
Practical Cluster Models for a Layered β-NiOOH Material

Abstract
:
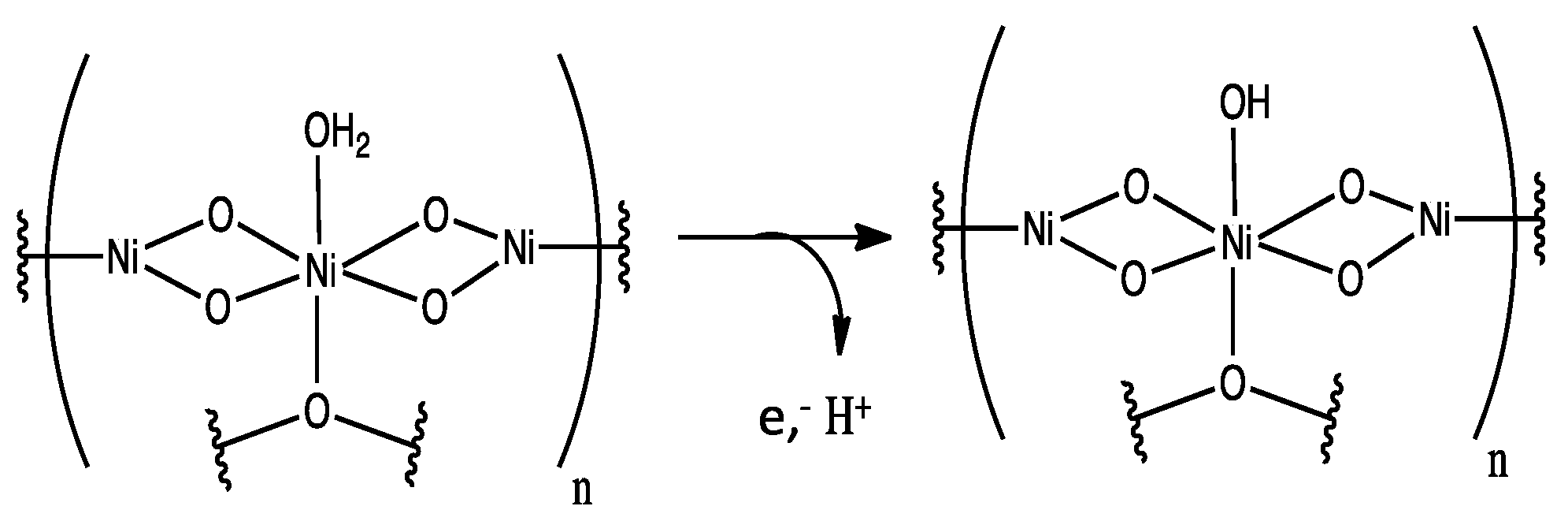
1. Introduction
2. Methods and Computational Protocol
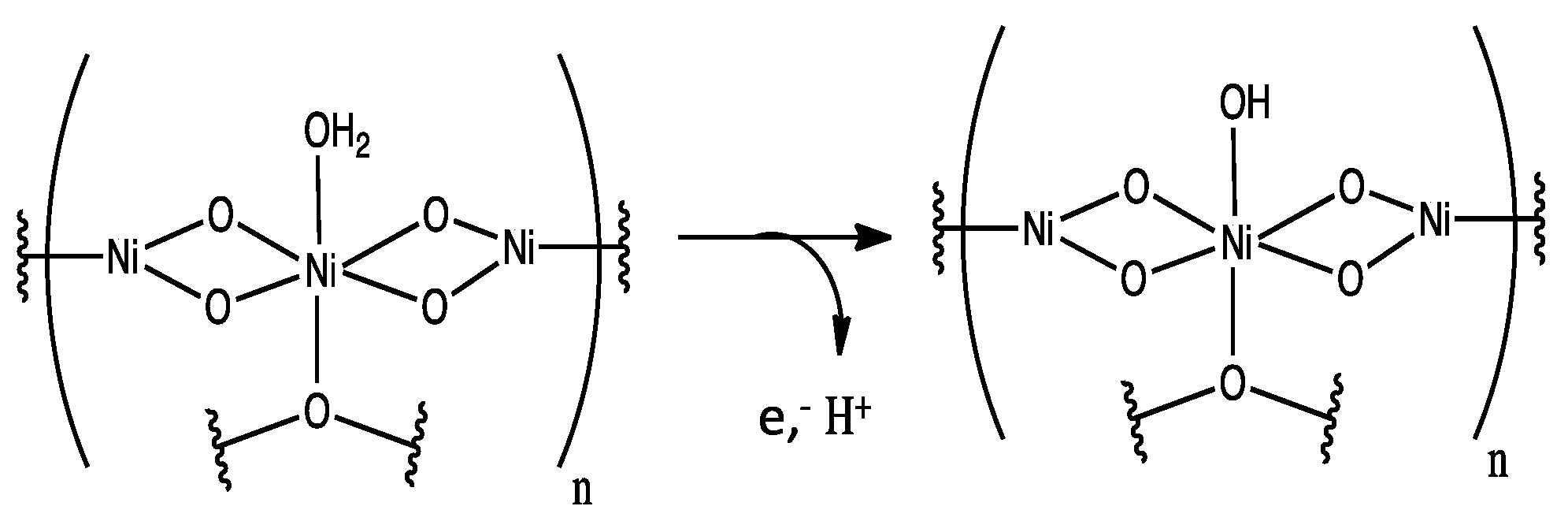
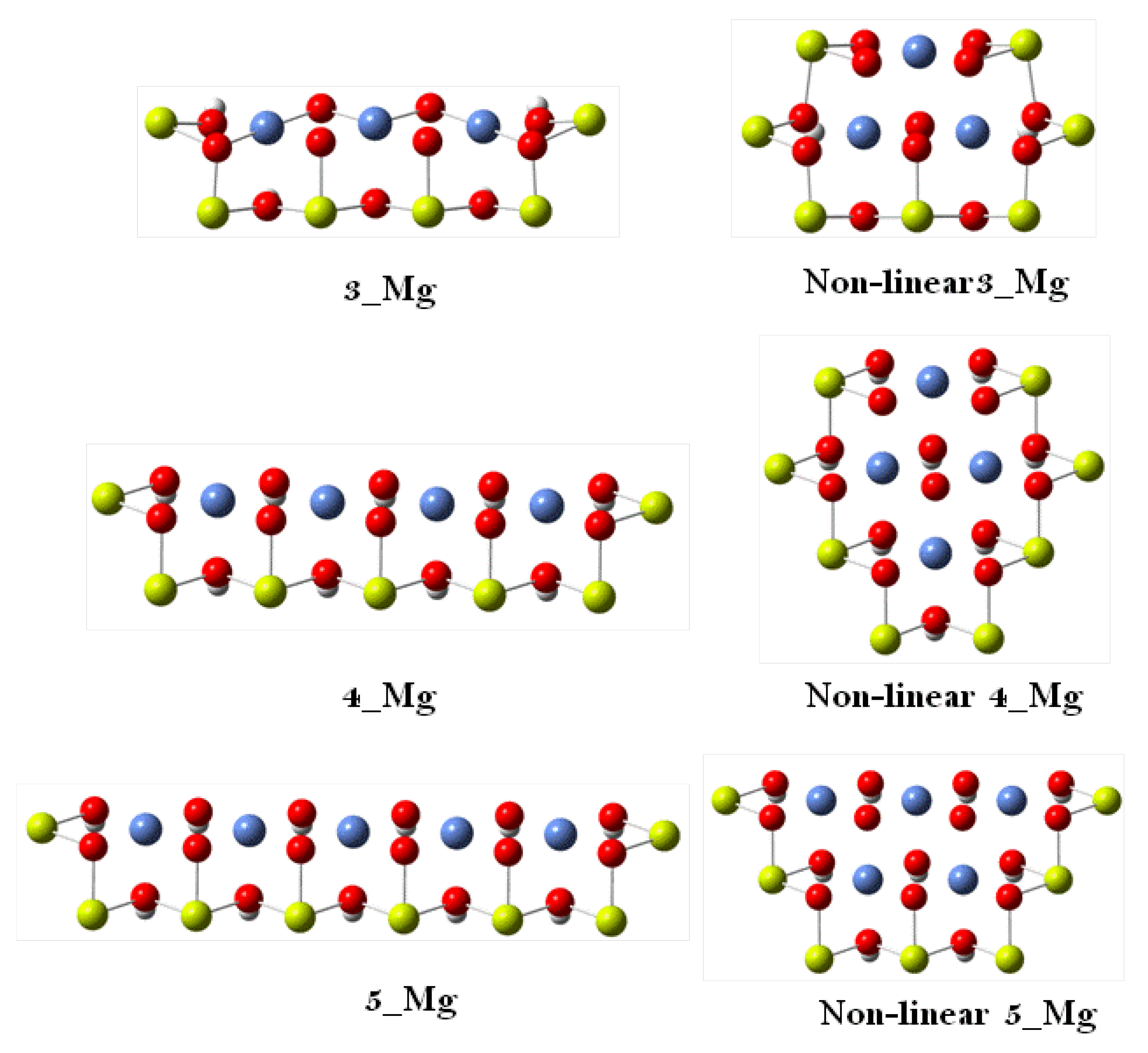
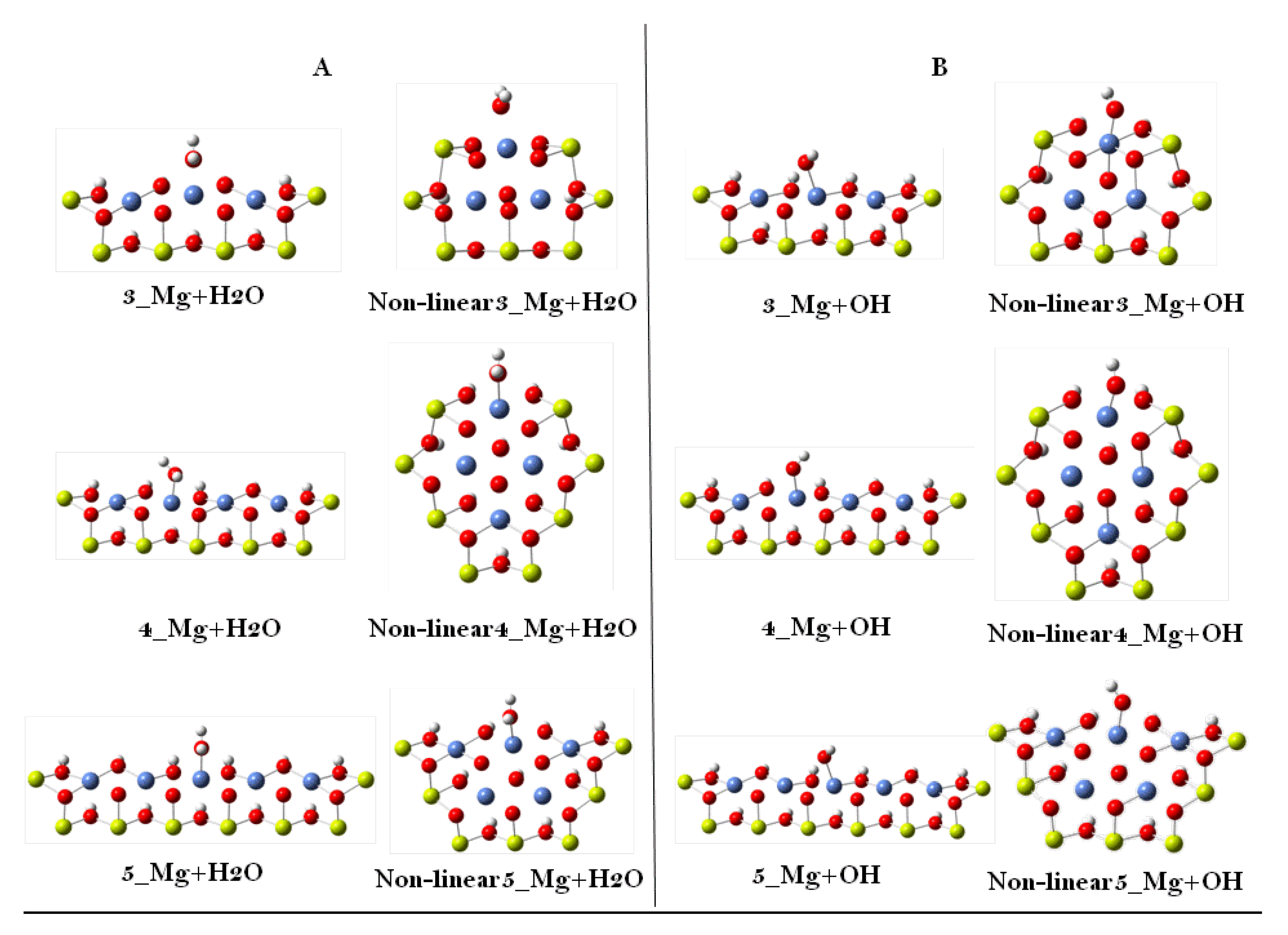
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dresselhaus, M.S.; Thomas, I.L. Alternative energy technologies. Nature 2001, 414, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Gratzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Schlapbach, L.; Zuttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Steele, B.C.H.; Heinzel, A. Materials for fuel-cell technologies. Nature 2001, 414, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Cook, T.R.; Dogutan, D.K.; Reece, S.Y.; Surendranath, Y.; Teets, T.S.; Nocera, D.G. Solar Energy Supply and Storage for the Legacy and Non Legacy Worlds. Chem. Rev. 2010, 110, 6474–6502. [Google Scholar] [CrossRef] [PubMed]
- McCrory, C.C.L.; Jung, S.H.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed]
- Suntivich, J.; May, K.J.; Gasteiger, J.B.; Goodenough, Y.A.; Shao-Horn, Y. Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles. Science 2013, 334, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Man, I.C.; Su, H.Y.; Calle-Vallejo, F.; Hansen, H.A.; Martíne, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Nørskov, J.K.; Rossmeisl, J. Universality in Oxygen Evolution Electrocatalysis on Oxides Surface. ChemCatChem 2010, 3, 1159–1165. [Google Scholar] [CrossRef]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef] [PubMed]
