5.09 | 3.78 | 4.19 | 4.26 |
| RF | 1.92 | 1.79 | 2.59 | 3.05 | 2.02 | 2.30 | 2.17 |
| a0/c0 | 0.6240 | 0.6188 | 0.6250 | 0.6251 | 0.6250 | 0.6255 | 0.6252 |
| W.-H. intercept 1 | 0.0033 | 0.0048 | 0.0069 | 0.0104 | 0.0063 | 0.011 | 0.0088 |
| W.-H. slope 1 | 0.0003 | 0.0013 | 0.0023 | 0.0006 | 0.0045 | 0.0031 | 0.0046 |
| Crystallite size (nm) | 42 | 29 | 20 | 13 | 22 | 13 | 16 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giuli, G.; Eisenmann, T.; Bresser, D.; Trapananti, A.; Asenbauer, J.; Mueller, F.; Passerini, S. Structural and Electrochemical Characterization of Zn1−xFexO—Effect of Aliovalent Doping on the Li+ Storage Mechanism. Materials 2018, 11, 49. https://doi.org/10.3390/ma11010049
Giuli G, Eisenmann T, Bresser D, Trapananti A, Asenbauer J, Mueller F, Passerini S. Structural and Electrochemical Characterization of Zn1−xFexO—Effect of Aliovalent Doping on the Li+ Storage Mechanism. Materials. 2018; 11(1):49. https://doi.org/10.3390/ma11010049
Chicago/Turabian StyleGiuli, Gabriele, Tobias Eisenmann, Dominic Bresser, Angela Trapananti, Jakob Asenbauer, Franziska Mueller, and Stefano Passerini. 2018. "Structural and Electrochemical Characterization of Zn1−xFexO—Effect of Aliovalent Doping on the Li+ Storage Mechanism" Materials 11, no. 1: 49. https://doi.org/10.3390/ma11010049
APA StyleGiuli, G., Eisenmann, T., Bresser, D., Trapananti, A., Asenbauer, J., Mueller, F., & Passerini, S. (2018). Structural and Electrochemical Characterization of Zn1−xFexO—Effect of Aliovalent Doping on the Li+ Storage Mechanism. Materials, 11(1), 49. https://doi.org/10.3390/ma11010049