Abstract
In this work, oxygen vacancies were introduced onto the surface of BiFeO3 nanoparticles by NaBH4 reduction method to yield oxygen-deficient BiFeO3−x samples. Comprehensive analysis on the basis of high-resolution transmission electron microscopy (HRTEM) observation and X-ray photoelectron spectrum (XPS) confirms the existence of surface oxygen vacancies on the BiFeO3−x nanoparticles. The photocatalytic activity of as-prepared BiFeO3−x samples was evaluated by the decolorization of rhodamine B (RhB) under simulated sunlight irradiation. The experimental results indicate that the photocatalytic activity of samples is highly related to the NaBH4 reduction time, and the BiFeO3−x sample reduced for 40 min exhibits the highest photocatalytic efficiency, which is much higher than that of pristine BiFeO3 nanoparticles. This can be explained by the fact that the surface oxygen vacancies act as photoinduced charges acceptors and adsorption sites suppress the recombination of photogenerated charges, leading to an increasing availability of photogenerated electrons and holes for photocatalytic reaction. In addition, the obtained BiFeO3−x sample exhibits good photocatalytic reusability.
1. Introduction
Semiconductor photocatalysis has attracted tremendous interest because of its potential applications in solar energy conversion and environmental purification [,]. As a famous photocatalyst, TiO2 has been widely investigated due to its low cost and powerful photocatalytic capacity. However, TiO2 can be only excited under UV light irradiation, which accounts for 4% of the total solar energy. This limits the practical application of TiO2 as a photocatalyst. To make better use of solar energy that consists largely of visible light, it is essential to explore visible-light-driven photocatalysts [,,].
BiFeO3 is an important perovskite-type oxide with outstanding multiferroic property. In addition to this excellent property, BiFeO3, as a narrow band gap semiconductor (~2.1 eV), exhibits visible light photocatalytic activity for the degradation of organic dyes and benzene [,,,,]. However, its catalytic efficiency is not high enough for practical applications. It is well known that the catalytic activity of photocatalyst is closely related to various factors [,]. Among them, the effective separation of photogenerated electron-hole (e−-h+) pairs is very important in improving the photocatalytic activity. Up to now, many strategies have been used to modify BiFeO3, aiming to promote the separation of photogenerated charges [,,,,].
Recently, it is reported that the introduction of oxygen vacancies on the surface of photocatalysts is demonstrated to be an efficient way to enhance their photocatalytic activities [,,,,]. Generally, the surface oxygen vacancies can serve as the photogenerated charge traps and the adsorption sites, where the photoinduced charges can readily migrate to the adsorbed species. This process is expected to suppress the recombination of photogenerated electron-hole pairs, leading to an increased availability of electrons and holes for the photocatalytic reaction. Up to now, relatively little work has been devoted to the investigation of the photocatalytic property of BiFeO3−x with surface oxygen vacancies. Most recently, Zhang and Wang et al. reported the preparation of BiFeO3−x samples via the hydrogenation method and their enhanced photocatalytic performance [,]. However, this method involves harsh synthetic conditions, and furthermore expensive facilities are required. Compared with the hydrogenation method, the chemical reduction route has the main advantages of simplicity and low cost []. In this work, we develop a chemical reduction route, which is based on the NaBH4 reduction process, for the preparation of BiFeO3−x with surface oxygen vacancies. The photocatalytic activity of products was evaluated by the decolorization of RhB under simulated sunlight irradiation, and the involved photocatalytic mechanism was proposed.
2. Experimental
BiFeO3 nanoparticles were synthesized by a polyacrylamide gel route as reported in the literature []. Stoichiometric amounts of Bi(NO3)3·5H2O and Fe(NO3)3·9H2O were dissolved into diluted HNO3 to form the transparent solution. Subsequently, the ethylenediamine-tetraacetic acid (EDTA) (in a 1.5:1 molar ratio with respect to the cations) was added into the above solution. After that, a certain amount of glucose was dissolved (20 g/100 mL). To the solution were added acrylamide and N,N′-methylene-bisacrylamide monomers with molar ratio of acrylamide/bisacrylamide (25/1), followed by adjusting the pH value to 3 by the addition of ammonia. The resulted solution was heated at 80 °C to initiate the polymerization reaction. The obtained gel was dried at 120 °C for 24 h, and then calcined at 600 °C for 3 h to obtain final BiFeO3 nanoparticles.