- Risch, M.; Grimaud, A.; May, K.J.; Stoerzinger, K.A.; Chen, T.J.; Mansour, A.N.; Shao-Horn, Y. Structural Changes of Cobalt-Based Perovskites upon Water Oxidation Investigated by EXAFS. J. Phys. Chem. C 2013, 117, 8628–8635. [Google Scholar] [CrossRef]
- Landon, J.; Demeter, E.; Inoglu, N.; Keturakis, C.; Wachs, I.E.; Vasic, R.; Frenkel, A.I.; Kitchin, J.R. Spectroscopic Characterization of Mixed Fe–Ni Oxide Electrocatalysts for the Oxygen Evolution Reaction in Alkaline Electrolytes. Am. Chem. Soc. Catal. 2012, 2, 1793–1801. [Google Scholar] [CrossRef]
- Bajdich, M.; García-Mota, M.; Vojvodic, A.; Nørskov, J.K.; Bell, A.T. Theoretical Investigation of the Activity of Cobalt Oxides for the Electrochemical Oxidation of Water. J. Am. Chem. Soc. 2013, 135, 13521–13530. [Google Scholar] [CrossRef] [PubMed]
- Subbaraman, R.; Tripkovic, D.; Chang, K.C.; Strmcnik, D.; Paulikas, A.P.; Hirunsit, P.; Chan, M.; Greeley, J.; Stamenkovic, V.; Markovic, N.M. Trends in activity for the water electrolyser reactions on 3d M(Ni,Co,Fe,Mn) hydr(oxy)oxide catalysts. Nat. Mater. 2012, 11, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Trotochaud, L.; Ranney, J.K.; Williams, K.N.; Boettcher, S.W. Solution-Cast Metal Oxide Thin Film Electrocatalysts for Oxygen Evolution. J. Am. Chem. Soc. 2012, 134, 17253–17261. [Google Scholar] [CrossRef] [PubMed]
- Bode, H.; Dehmelt, K.; Witte, J. Zurkenntnis der nickelhydroxidelektrode—I.Über das nickel (II)-hydroxidhydrat. Electrochimica Acta 1966, 11, 1079–1087. [Google Scholar] [CrossRef]
