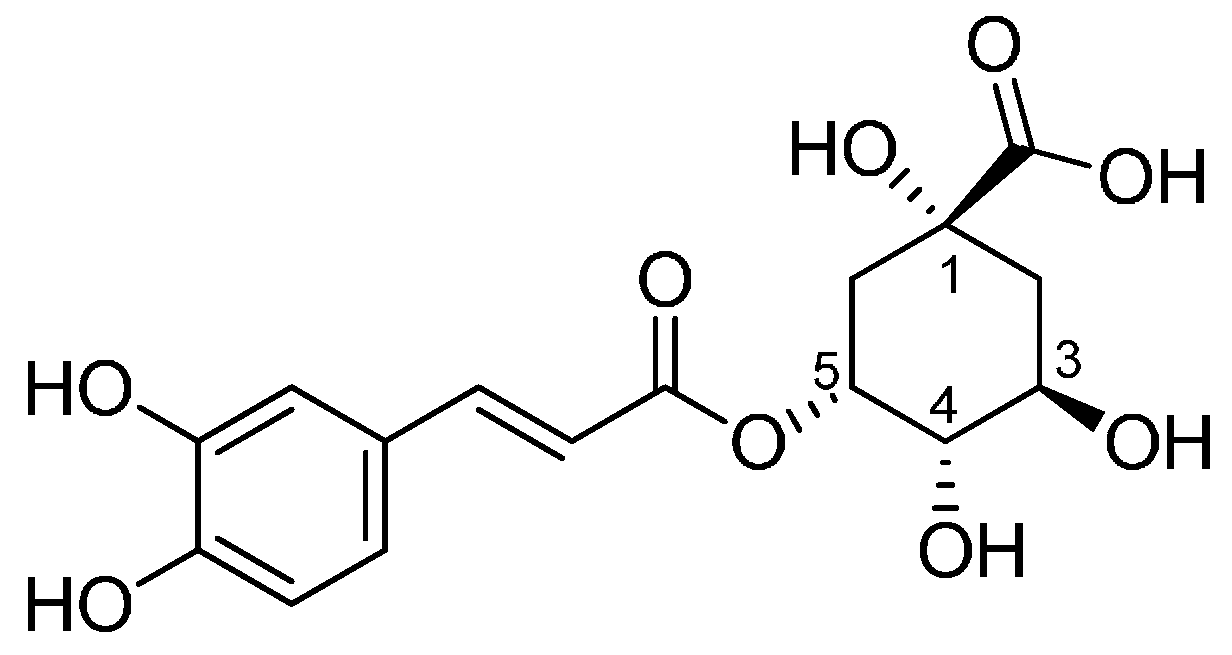
Synthesis of Bioactive Chlorogenic Acid-Silica Hybrid Materials via the Sol–Gel Route and Evaluation of Their Biocompatibility



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
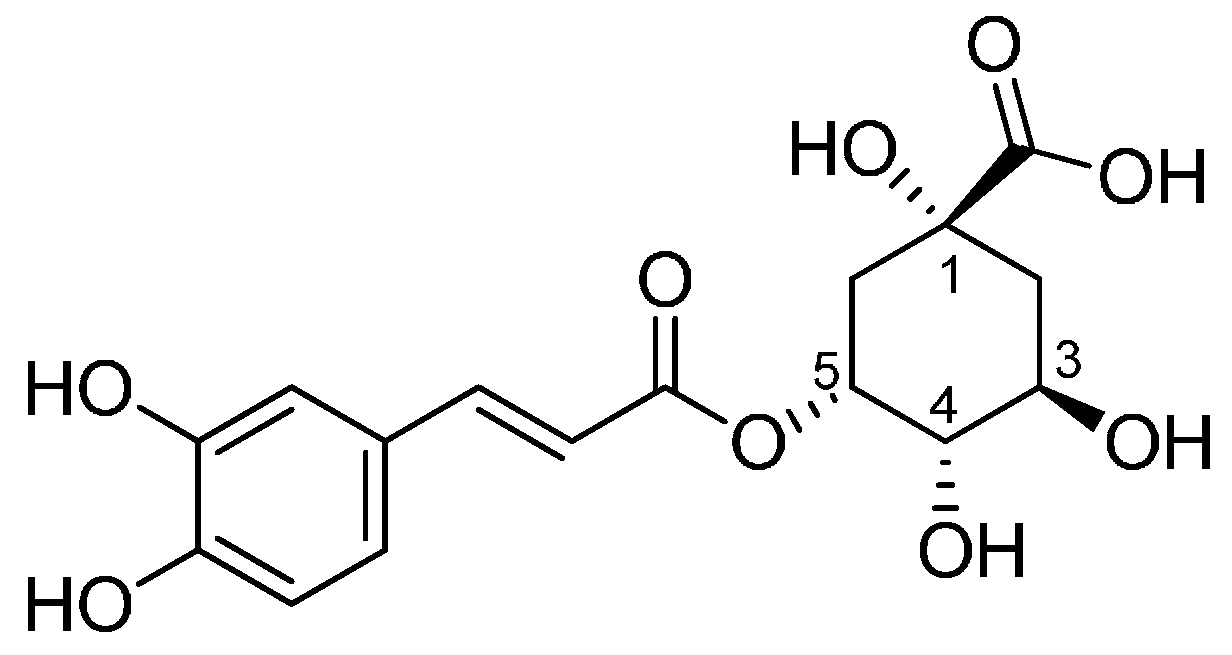
:1. Introduction
2. Results and Discussion
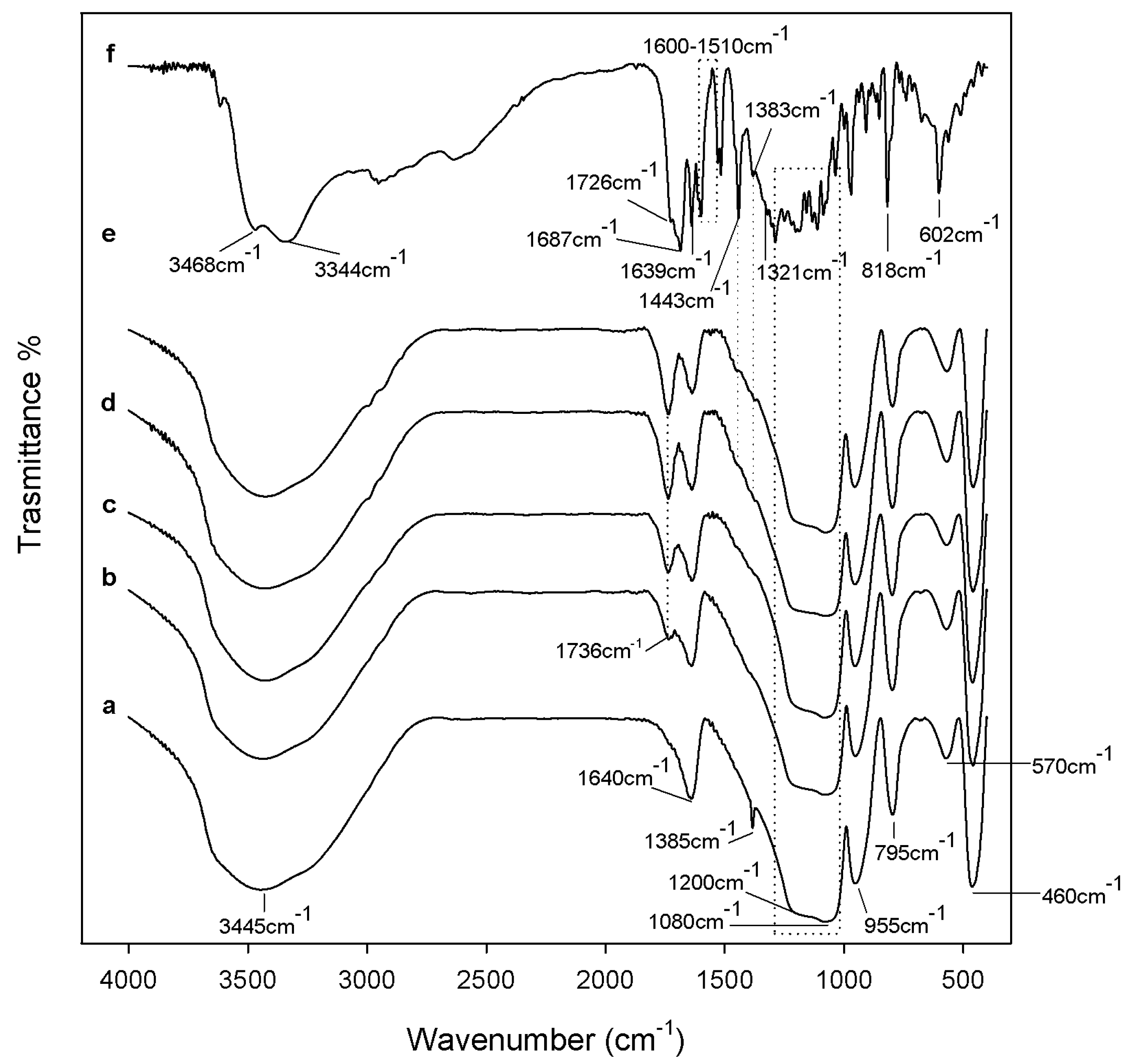
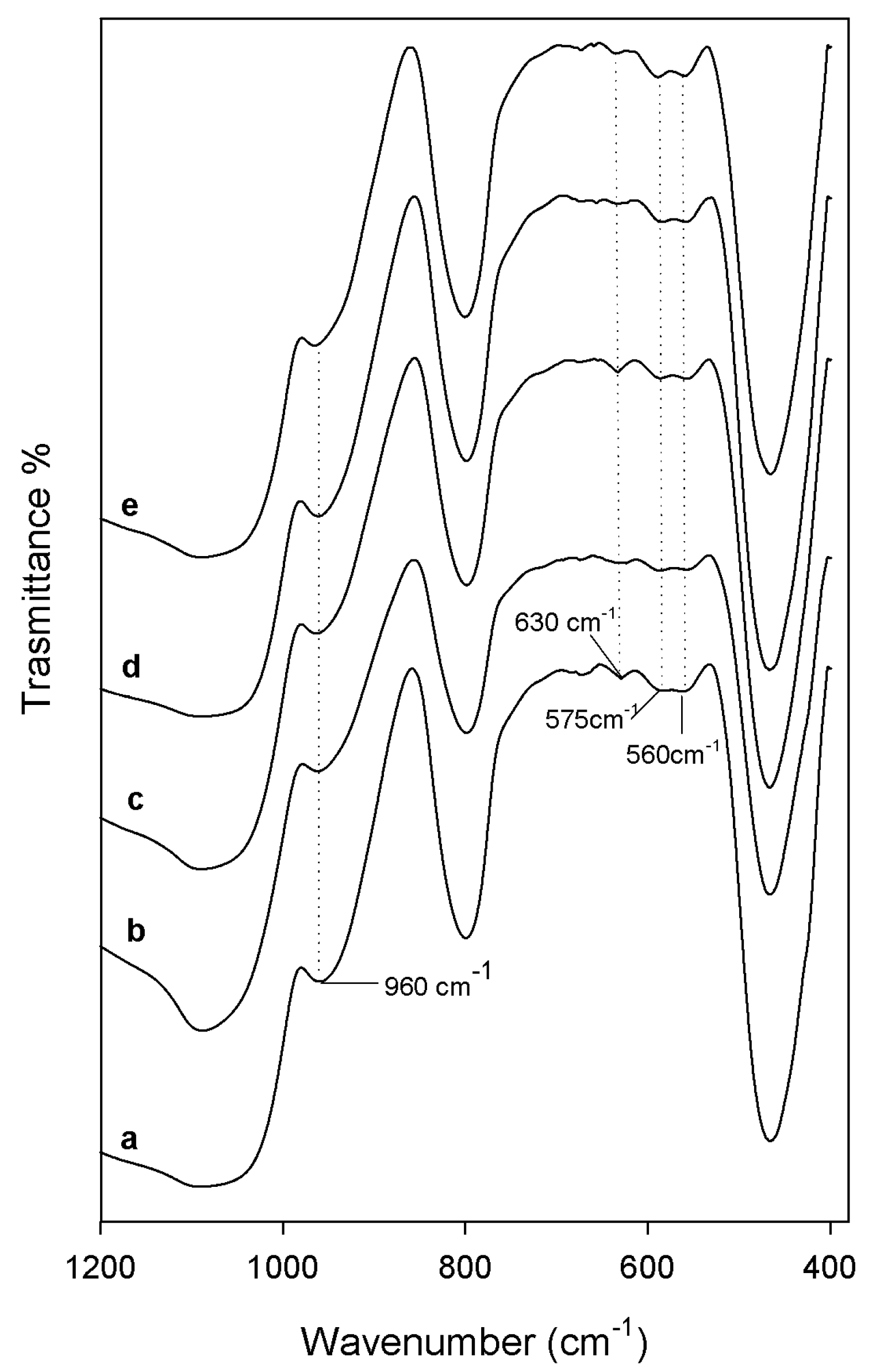
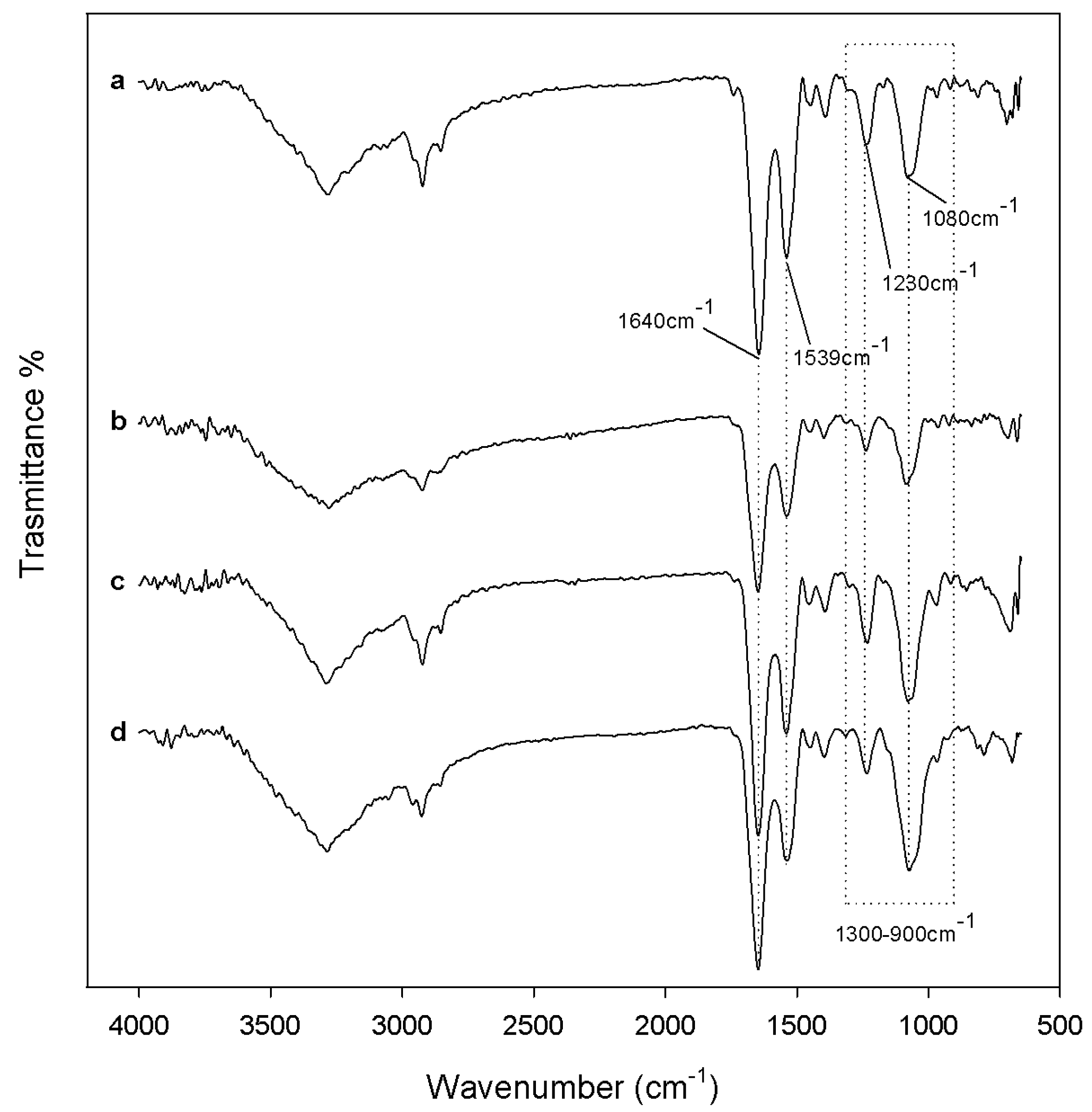
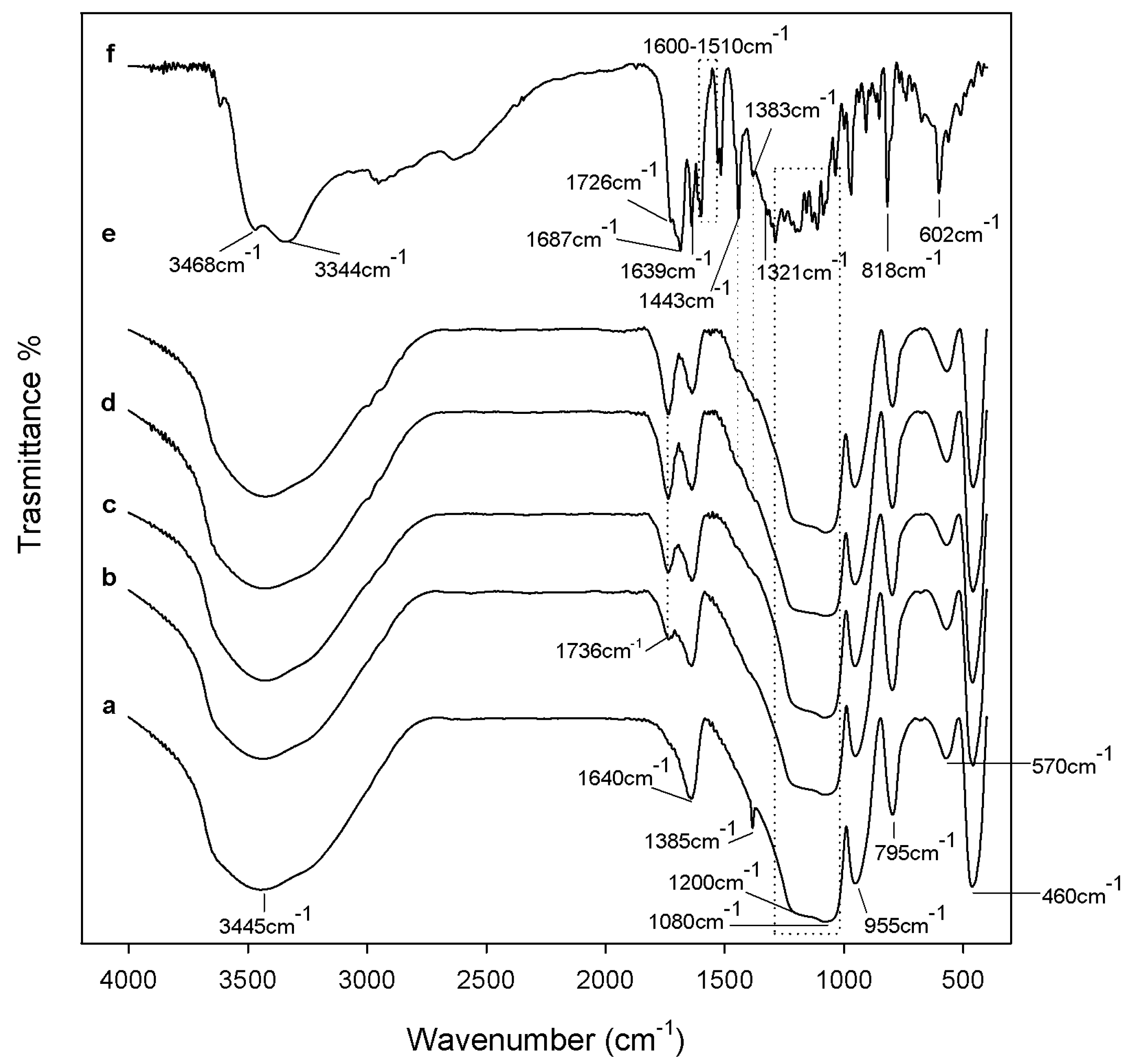
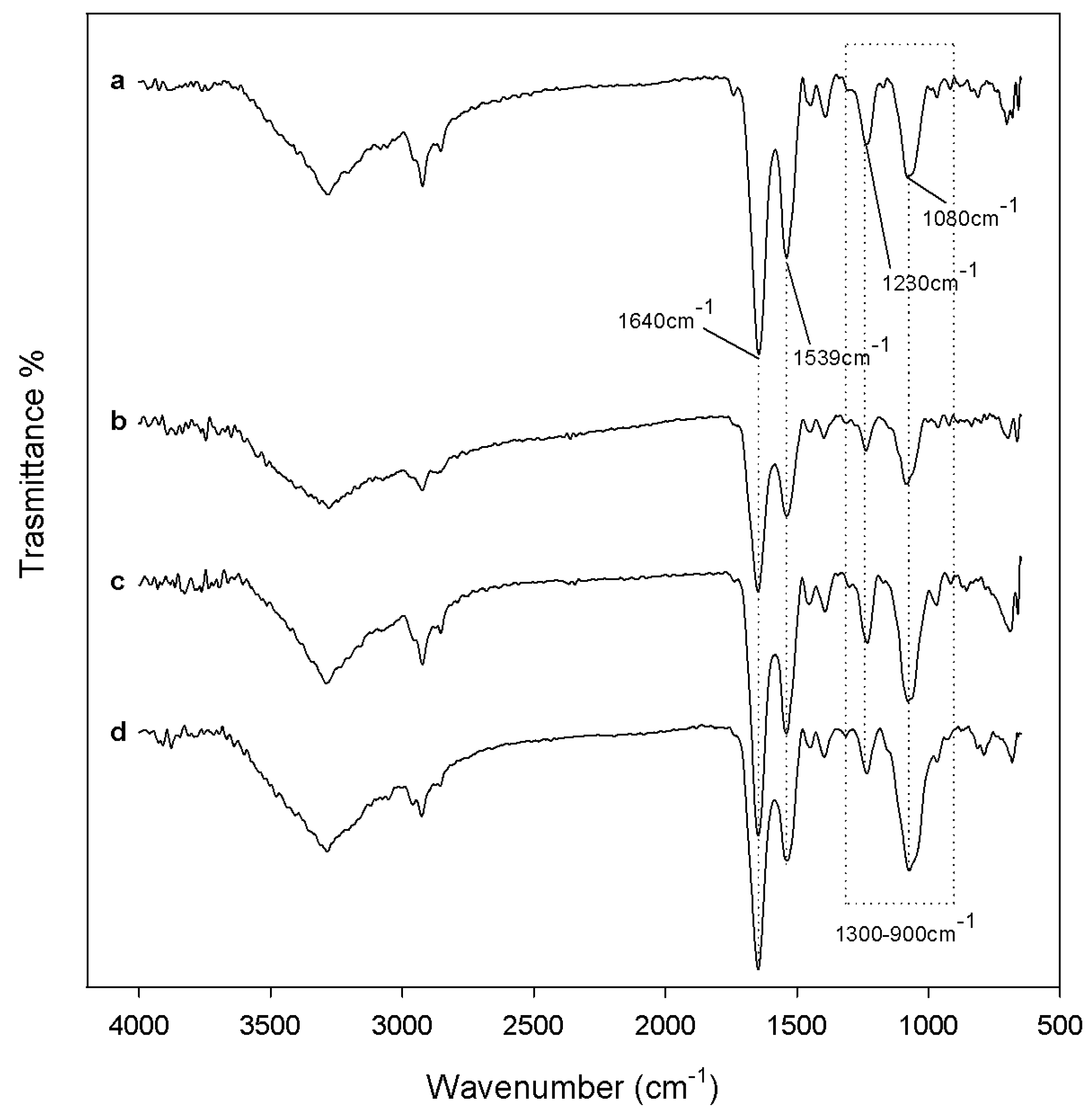
2.1. Characterization of Synthetized Organic-Inorganic Hybrid Materials
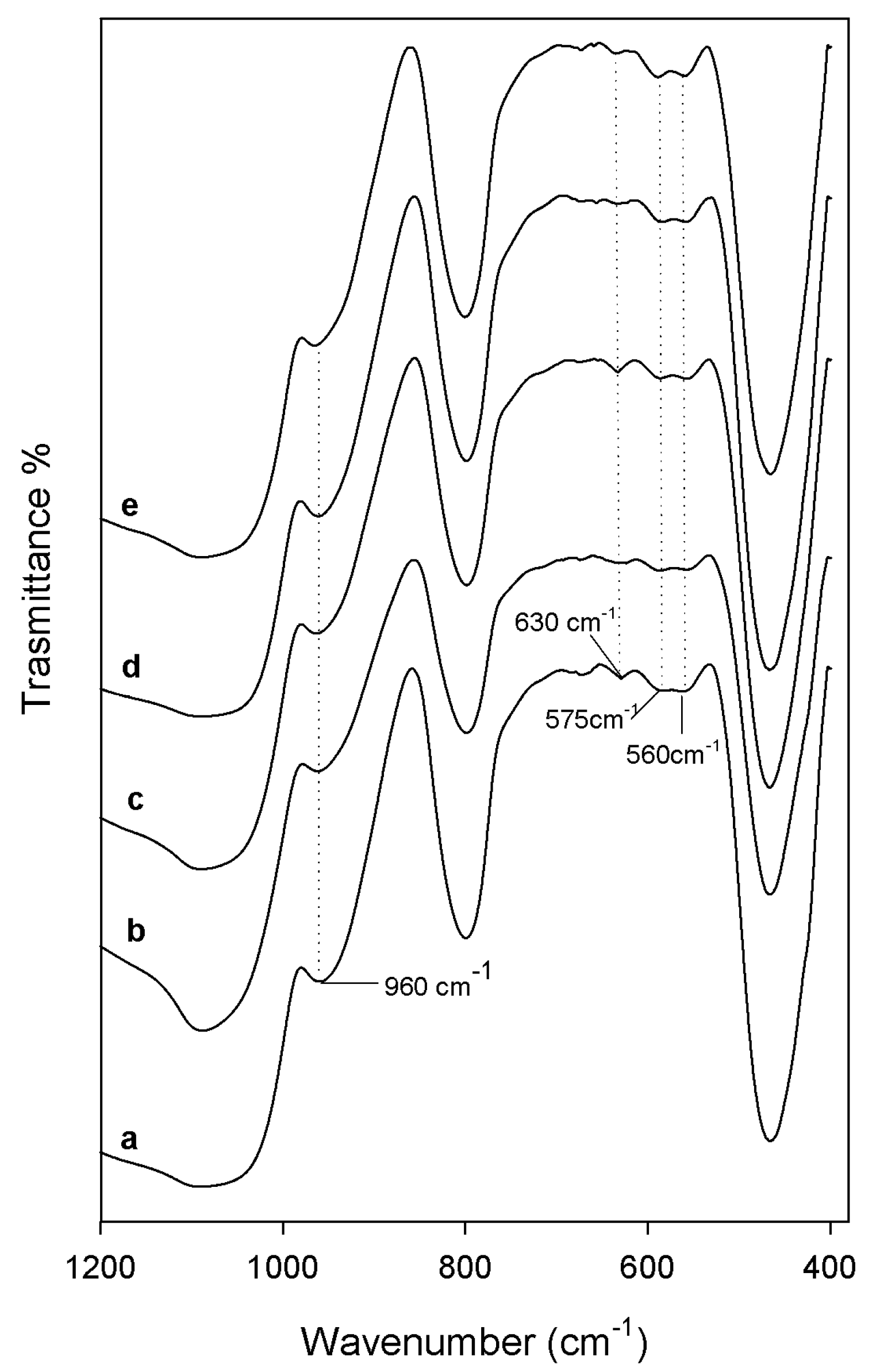
2.2. Bioactivity Test
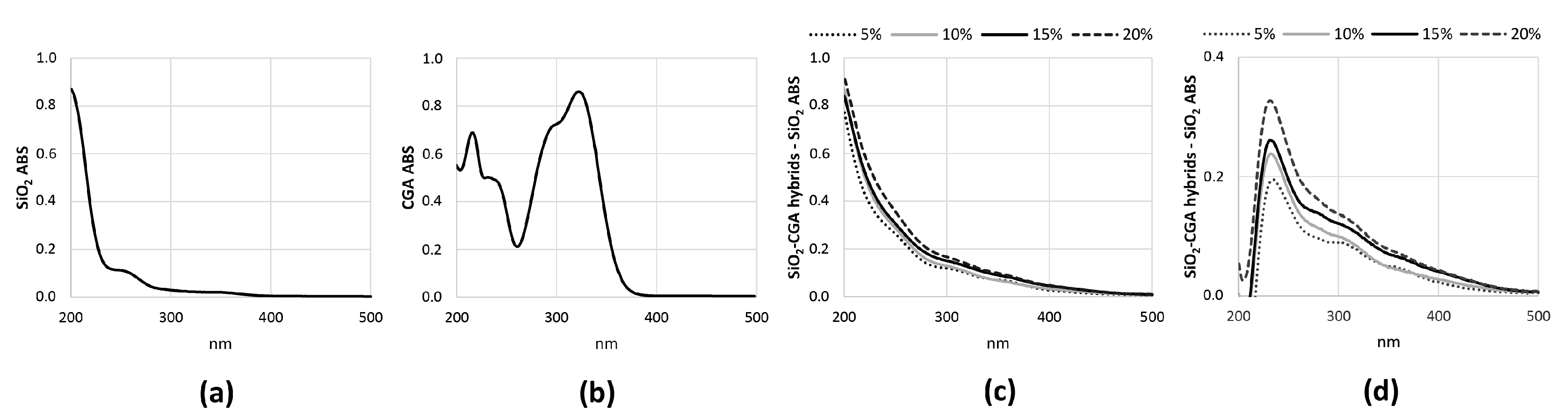
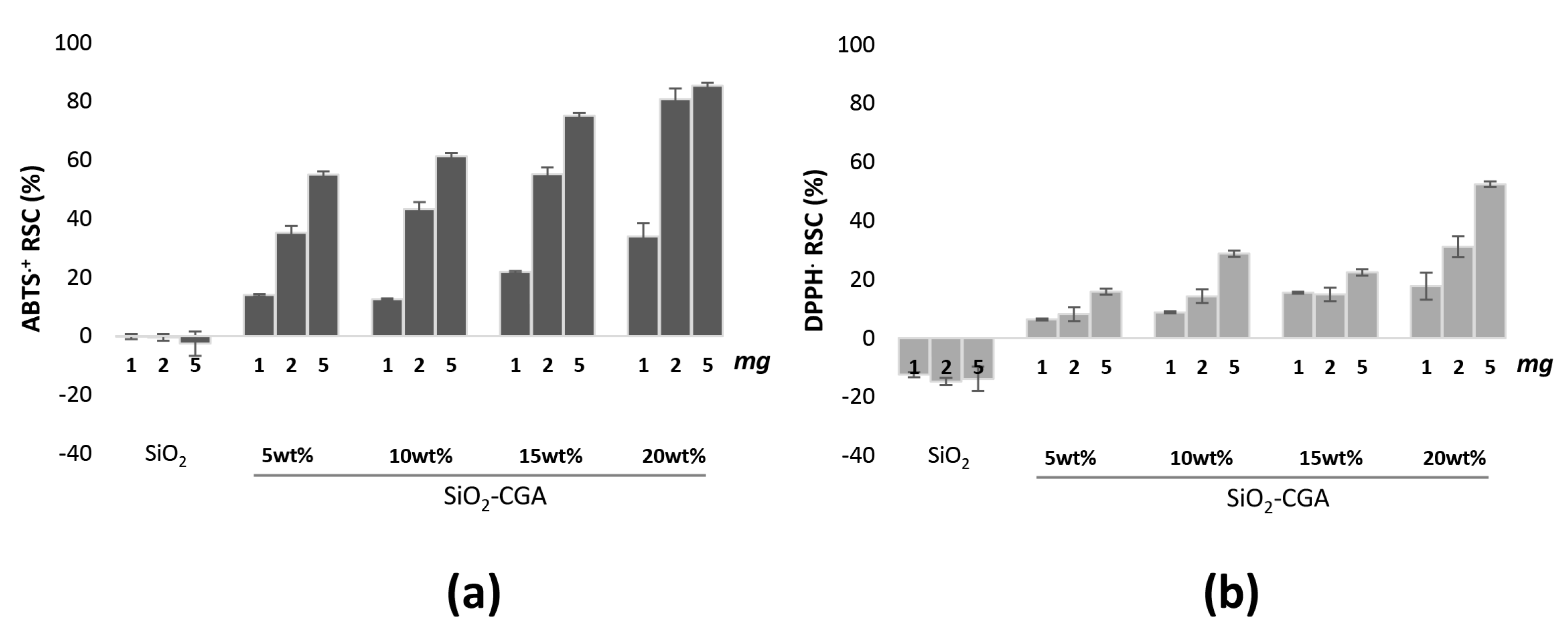
2.3. Antiradical Capability of SiO2-CGA Hybrids
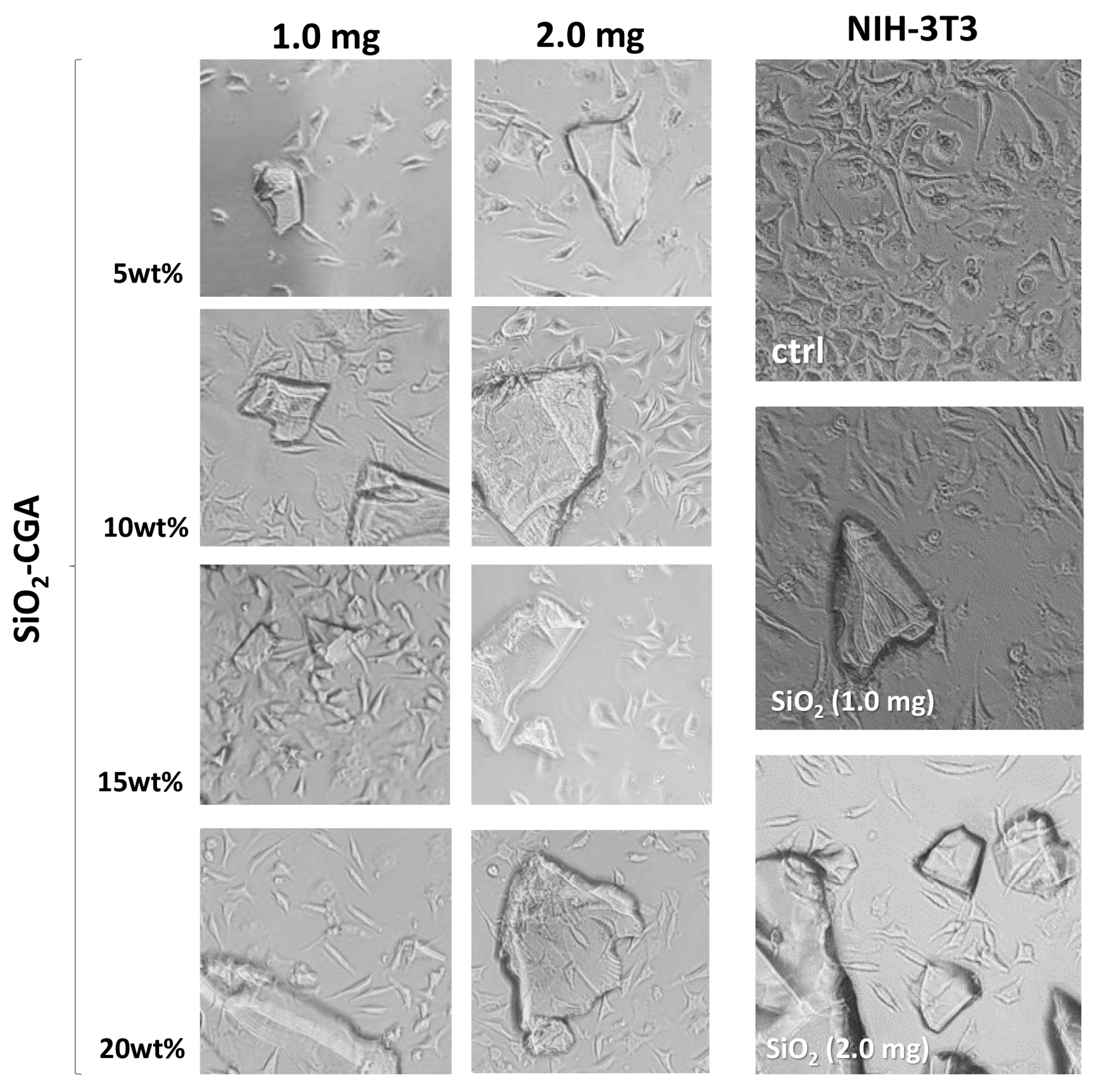
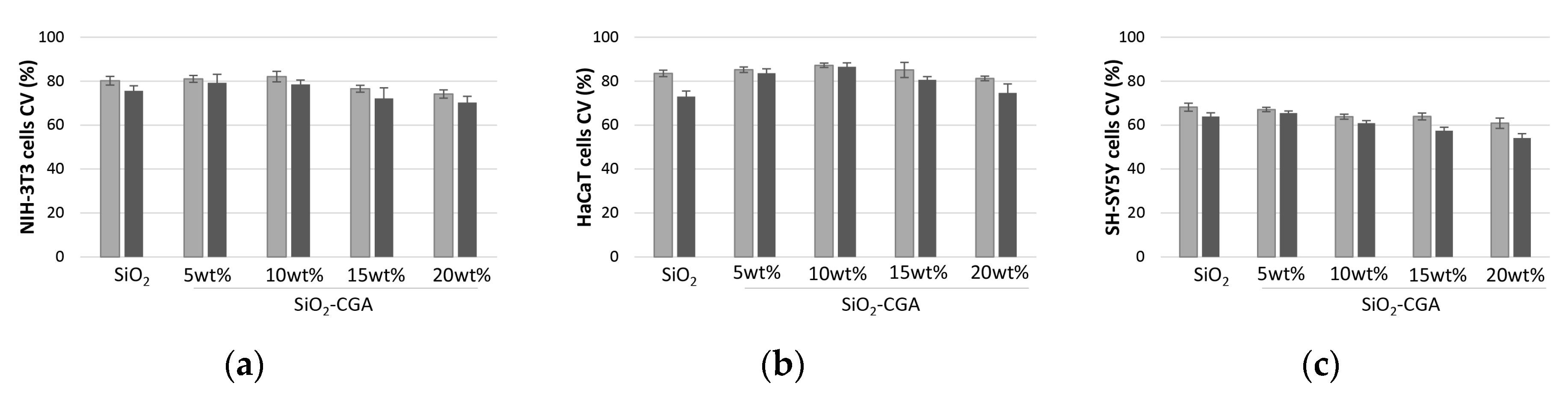
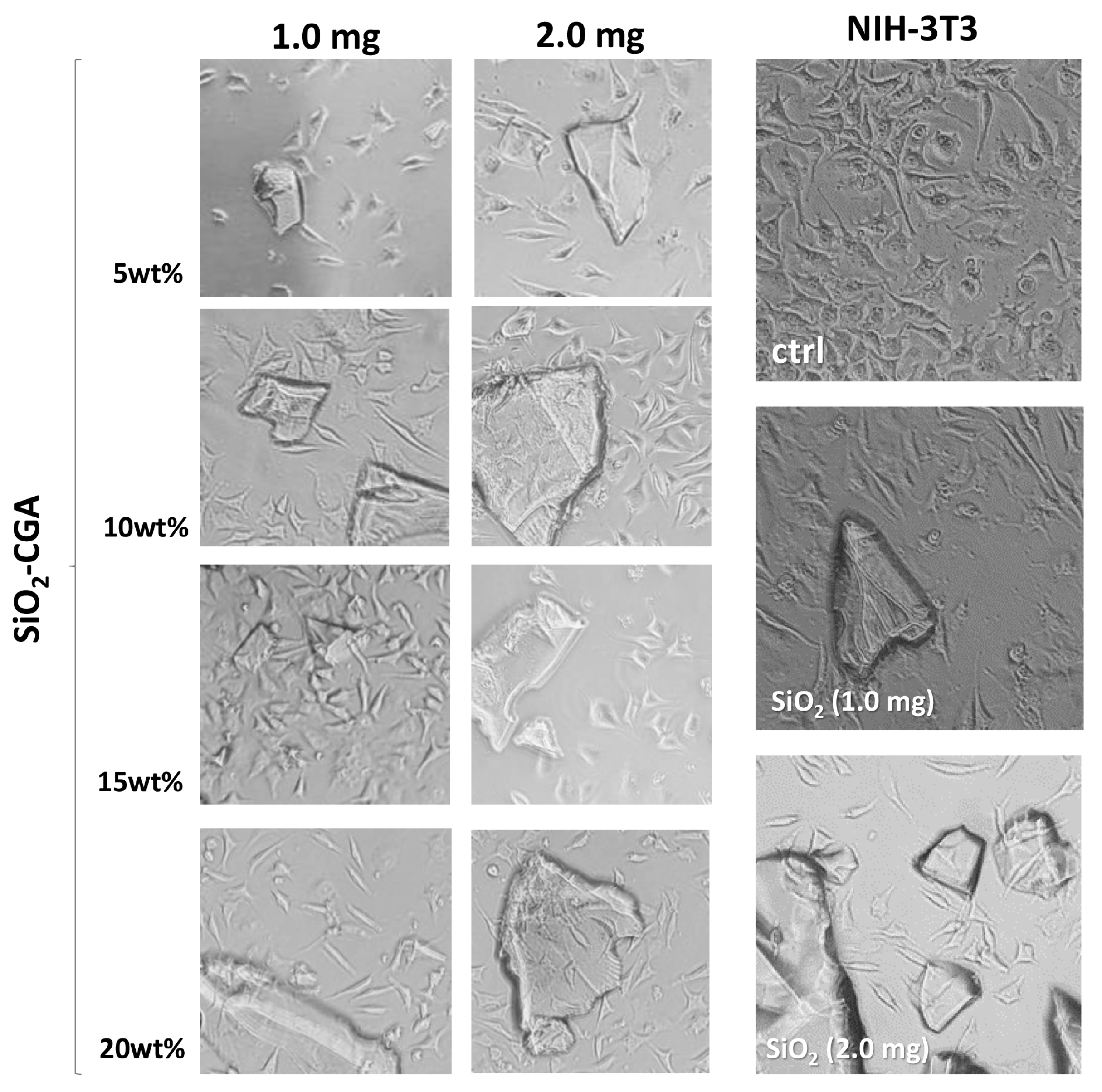
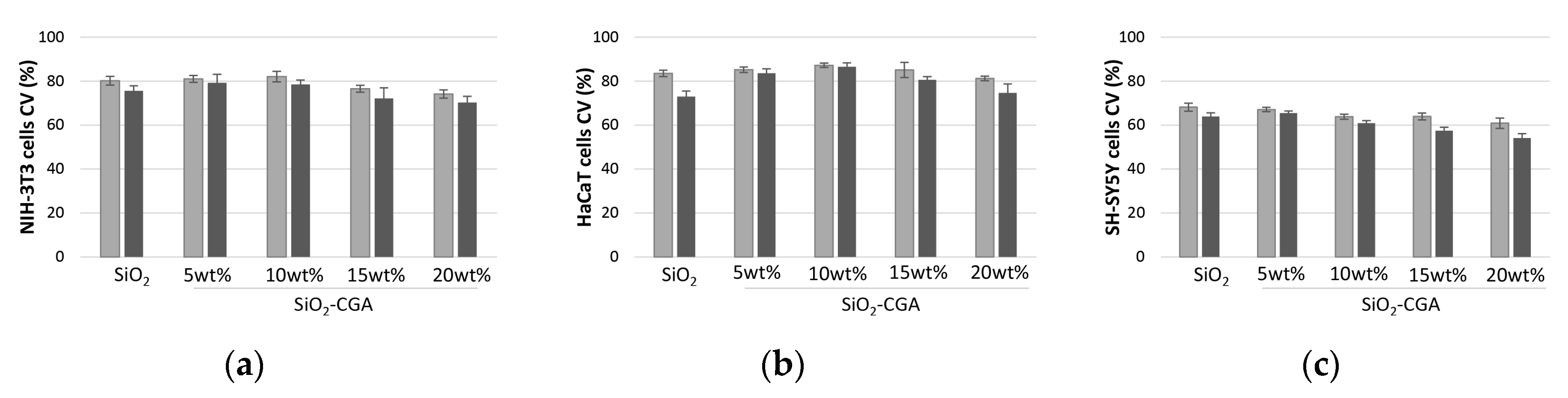
2.4. Cytotoxicity of SiO2-CGA Hybrids
3. Materials and Methods
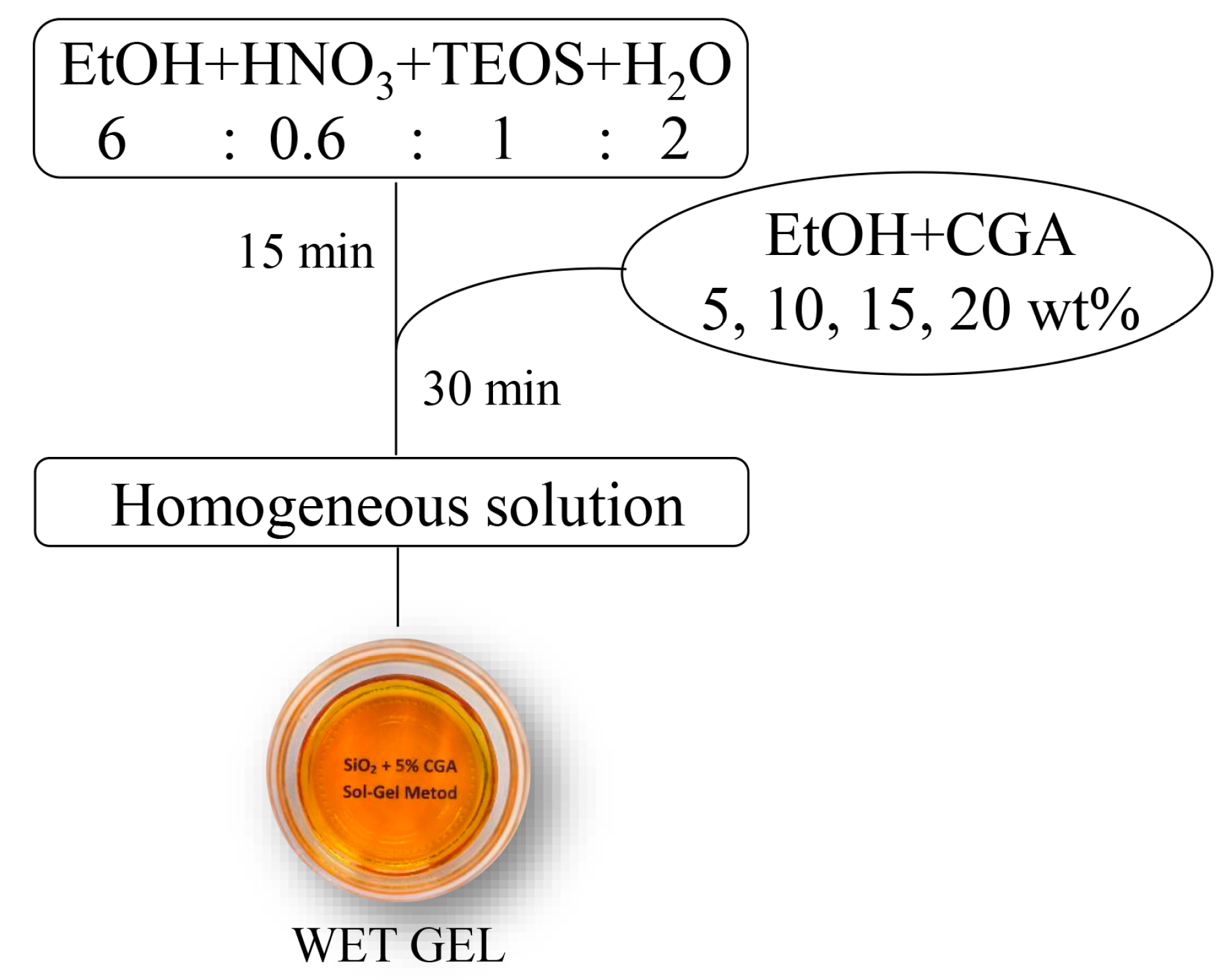
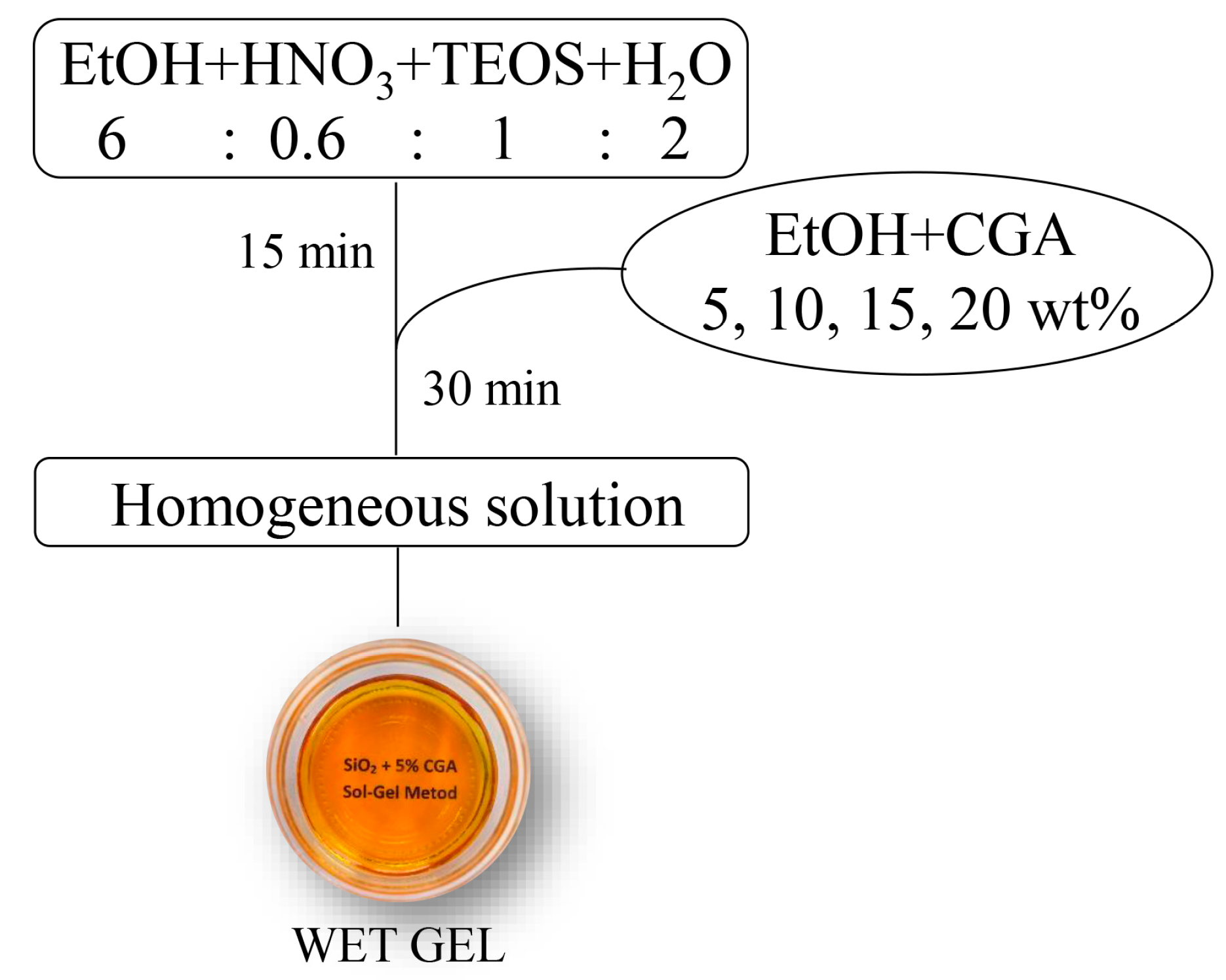
3.1. Sol–gel Synthesis
3.2. Materials Characterization
3.3. Bioactivity Test
3.4. Determination of DPPH Scavenging Capacity
3.5. Determination of ABTS•+ Scavenging Capacity
3.6. Cell Culture and Cytotoxicity Assessment