BiFeO3−x samples were prepared via NaBH4 reduction method. 0.1 g BiFeO3 nanoparticles were dispersed into 20 mL NaBH4 solution (0.1 M) under magnetic stirring in ice-water bath. After reaction for a certain time, the product was separated by centrifugation, washed with distilled water and ethanol several times, and then dried in a vacuum drying oven at 60 °C for 4 h to obtain BiFeO3−x samples. To study the effect of reduction time on the photocatalytic activity of BiFeO3−x sample, a series of samples were prepared for different reduction times of 20, 40, and 60 min and termed as samples R20-BiFeO3−x, R40-BiFeO3−x, and R60-BiFeO3−x, respectively.
The photocatalytic activities of the samples were examined by the decolorization of rhodamine B (RhB) under simulated sunlight irradiation of a 300 W xenon lamp. In a typical photocatalytic process, the initial RhB concentration was 5 mg L−1 with a photocatalyst loading of 1 g L−1. Before irradiation, the suspension was magnetically stirred in the dark for 30 min to establish the adsorption-desorption equilibrium of RhB molecule on the surface of photocatalysts. Then the suspension was exposed to simulated sunlight irradiation under stirring. During the illumination, a small amount of reaction solution was taken every 1 h for measuring the concentration of RhB. Before the measurement, the suspension was centrifuged to separate the photocatalysts and obtain supernatant. The concentration of RhB was determined by detecting the absorbance of the supernatant at the wavelength 553 nm using an UV-VIS spectrophotometer. In order to evaluate the photocatalytic stability of samples, the recycling photocatalytic experimental was carried out. After the first cycle, the photocatalyst particles was collected by centrifugation and washed with water, and then dried in an oven. The recovered photocatalyst was added into the fresh RhB solution for the next cycle of the photocatalytic decolorization reaction under the same conditions.
The phase purity of products was examined by X-ray diffractometer (XRD). The morphology and structure of samples were observed using a transmission electron microscope (TEM). The UV-VIS diffuse reflectance spectra (DRS) of the samples were recorded by a UV-VIS spectrophotometer. The electron binding energies for the elements were measured by X-ray photoelectron spectrometer (XPS). The BET specific surface area of the sample is measured by the N2 adsorption-desorption technique on an ASAP2020M system (Micromeritics, Tristar II 3020, Norcross, GA, USA). The steady state photoluminescence spectra of samples were recorded by fluorescence lifetime and steady state spectroscopy (FLS920, Edinburgh Instrument, Livingston, Scotland, UK) with the excitation wavelength of ~350 nm.
3. Results and Discussion
Figure 1 shows the XRD patterns of BiFeO3 and R40-BiFeO3−x samples. It can be seen that all the diffraction peaks of samples can be indexed to the rhombohedral structure of BiFeO3, and no traces of impurity phases are detected. This suggests that the NaBH4 reduction treatment has no remarkable influence on the phase purity of BiFeO3.
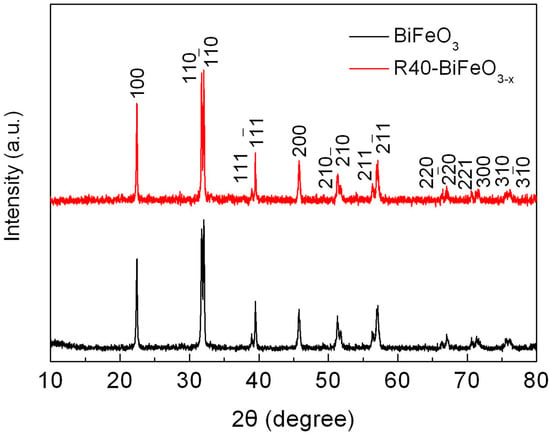
Figure 1.
XRD patterns of BiFeO3 and R40-BiFeO3−x samples.
Figure 2a,b show the TEM images of BiFeO3 and R40-BiFeO3−x samples, respectively. Both the samples display sphere-like shape with size ranging from 80 nm to 110 nm, indicating that the morphology and size of BiFeO3 sample did not show obvious change after NaBH4 treatment. To further observe crystal structure of the products, the HRTEM images of BiFeO3, R40-BiFeO3−x, and R60-BiFeO3−x samples are provided in Figure 2c–e, respectively. Figure 2c clearly presents the two-dimensional lattice fringes, revealing that the BiFeO3 nanoparticles are highly crystalline. For the R40-BiFeO3−x sample, as shown in Figure 2d, the sample displays disordered edge with 15–20 nm thickness, while the inner part of sample is still well-crystallized. This suggests that the NaBH4 reduction leads to the creation of defect layer on the surface of BiFeO3 nanoparticles. In the case of R60-BiFeO3−x sample, it is found that the thickness of disordered edge is about 20–30 nm (Figure 2e). This indicates that with increasing the reduction time, the thickness of the defect layer in the BiFeO3 exhibits an increasing trend.