- Bode, H.; Dehmelt, K.; Witte, J. Über die Oxydationsprodukte von Nickel (II)-hydroxiden. Z. Anorg. Allg. Chem. 1969, 366, 1–21. [Google Scholar] [CrossRef]
- Van der Ven, A.; Morgan, D.; Meng, Y.S.; Ceder, G. Phase Stability of Nickel Hydroxides and Oxyhydroxides. J. Electrochem. Soc. 2006, 153, A210–A215. [Google Scholar] [CrossRef]
- Portemer, F.; Figlarz, M.J. Characterization of Active Material Deposited at the Nickel Hydroxide Electrode by Electrochemical Impregnation. Electrochem. Soc. 1992, 139, 671–678. [Google Scholar] [CrossRef]
- Park, S.; Lee, Y.; Elcombe, M.; Vogt, T. Synthesis and Structure of the Bilayer Hydrate Na0.3 NiO2·1.3D2O. Inorg. Chem. 2006, 45, 3490–3492. [Google Scholar] [CrossRef] [PubMed]
- Guerlou-Demourgues, L.; Fournes, L.; Delmas, C. On the Iron Oxidation State in the Iron-Substituted γ Nickel Oxyhydroxides. J. Solid State Chem. 1995, 114, 6–14. [Google Scholar] [CrossRef]
- Bartl, H.; Bode, H.; Witte, G. Zur Kenntnis Nickel hydroxide elektrode-IV: Kristall strukturuntersuchung des Hochoxidierten γ-Nickelhydroxids. Electrochimica Acta 1971, 16, 615–621. [Google Scholar] [CrossRef]
- Yang, X.; Takada, K.; Itose, M.; Ebina, Y.; Ma, R.; Fukuda, K.; Sasak, T. Highly Swollen Layered Nickel Oxide with a Trilayer Hydrate Structure. Chem. Mater. 2008, 20, 479–485. [Google Scholar] [CrossRef]
- Lyons, M.E.G.; Brandon, M.P. The Oxygen Evolution Reaction on Passive Oxide Covered Transition Metal Electrodes in Aqueous Alkaline Solution: Part 1-Nickel. Int. J. Electrochem. Soc. 2008, 3, 1386–1424. [Google Scholar]