3.7. ATR-FTIR Analysis
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Piccolella, S.; Pacifico, S. Chapter five—Plant-derived polyphenols: A chemopreventive and chemoprotectant worth-exploring resource in toxicology. In Advances in Molecular Toxicology; James, C.F., Jacqueline, M.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 9, pp. 161–214. [Google Scholar]
- Galasso, S.; Pacifico, S.; Kretschmer, N.; Pan, S.-P.; Marciano, S.; Piccolella, S.; Monaco, P.; Bauer, R. Influence of seasonal variation on Thymus longicaulis C. Presl chemical composition and its antioxidant and anti-inflammatory properties. Phytochemistry 2014, 107, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.L.; Tedesco, I.; Spagnuolo, C.; Russo, M. Antioxidant polyphenols in cancer treatment: Friend, foe or foil? Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Guo, Y.; Zhang, C.; Wu, R.; Yang, A.Y.; Gaspar, J.; Kong, A.-N.T. Dietary phytochemicals and cancer chemoprevention: A perspective on oxidative stress, inflammation, and epigenetics. Chem. Res. Toxicol. 2016, 29, 2071–2095. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Park, J.-Y.; Lambert, J.D. Differential prooxidative effects of the green tea polyphenol, (–)-epigallocatechin-3-gallate, in normal and oral cancer cells are related to differences in sirtuin 3 signaling. Mol. Nutr. Food Res. 2015, 59, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Santana-Gálvez, J.; Cisneros-Zevallos, L.; Jacobo-Velázquez, D. Chlorogenic acid: Recent advances on its dual role as a food additive and a nutraceutical against metabolic syndrome. Molecules 2017, 22, 358. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Rashid, S.; Nafees, S.; Hasan, S.K.; Shahid, A.; Majed, F.; Sultana, S. Protective effect of chlorogenic acid against methotrexate induced oxidative stress, inflammation and apoptosis in rat liver: An experimental approach. Chem.-Biol. Interact. 2017, 272, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Nallamuthu, I.; Devi, A.; Khanum, F. Chlorogenic acid loaded chitosan nanoparticles with sustained release property, retained antioxidant activity and enhanced bioavailability. Asian J. Pharm. Sci. 2015, 10, 203–211. [Google Scholar] [CrossRef]
- Fu, S.; Wu, C.; Wu, T.; Yu, H.; Yang, S.; Hu, Y. Preparation and characterisation of chlorogenic acid-gelatin: A type of biologically active film for coating preservation. Food Chem. 2017, 221, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Salimi, A.; Hallaj, R. Adsorption and reactivity of chlorogenic acid at a hydrophobic carbon ceramic composite electrode: Application for the amperometric detection of hydrazine. Electroanalysis 2004, 16, 1964–1971. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Mozzati, M.C.; Ferrara, C.; Mustarelli, P. Structure and magnetic properties of SiO2/PCL novel sol-gel organic-inorganic hybrid materials. J. Solid State Chem. 2013, 203, 92–99. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Papale, F.; Leonelli, C. Influence of the drying treatment on the performance of V-Nb mixed oxides catalysts synthesised via sol-gel. J. Non-Cryst. Solids 2013, 380, 1–5. [Google Scholar] [CrossRef]
- Catauro, M.; Papale, F.; Bollino, F. Characterization and biological properties of TiO2/PCL hybrid layers prepared via sol-gel dip coating for surface modification of titanium implants. J. Non-Cryst. Solids 2015, 415, 9–15. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Nocera, P.; Piccolella, S.; Pacifico, S. Entrapping quercetin in silica/polyethylene glycol hybrid materials: Chemical characterization and biocompatibility. Mater. Sci. Eng. C 2016, 68, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Catauro, M.; Bollino, F.; Papale, F.; Piccolella, S.; Pacifico, S. Sol-gel synthesis and characterization of SiO2/PCL hybrid materials containing quercetin as new materials for antioxidant implants. Mater. Sci. Eng. C 2016, 58, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Catauro, M.; Papale, F.; Bollino, F.; Piccolella, S.; Marciano, S.; Nocera, P.; Pacifico, S. Silica/quercetin sol-gel hybrids as antioxidant dental implant materials. Sci. Technol. Adv. Mater. 2015, 16, 035001. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Adeogun, M.J.; Hay, J.N. Structure control in sol-gel silica synthesis using ionene polymers. 2: Evidence from spectroscopic analysis. J. Sol-Gel Sci. Technol. 2001, 20, 119–128. [Google Scholar] [CrossRef]
- Innocenzi, P. Infrared spectroscopy of sol-gel derived silica-based films: A spectra-microstructure overview. J. Non-Cryst. Solids 2003, 316, 309–319. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Papale, F.; Ferrara, C.; Mustarelli, P. Silica-polyethylene glycol hybrids synthesized by sol-gel: Biocompatibility improvement of titanium implants by coating. Mater. Sci. Eng. C 2015, 55, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Hou, W.; Zhang, C.; Sun, D.; Huang, X.; Gouting Wang, A. Structure and the point of zero charge of magnesium aluminium hydroxide. J. Chem. Soc. Faraday Trans. 1998, 94, 915–918. [Google Scholar] [CrossRef]
- Nedelec, J.M.; Hench, L.L. Ab initio molecular orbital calculations on silica rings. J. Non-Cryst. Solids 1999, 255, 163–170. [Google Scholar] [CrossRef]