Figure 2.
(a,b) TEM images of BiFeO3 and R40-BiFeO3−x samples, respectively; (c–e) HRTEM images of BiFeO3, R40-BiFeO3−x, and R60-BiFeO3−x samples, respectively; and (d,e), the black dash line shows the boundary between the crystalline core and the disordered layer (pointed out by white arrows).
To further analyze the defect layer, the XPS detection is performed to investigate the surface chemical bonding of the BiFeO3 and reduced BiFeO3 samples. Figure 3a,b present the high-resolution XPS spectra for Bi 4f and Fe 2p in BiFeO3 and R40-BiFeO3−x, respectively. Two signals at binding energies of 164.1 eV and 159.2 eV for both samples correspond to the Bi 4f5/2 and Bi 4f7/2, respectively (Figure 3a), which are consistent with the chemical states of Bi3+ []. In Figure 3b, the intense peaks positioned at 724.1 eV in both samples are assigned to the 2p1/2 peaks of Fe3+. The other main peaks at 710.7 eV ascribing to Fe 2p3/2 are fitted into two peaks. These peaks situated on 711.5 eV and 710.2 eV are caused by the +3 and +2 oxidation state of Fe ion, respectively. Additionally, the satellite peaks centered at 718.3 eV for the two samples are observed, which is due to the multiple oxidation states of Fe. The XPS analysis of Fe element indicates the coexistence of Fe2+ and Fe3+ in the two samples. Furthermore, according to the analysis of the peak area in Figure 3b, the ratios of Fe2+ to Fe3+ in the BiFeO3 and R40-BiFeO3−x are 23/77 and 48/52, respectively, which reveals that the concentration of Fe2+ in the sample is increased after the reduction treatment.
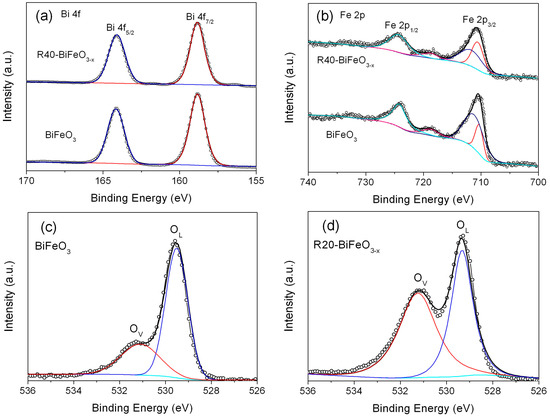
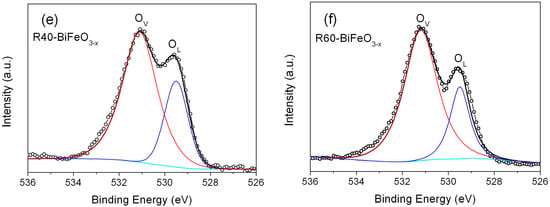
Figure 3.
High-resolution XPS spectra of BiFeO3 and R40-BiFeO3−x samples: (a) Bi 4f, (b) Fe 2p; O 1s high-resolution XPS spectra of BiFeO3 (c), R20-BiFeO3−x (d), R40-BiFeO3−x (e), and R60-BiFeO3−x (f).