- Lu, P.W.T.; Srinivasan, S. Electrochemical-Ellipsometric Studies of Oxide Film Formed on Nickel during Oxygen Evolution. J. Electrochem. Soc. 1978, 125, 1416–1422. [Google Scholar] [CrossRef]
- Bediako, D.K.; Lassalle-Kaiser, B.; Surendranath, Y.; Yano, J.; Yachandra, V.K.; Nocera, D.G. Structure–Activity Correlations in a Nickel–Borate Oxygen Evolution Catalyst. J. Am. Chem. Soc. 2012, 134, 6801–6809. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Selloni, A. Mosaic Texture and Double c-Axis Periodicity of β-NiOOH: Insights from First-Principles and Genetic Algorithm Calculations. J. Phys. Chem. Lett. 2014, 5, 3981–3985. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Selloni, A. Mechanism and Activity of Water Oxidation on Selected Surfaces of Pure and Fe-Doped NiOx. Am. Chem. Soc. Catal. 2014, 4, 1148–1153. [Google Scholar] [CrossRef]
- Friebel, D.; Louie, M.W.; Bajdich, M.; Sanwald, K.E.; Cai, Y.; Wise, A.M.; Cheng, M.J.; Sokaras, D.; Weng, T.C.; Alonso, M.R.; et al. Identification of Highly Active Fe Sites in (Ni,Fe)OOH for Electrocatalytic Water Splitting. J. Am. Chem. Soc. 2015, 137, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Li, Y.; Wang, H.; Liang, Y.; Wu, J.Z.; Zhou, J.; Wang, J.; Regier, T.; Wei, F.; Dai, H. An Advanced Ni–Fe Layered Double Hydroxide Electrocatalyst for Water Oxidation. J. Am. Chem. Soc. 2013, 135, 8452–8455. [Google Scholar] [CrossRef] [PubMed]