- Mishra, S.; Tandon, P.; Eravuchira, P.J.; El-Abassy, R.M.; Materny, A. Vibrational spectroscopy and density functional theory analysis of 3-O-caffeoylquinic acid. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 104, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Bajko, E.; Kalinowska, M.; Borowski, P.; Siergiejczyk, L.; Lewandowski, W. 5-O-caffeoylquinic acid: A spectroscopic study and biological screening for antimicrobial activity. LWT Food Sci. Technol. 2016, 65, 471–479. [Google Scholar] [CrossRef]
- Angélica Alvarez Lemus, M.; Castañeda, O.J.O.; Hernández Pérez, A.D.; González, R.L. An alcohol-free SiO2 sol-gel matrix functionalized with acetic acid as drug reservoir for the controlled release of pentoxifylline. J. Nanomater. 2014, 2014, 108. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Renella, R.A.; Papale, F. Sol-gel synthesis of SiO2–CaO–P2O5 glasses: Influence of the heat treatment on their bioactivity and biocompatibility. Ceram. Int. 2015, 41, 12578–12588. [Google Scholar] [CrossRef]
- Catauro, M.; Renella, R.A.; Papale, F.; Vecchio Ciprioti, S. Investigation of bioactivity, biocompatibility and thermal behavior of sol-gel silica glass containing a high peg percentage. Mater. Sci. Eng. C 2016, 61, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Takadama, H. How useful is sbf in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.