Figure 3c–f shows the high-resolution XPS spectra for O 1s in BiFeO3 and reduced BiFeO3 samples. One can see that the broad XPS peaks of O 1s can be divided into two peaks located at 531.1 eV and 529.6 eV, revealing two different kinds of O chemical state in the as-prepared samples. The peaks at 529.6 eV are ascribed to the lattice oxygen of BiFeO3 (named as OL), and the peaks at 531.1 eV are generally attributed to the chemisorbed oxygen caused by oxygen vacancies (named as Ov) []. In nanosized BiFeO3, the long-range order of the lattice is commonly destroyed at the surface of sample, making the generation of oxygen vacancies. Compared to BiFeO3 nanoparticles, it is worth noting that the reduced BiFeO3 samples present a much higher peak at 531.1 eV compared with BiFeO3. Moreover, the analysis of the peak areas indicates that the concentration of the oxygen vacancies in the R20-BiFeO3−x, R40-BiFeO3−x, and R60-BiFeO3−x are 0.53, 0.70, and 0.75, respectively. The results suggest that the concentration of oxygen vacancies increases with increasing the reduction time. It is well known that the detection depth of XPS is about 5 nm, therefore, the XPS spectrum information of reduced sample comes from the surface defect layer in the Figure 2d,e. Furthermore, it is generally accepted that the surface oxygen vacancies of oxides can destroy their surface lattice structure, and induce the generation of defect edge. Combined with the XPS and HRTEM analysis, it can be concluded that the defect layer on the NaBH4 reduced BiFeO3 nanoparticles is mainly attributed to the creation of a great amount of surface oxygen vacancies. On the other hand, the BET specific surface areas of the BiFeO3, R20-BiFeO3−x, R40-BiFeO3−x, and R60-BiFeO3−x samples, measured by the N2 adsorption-desorption technique, are 6.52 m2/g, 6.49 m2/g, 6.55 m2/g, and 6.57 m2/g, respectively. This suggests that the surface area of BiFeO3 undergoes no obvious change after NaBH4 reduction.
Figure 4a shows the UV-VIS diffuse reflectance spectra of BiFeO3 and BiFeO3−x samples. Compared with pristine BiFeO3 nanoparticles, the BiFeO3−x samples show an enhanced light absorbance in the range of 550–800 nm. In order to exactly determine the band gap of samples, the steady state photoluminescence spectra of samples were carried out. As shown in Figure 4b, the strong and sharp emission peaks at 515 nm are observed for BiFeO3 and R40-BiFeO3−x. These emission peaks are attributed to the recombination of photogenerated charges between valence band (VB) and conduction band (CB), from which the band gap energy (Eg) of BiFeO3 and R40-BiFeO3−x is obtained to be 2.4 eV. In comparison to BiFeO3, it is worth noting that the R40-BiFeO3−x exhibits an obvious emission peak centered at ~635 nm, which is considered to be the recombination of photoinduced charges between valence band and oxygen vacancy state. It is widely accepted that surface oxygen vacancies of semiconductor-based photocatalyst generally introduce an oxygen vacancy state within its forbidden gap [].
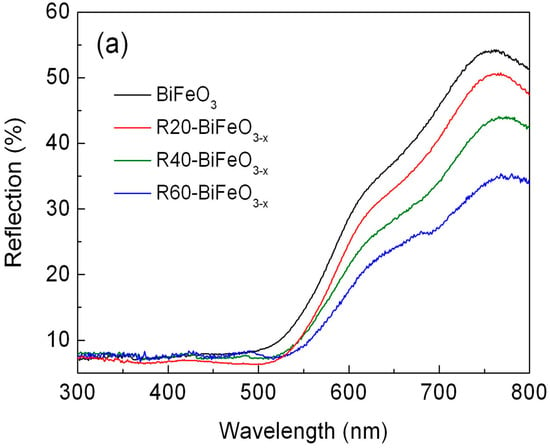
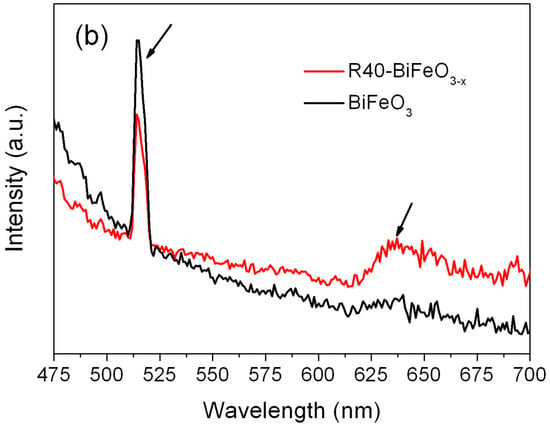
Figure 4.
(a) UV-VIS diffuse reflectance spectra of BiFeO3 and BiFeO3−x samples; and (b) the steady state photoluminescence spectra of BiFeO3 and R40-BiFeO3−x samples.