- Louie, M.W.; Bell, A.T. An Investigation of Thin-Film Ni–Fe Oxide Catalysts for the Electrochemical Evolution of Oxygen. J. Am. Chem. Soc. 2013, 135, 12329–12337. [Google Scholar] [CrossRef] [PubMed]
- Oliver-Tolentino, M.A.; Vázquez-Samperio, J.; Manzo-Robledo, A.; de Guadalupe González-Huerta, R.; Flores-Moreno, J.L.; Ramírez-Rosales, D.; Guzmán-Vargas, A. An Approach to Understanding the Electrocatalytic Activity Enhancement by Superexchange Interaction toward OER in Alkaline Media of Ni−Fe LDH. J. Phys. Chem. C 2014, 118, 22432–22438. [Google Scholar] [CrossRef]
- Zhiyi, L.; Xu, W.; Zhu, W.; Yang, Q.; Lei, X.; Liu, J.; Li, Y.; Sun, X. Three-dimensional NiFe layered double hydroxide film for high-efficiency oxygen evolution reaction. Chem. Commun. 2014, 50, 6479–6482. [Google Scholar]
- Zaffran, J.; Toroker, M.C. Metal-Oxygen Bond Ionicity as an Efficient Descriptor for Doped NiOOH Photocatalytic Activity. Chem. Phys. Chem. 2016, 17, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Zaffran, J.; Toroker, M.C. Benchmarking Density Functional Theory Based Methods to Model NiOOH Material Properties: Hubbard and van der Waals Corrections vs Hybrid Functionals. J. Chem. Theory Comput. 2016, 12, 3807–3812. [Google Scholar] [CrossRef] [PubMed]
- Butera, V.; Toroker, M.C. Electronic Properties of Pure and Fe-Doped β-Ni(OH)2: New Insights Using Density Functional Theory with a Cluster Approach. J. Phys. Chem. C 2016, 120, 12344–12350. [Google Scholar] [CrossRef]