-W.; Bai, J.-P.; Zhang, Q.; Hu, X.-L.; Tian, X.; Zhu, J.; Liu, J.; Meng, W.-H.; Zhao, Q.-C. Caffeoylquinic acid derivatives from the roots of Arctium lappa L. (burdock) and their structure–activity relationships (sars) of free radical scavenging activities. Phytochem. Lett. 2016, 15, 159–163. [Google Scholar] [CrossRef]
- Messer, R.L.W.; Lockwood, P.E.; Wataha, J.C.; Lewis, J.B.; Norris, S.; Bouillaguet, S. In vitro cytotoxicity of traditional versus contemporary dental ceramics. J. Prosthet. Dent. 2003, 90, 452–458. [Google Scholar] [CrossRef]
- Xue, N.; Zhou, Q.; Ji, M.; Jin, J.; Lai, F.; Chen, J.; Zhang, M.; Jia, J.; Yang, H.; Zhang, J.; et al. Chlorogenic acid inhibits glioblastoma growth through repolarizating macrophage from M2 to M1 phenotype. Sci. Rep. 2017, 7, 39011. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.R.; Appel, C.L. Polyphenols as dietary supplements: A double-edged sword. Nutr. Diet. Suppl. 2010, 2, 1–12. [Google Scholar] [CrossRef]