Figure 5 presents the photocatalytic decolorization of RhB under simulated sunlight irradiation as a function of reaction time in the presence of BiFeO3 and BiFeO3−x samples. Before examining the photocatalytic properties, the blank and adsorption experiments are carried out. It is seen that no obvious decolorization of dye is observed in the absence of either photocatalysts or simulated sunlight, indicating that the impact of self-decolorization and adsorption on the photocatalytic effect can be ignored. In the presence of BiFeO3 nanoparticles, about 40% of RhB is decolored within 6 h. This illustrates that BiFeO3 nanoparticles process a moderate photocatalytic activity for the decolorization of RhB under simulated sunlight illumination. When the BiFeO3 sample is reduced by NaBH4, the reduction time exhibits an important influence on the photocatalytic performance of samples. With increasing the treatment time, the decolorization percentage of RhB is seen to gradually increase, from ~40% for ttreatment time = 0 min to ~57% for ttreatment time = 40 min. However, when the treatment time is further increased up to 60 min, the photocatalytic efficiency sharply decreases.
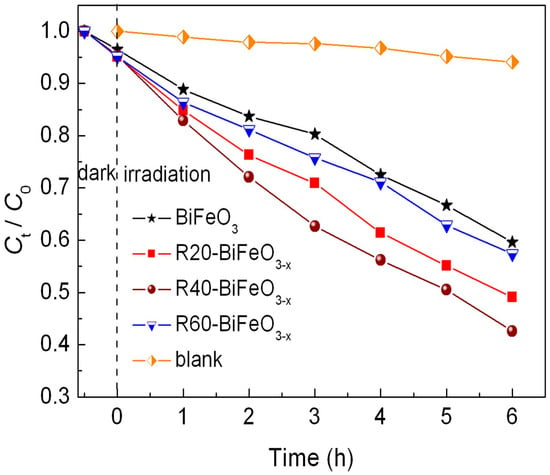
Figure 5.
Photocatalytic degradation of RhB versus irradiation time in the presence of BiFeO3 and BiFeO3−x samples, along with the blank and adsorption experiment results.
It is believed that the reusability of photocatalysts is crucial for their practical application. The recycling photocatalytic experiment is carried out under the same conditions to evaluate the stability of R40-BiFeO3−x sample, as shown in Figure 6. After five recycles, R40-BiFeO3−x sample maintains a high photocatalytic activity, indicating the stable photocatalytic activity of NaBH4 reduced BiFeO3 nanoparticles.
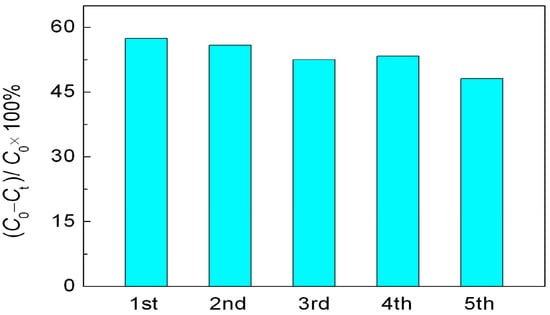
Figure 6.
Cycling experiments in the photocatalytic degradation of RhB over R40-BiFeO3−x sample for 6 h.
To clarify the photocatalytic mechanism of NaBH4 reduced BiFeO3, the active species trapping experiments were performed. As shown in Figure 7, after adding AgNO3 (a scavenger of photogenerated electron (e−), 2 mM), the decolorization percentage of RhB is slightly increased compared to that without introduction of scavenger. This is mainly attributed to the efficient separation of photoinduced electron-hole pairs, resulting from the consumption of photogenerated electrons by AgNO3. When ethanol (a scavenger of hydroxyl radicals (•OH), 10% by volume) is introduced, the decolorization percentage of dye is significantly decreased, indicating that •OH is a main active species involved in this photocatalytic reaction. When ethylene diamine tetraacetic acid (EDTA, a scavenger of photogenerated holes (h+), 2 mM) is added, the decolorization efficiency of dye is also obviously suppressed, implying that h+ plays an important role in this photocatalysis. In addition to h+ and •OH, •O2 and H2O2 are considered to be another active species in the photocatalytic reaction. The effect of •O2 and H2O2, which are derived from the reaction between dissolved O2 and photogenerated electrons (e−), on the photocatalytic reaction can be detected by investigating the influence of N2 on the photocatalytic efficiency since the O2 molecules dissolved in reaction solution can be expelled from the solution by the N2-purging procedure. Upon bubbling with N2 (0.1 L/min), the decolorization percentage of dye undergoes a slight decrease, indicating relatively minor role of •O2 and/or H2O2 responsible for the dye decolorization.