- Rosso, K.M.; Smith, D.M.A.; Dupuis, M. An ab initio model of electron transport in hematati α-Fe2O3 basal planes. J. Chem. Phys. 2003, 118, 6455–6466. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations: Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Li, X.; Frisch, M.J. Energy-represented DIIS within a hybrid geometry optimization method. J. Chem. Theory Comput. 2006, 2, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Alidousta, N.; Carter, E.A. First-principles Assessment of Hole Transport in Pure and Li-doped NiO. Phys. Chem. Chem. Phys. 2015, 17, 18098–18110. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H., Jr.; Hay, P.J. Modern Theoretical Chemistry; Schaefer, H.F., III, Ed.; Plenum: New York, NY, USA, 1977; Volume 8, pp. 1–28. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations—Potentials for the transition-metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations—Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Errata: Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396–1399. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods Without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Tkalych, A.J.; Yu, K.; Carter, E. A Structural and Electronic Features of β-Ni(OH)2 and β-NiOOH from First Principles. J. Phys. Chem. C 2015, 119, 24315–24322. [Google Scholar] [CrossRef]
- Casas-Cabanas, M.; Canales-Vázquez, J.; Rodríguez-Carvajal, J.; Palacín, M.R. Deciphering the Structural Transformations during Nickel Oxyhydroxide Electrode Operation. J. Am. Chem. Soc. 2007, 129, 5840–5842. [Google Scholar] [CrossRef] [PubMed]