- Gasparri, F.; Muzio, M. Monitoring of apoptosis of hl60 cells by fourier-transform infrared spectroscopy. Biochem. J. 2003, 369, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Goormaghtigh, E.; Cabiaux, V.; Ruysschaert, J.-M. Determination of soluble and membrane protein structure by fourier transform infrared spectroscopy. In Physicochemical Methods in the Study of Biomembranes; Hilderson, H.J., Ralston, G.B., Eds.; Springer: Boston, MA, USA, 1994; pp. 405–450. [Google Scholar]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catauro, M.; Pacifico, S. Synthesis of Bioactive Chlorogenic Acid-Silica Hybrid Materials via the Sol–Gel Route and Evaluation of Their Biocompatibility. Materials 2017, 10, 840. https://doi.org/10.3390/ma10070840
Catauro M, Pacifico S. Synthesis of Bioactive Chlorogenic Acid-Silica Hybrid Materials via the Sol–Gel Route and Evaluation of Their Biocompatibility. Materials. 2017; 10(7):840. https://doi.org/10.3390/ma10070840
Chicago/Turabian StyleCatauro, Michelina, and Severina Pacifico. 2017. "Synthesis of Bioactive Chlorogenic Acid-Silica Hybrid Materials via the Sol–Gel Route and Evaluation of Their Biocompatibility" Materials 10, no. 7: 840. https://doi.org/10.3390/ma10070840
APA StyleCatauro, M., & Pacifico, S. (2017). Synthesis of Bioactive Chlorogenic Acid-Silica Hybrid Materials via the Sol–Gel Route and Evaluation of Their Biocompatibility. Materials, 10(7), 840. https://doi.org/10.3390/ma10070840