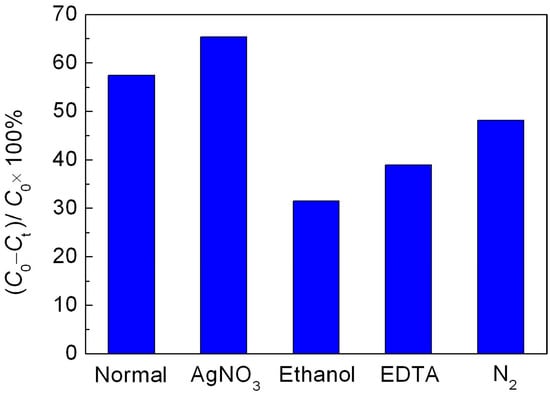
Figure 7.
Effect of AgNO3, ethanol, EDTA and N2 on the photocatalytic decolorization of RhB over the R40-BiFeO3−x sample.
On the basis of above experimental results, a possible promotion mechanism of surface oxygen vacancies on the simulated sunlight photocatalytic activity of R40-BiFeO3−x is proposed, as shown in Figure 8. Under the simulated sunlight irradiation, BiFeO3 is excited to generate photoexcited electron-hole pairs (Equation (3)). Unfortunately, the recombination rate of the photogenerated carries is high, leading to a moderate photocatalytic activity of BiFeO3. When oxygen vacancies are introduced onto the surface of BiFeO3 sample after NaBH4 reduction, an oxygen vacancy state appears in the forbidden gap of the BiFeO3, resulting in the electron transition from the valence band to oxygen vacancy state. This is beneficial to extend the light response region of BiFeO3. On the other hand, it is noted that the surface oxygen vacancies, which are excellent charge carrier acceptors and adsorption sites, can readily capture the photogenerated electron and promote the transfer of photoinduced charges to adsorbed species []. Consequently, the recombination of photogenerated charges can be suppressed, which results in an increasing availability of photogenerated electrons and holes participating in the photocatalytic redox reactions. However, it can be seen from Figure 5 that when the NaBH4 treatment time reaches 60 min, the photocatalytic efficiency exhibits an obvious decrease. The main reason is that excessive NaBH4 reduction is more likely to induce bulk oxygen vacancies. These bulk defects may serve as the new recombination centers for photogenerated electron-hole pairs and will lead to the reduction of photocatalytic efficiency.
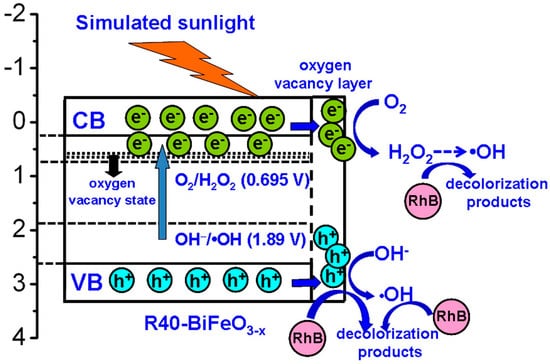
Figure 8.
Schematic illustration of the possible promotion mechanism of surface oxygen vacancies on the simulated sunlight photocatalytic activity of reduced BiFeO3.
To further investigate the photocatalytic redox reactions of photogenerated carriers, it is necessary to determine the energy-band potentials of photocatalysts. The valence band (VB) and conduction band (CB) potentials of the R40-BiFeO3−x can be calculated according to the following equations:
where X and Ee are the absolute electronegativity of materials (defined as the arithmetic mean of the electron affinity and the first ionization of the constituent atoms) and energy of free electrons on the hydrogen scale (~4.5 eV), respectively. Eg is the band gap value of the R40-BiFeO3−x. The X value of BiFeO3 is calculated to be 5.93 eV based on the data reported in literatures [,]. As a result, the VB potential of R40-BiFeO3−x is estimated to be 2.63 V vs. NHE, and the CB potential of R40-BiFeO3−x is calculated to be 0.23 V vs. NHE. It can be seen that the VB potential of R40-BiFeO3−x is positive to the redox potential of OH−/•OH (+1.89 V vs. NHE), indicating that photogenerated h+ can oxidize OH− to form •OH (Equation (4)). On the other hand, the CB potential of R40-BiFeO3−x is positive to the redox potential of O2/•O2 (−0.13 V vs. NHE), but negative to that of O2/H2O2 (+0.695 vs. NHE). This suggests that the photogenerated e− can reduce O2 to generate H2O2 instead of •O2 (Equation (5)). Furthermore, H2O2 can undergoes a series of reactions to generate •OH radicals (Equation (6)). As a result, it is inferred that the h+, •OH, and H2O2 work together for the decolorization of RhB in the present photocatalytic reaction (Equation (7)), which is consistent with the trapping experimental results (Figure 7).