- Kazimirov, V.Y.; Smirnov, M.B.; Bourgeois, L.; Guerlou-Demourgues, L.; Servant, L.; Balagurov, A.M.; Natkaniec, I.; Khasanova, N.R.; Antipov, E.V. Atomic structure and lattice dynamics of Ni and Mg hydroxides. Solid State Ionics 2010, 181, 1764–1770. [Google Scholar] [CrossRef]
- Oliva, P.; Leonardi, J.; Laurent, J.F.; Delmas, C.; Braconnier, J.J.; Figlarz, M.; Fievet, F.; Guibert, A. Review of the structure and the electrochemistry of nickel hydroxides and oxy-hydroxides. J. Power Sources 1982, 8, 229–255. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Barnard, R.; Randell, C.F.; Tye, F.L. Studies concerning charged nickel hydroxide electrodes I Measurement of reversible potentials. J. Appl. Electrochem. 1980, 10, 109–125. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Lau, L.W.M.; Gerson, A.; Smart, R.S.C. X-ray Photoelectron Spectroscopic Chemical State Quantification of Mixed Nickel Metal, Oxide and Hydroxide Systems. Surf. Interface Anal. 2009, 41, 324–332. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Biesinger, M.C.; Smart, R.S.C.; McIntyre, N.S. New Interpretations of XPS Spectra of Nickel Metal and Oxides. Surf. Sci. 2006, 600, 1771–1779. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, version 5.0.8.; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Kerisit, S.; Rosso, K.M. Charge Transfer in FeO: A combined Molecular-Dynamics and Ab Initio Study. J. Chem. Phys. 2005, 123, 224712. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Toroker, M.C.; Carter, E.A. Electron Transport in pure and doped hematite. Nano Lett. 2011, 11, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
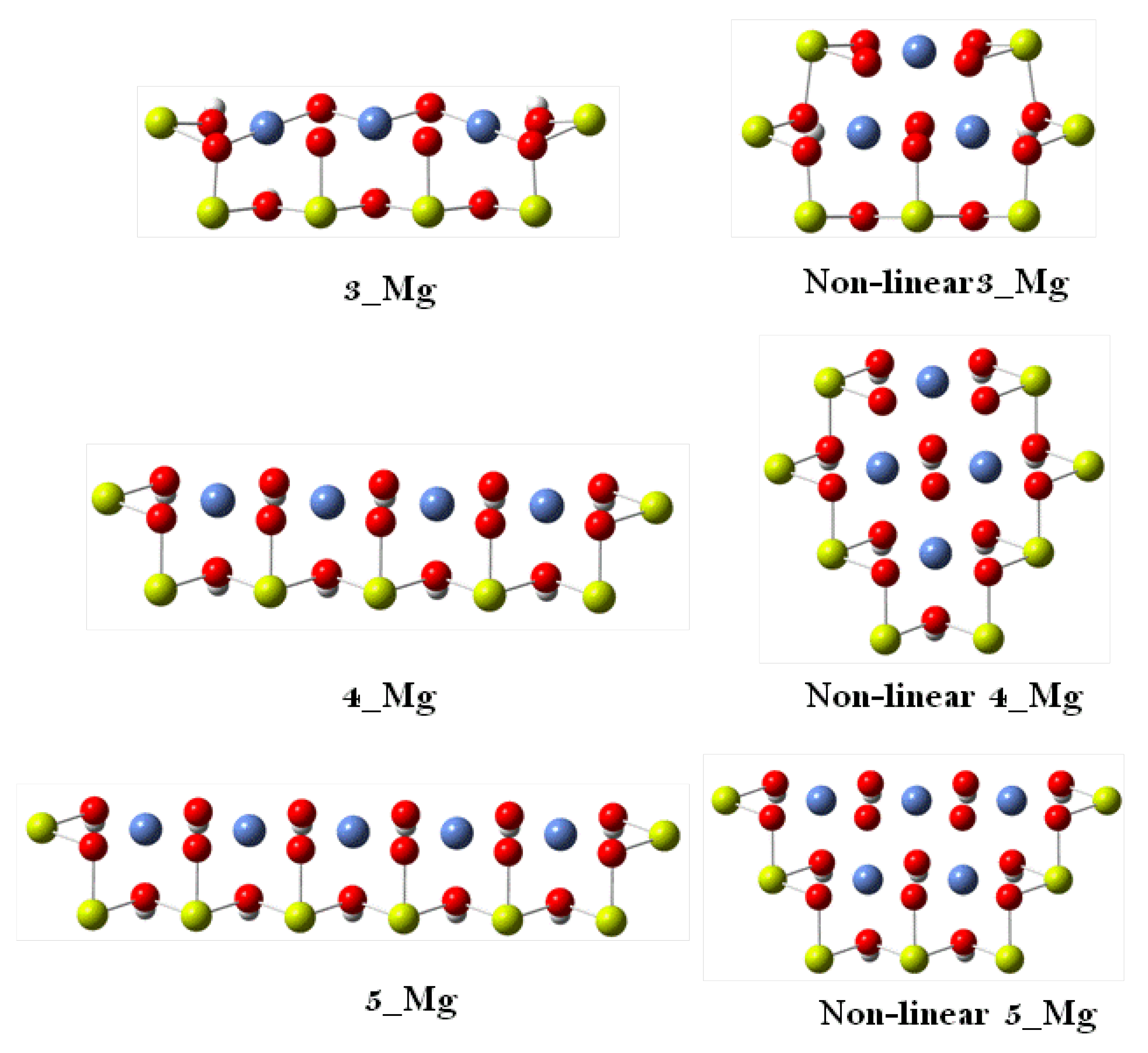
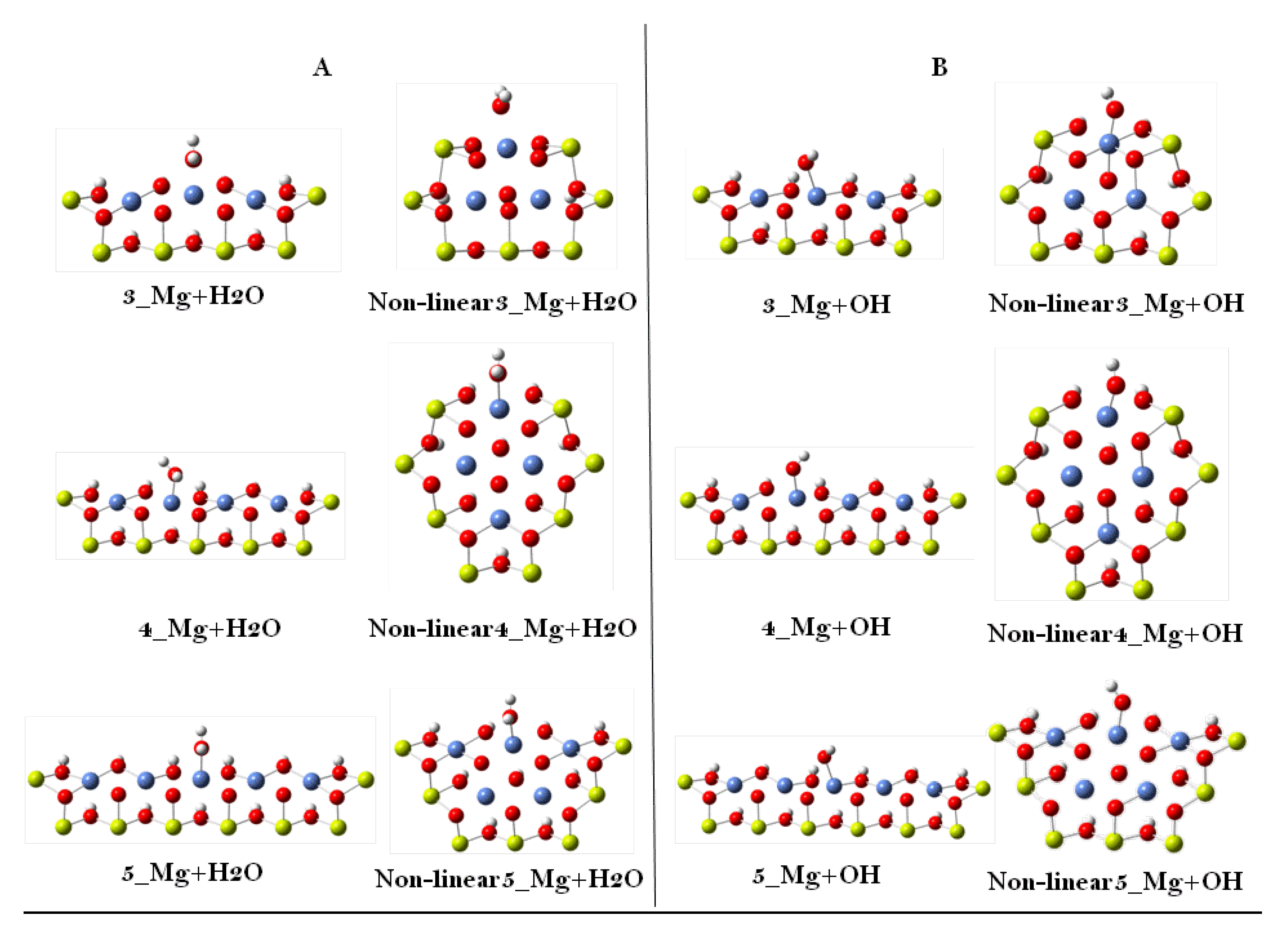

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 | 4 | 5 | Slab Model | Experimental | ||||
|---|---|---|---|---|---|---|---|---|
| Linear | Non-Linear | Linear | Non-Linear | Linear | Non-Linear | |||
| Ni–OH | 2.243 | 2.075 | 2.191 | 2.038 | 2.228 | 2.066 | 1.990 | 2.060 |
| Ni–O | 1.912 | 1.947 | 1.909 | 1.961 | 1.914 | 1.938 | 1.981 | 1.875 |
| Cluster Model | Adsorption Energy (eV) | |||
|---|---|---|---|---|
| Cluster 3_Mg | Cluster 4_Mg | Cluster 5_Mg | ||
| Linear | H2O | 1.868 | 1.264 | 1.406 |
| OH | 1.412 | 1.455 | 1.441 | |
| Non-linear | H2O | 1.744 | 1.545 | 1.555 |
| OH | 1.818 | 1.130 | 1.177 | |
| Cluster Model | Deprotonation Energy (eV) | ||
|---|---|---|---|
| Cluster 3 | Cluster 4 | Cluster 5 | |
| Linear | 2.889 | 2.772 | 2.656 |
| Non-linear | 3.419 | 3.379 | 3.341 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butera, V.; Toroker, M.C. Practical Cluster Models for a Layered β-NiOOH Material. Materials 2017, 10, 480. https://doi.org/10.3390/ma10050480
Butera V, Toroker MC. Practical Cluster Models for a Layered β-NiOOH Material. Materials. 2017; 10(5):480. https://doi.org/10.3390/ma10050480
Chicago/Turabian StyleButera, Valeria, and Maytal Caspary Toroker. 2017. "Practical Cluster Models for a Layered β-NiOOH Material" Materials 10, no. 5: 480. https://doi.org/10.3390/ma10050480
APA StyleButera, V., & Toroker, M. C. (2017). Practical Cluster Models for a Layered β-NiOOH Material. Materials, 10(5), 480. https://doi.org/10.3390/ma10050480