EVB = X − Ee + 0.5Eg
ECB = X − Ee − 0.5Eg
BiFeO3 + hν → BiFeO3 (e− + h+)
h+ + OH− → •OH
O2 + 2H+ + 2e− → H2O2
H2O2 + hν → 2•OH
h+, •OH or H2O2 + RhB → decolorization products
4. Conclusions
The BiFeO3−x nanoparticles with surface oxygen vacancies were successfully synthesized by a polyacrylamide gel method and subsequently reduced by NaBH4. Analysis results from HRTEM observations and XPS spectra reveal that the surface oxygen vacancies are introduced on the BiFeO3−x nanoparticles. The photocatalytic experiments indicate that the photocatalytic performance of BiFeO3−x nanoparticles for the decolorization of RhB under simulated sunlight irradiation depends highly on the NaBH4 reduction time. The BiFeO3−x sample reduced for 40 min possesses the highest photocatalytic activity, which is much higher than that of pristine BiFeO3 nanoparticles. This can be attributed to the enhanced photogenerated charge separation and transport caused by surface oxygen vacancies, resulting in an increasing availability of photogenerated electrons and holes participating in the photocatalytic reactions. Moverover, the BiFeO3−x nanoparticles exhibits good stability during the recycling photocatalytic experiment.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant nos. 51662027, 51602170), the Natural Science Foundation of Qinghai, China (grant no. 2016-ZJ-954Q), “ChunHui” Program of Ministry of Education of China (grant nos. Z2016074, Z2016075), and the Youth Science Foundation of Qinghai Normal University (15ZR07).
Author Contributions
Hua Yang and Lijing Di designed the experiment; Tao Xian and Lijing Di carried out the experiments; Hua Yang, Lijing Di, Tao Xian, and Xiujuan Chen analyzed the data; Lijing Di and Hua Yang drafted the manuscript; and all authors read and approved the final manuscript.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Fox, M.A.; Dulay, M.T. Heterogeneous photocatalysis. Chem. Rev. 1993, 93, 341–357. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.Q.; Wang, W.W.; Zong, W.J.; Xiong, S.M.; Zhang, Q.; Ji, F.Y.; Xu, X. Synthesis of Bi2S3/BiVO4 heterojunction with a one-step hydrothermal method based on pH control and the evaluation of visible-light photocatalytic performance. Materials 2017, 10, 891. [Google Scholar] [CrossRef] [PubMed]
- Chiang, T.H.; Chen, T.-M. Photocatalytic water splitting for O2 production under visible light irradiation using NdVO4-V2O5 hybrid powders. Materials 2017, 10, 331. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wang, L.; Zong, R.; Zhu, Y. Photocatalytic activity enhanced via g-C3N4 nanoplates to nanorods. J. Phys. Chem. C 2013, 117, 9952–9961. [Google Scholar] [CrossRef]
- Gao, F.; Chen, X.Y.; Yin, K.B.; Dong, S.A.; Ren, Z.F.; Yuan, F.; Yu, T.; Zou, Z.G.; Liu, J.M. Visible-light photocatalytic properties of weak magnetic BiFeO3 nanoparticles. Adv. Mater. 2007, 19, 2889–2892. [Google Scholar] [CrossRef]
- Xian, T.; Yang, H.; Dai, J.F.; Wei, Z.Q.; Ma, J.Y.; Feng, W.J. Photocatalytic properties of BiFeO3 nanoparticles with different sizes. Mater. Lett. 2011, 65, 1573–1575. [Google Scholar] [CrossRef]
- Bharathkumar, S.; Sakar, M.; Balakumar, S. Experimental evidence for the carrier transportation enhanced visible light driven photocatalytic process in bismuth ferrite (BiFeO3) one-dimensional fiber nanostructures. J. Phys. Chem. C 2016, 120, 18811–18821. [Google Scholar] [CrossRef]
- Sze-Mun, L.; Jin-Chung, S.; Abdul Rahman, M. A newly emerging visible light-responsive BiFeO3 perovskite for photocatalytic applications: A mini review. Mater. Res. Bull. 2017, 90, 15–30. [Google Scholar]
- Bai, X.; Wei, J.; Tian, B.; Liu, Y.; Reiss, T.; Guiblin, N.; Gemeiner, P.; Dkhil, B.; Infante, I.C. Size Effect on optical and photocatalytic properties in BiFeO3 nanoparticles. J. Phys. Chem. C 2016, 120, 3595–3601. [Google Scholar] [CrossRef]
- Navjot; Alexandr, T.; Singh, L.G. Plasmonic enhanced photocatalytic activity of Ag nanospheres decorated BiFeO3 nanoparticles. Catal. Lett. 2017, 147, 1640–1645. [Google Scholar] [CrossRef]
- Di, L.J.; Yang, H.; Hu, G.; Xian, T.; Ma, J.Y.; Jiang, J.L.; Li, R.S.; Wei, Z.Q. Enhanced photocatalytic activity of BiFeO3 particles by surface decoration with Ag nanoparticles. J. Mater. Sci. Mater. Electron. 2014, 25, 2463–2469. [Google Scholar] [CrossRef]
- Irfan, S.; Li, L.L.; Saleemi, A.S.; Nan, C.W. Enhanced photocatalytic activity of La3+ and Se4+ co-doped bismuth ferrite nanostructures. J. Mater. Chem. A 2017, 5, 11143–11151. [Google Scholar] [CrossRef]
- Fan, T.; Chen, C.; Tang, Z. Hydrothermal synthesis of novel BiFeO3/BiVO4 heterojunctions with enhanced photocatalytic activities under visible light irradiation. RSC Adv. 2016, 6, 9994–10000. [Google Scholar] [CrossRef]
- Dhanalakshmi, R.; Muneeswaran, M.; Shalini, K.; Giridharan, N.V. Enhanced photocatalytic activity of La-substituted BiFeO3 nanostructures on the degradation of phenol red. Mater. Lett. 2016, 165, 205–209. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Yu, P.Y.; Mao, S.S. Increasing solar absorption for photocatalysis with black hydrogenated titanium dioxide nanocrystals. Science 2011, 331, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Zhao, Z.; Zhu, W.-B.; Coker, E.N.; Li, B.; Zheng, M.; Yu, W.; Fan, H.; Sun, Z. Oxygen vacancy enhanced photocatalytic activity of pervoskite SrTiO3. ACS Appl. Mater. Interfaces 2014, 6, 19184–19190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, Z. Enhanced photoelectrochemical performance of the hierarchical micro/nano-structured TiO2 mesoporous spheres with oxygen vacancies via hydrogenation. RSC Adv. 2015, 5, 9482–9488. [Google Scholar] [CrossRef]
- Zou, X.; Liu, J.; Su, J.; Zuo, F.; Chen, J.; Feng, P. Facile Synthesis of thermal- and photostable titania with paramagnetic oxygen vacancies for visible-light photocatalysis. Chem. Eur. J. 2013, 19, 2866–2873. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Yang, M.-Q.; Fu, X.; Zhang, N.; Xu, Y.-J. Defective TiO2 with oxygen vacancies: Synthesis, properties and photocatalytic applications. Nanoscale 2013, 5, 3601–3614. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, Y.; Chu, M.; Rong, N.; Xiao, P.; Zhang, Y. Hydrogen-treated BiFeO3 nanoparticles with enhanced photoelectrochemical performance. RSC Adv. 2016, 6, 24760–24767. [Google Scholar] [CrossRef]
- Wang, S.; Chen, D.; Niu, F.; Zhang, N.; Qin, L.; Huang, Y. Hydrogenation-induced surface oxygen vacancies in BiFeO3 nanoparticles for enhanced visible light photocatalytic performance. J. Alloys Compd. 2016, 688, 399–406. [Google Scholar] [CrossRef]
- Kang, Q.; Cao, J.; Zhang, Y.; Liu, L.; Xu, H.; Ye, J. Reduced TiO2 nanotube arrays for photoelectrochemical water splitting. J. Mater. Chem. A 2013, 1, 5766–5774. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y.; Ding, X.; Jiang, J. Enhanced visible-light-response photocatalytic activity of bismuth ferrite nanoparticles. J. Alloys Compd. 2011, 509, 6585–6588. [Google Scholar] [CrossRef]
- Hotop, H.; Lineberger, W.C. Binding energies in atomic negative ions. J. Phys. Chem. Ref. Data 1975, 4, 539–576. [Google Scholar] [CrossRef]
- Andersen, T.; Haugen, H.K.; Hotop, H. Binding energies in atomic negative ions: III. J. Phys. Chem. Ref. Data 1999, 28, 1511–1533. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).