
Towards Renewable Iodide Sources for Electrolytes in Dye-Sensitized Solar Cells

Abstract
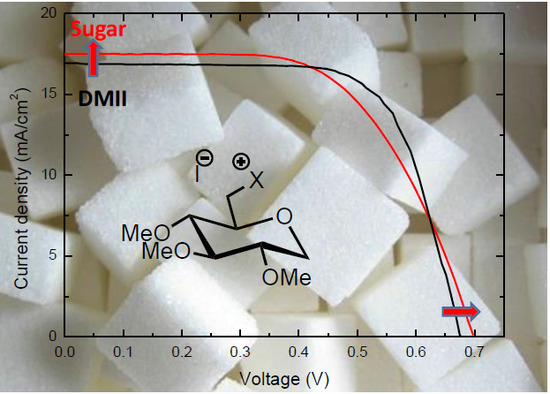
:
1. Introduction
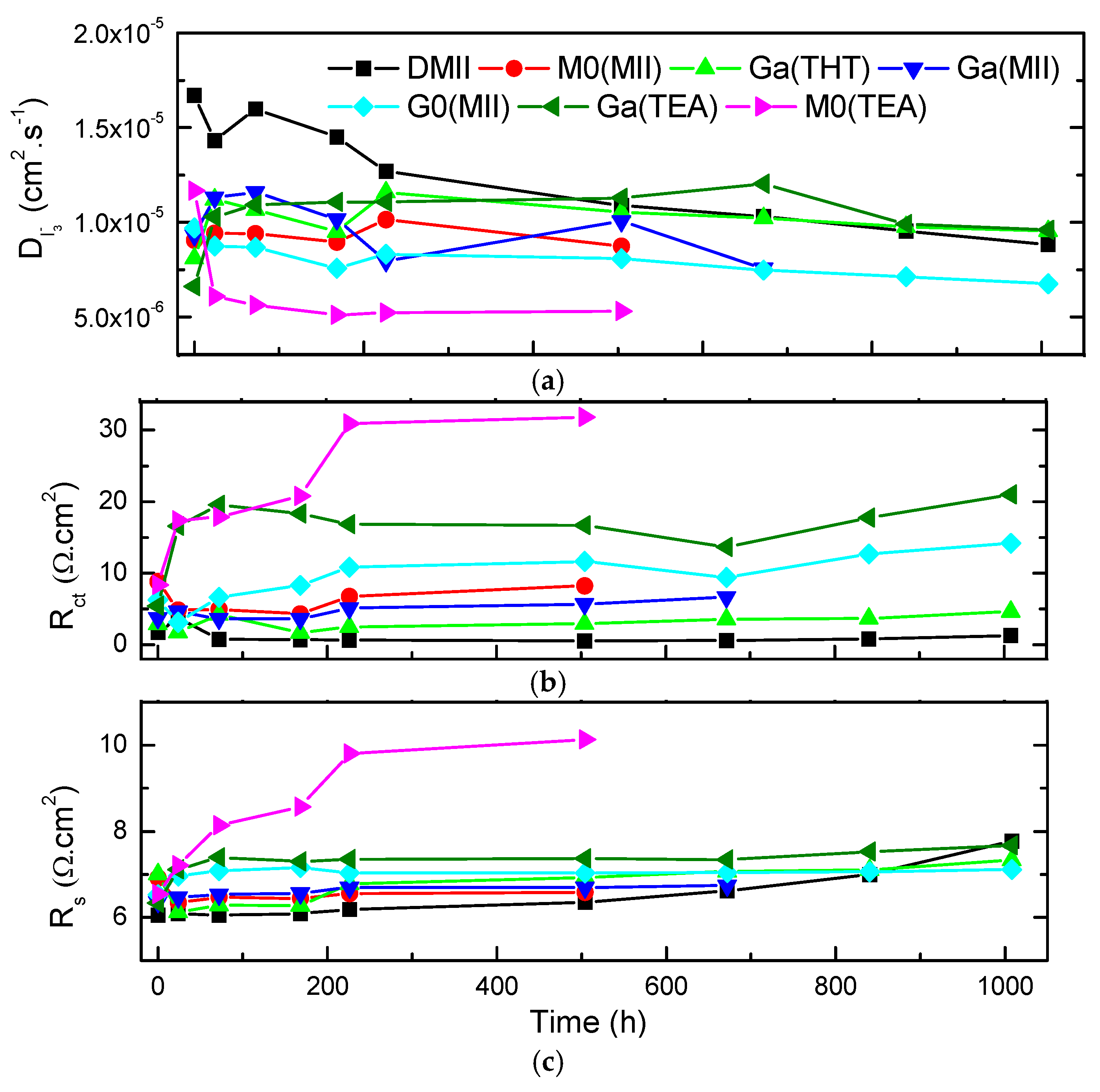
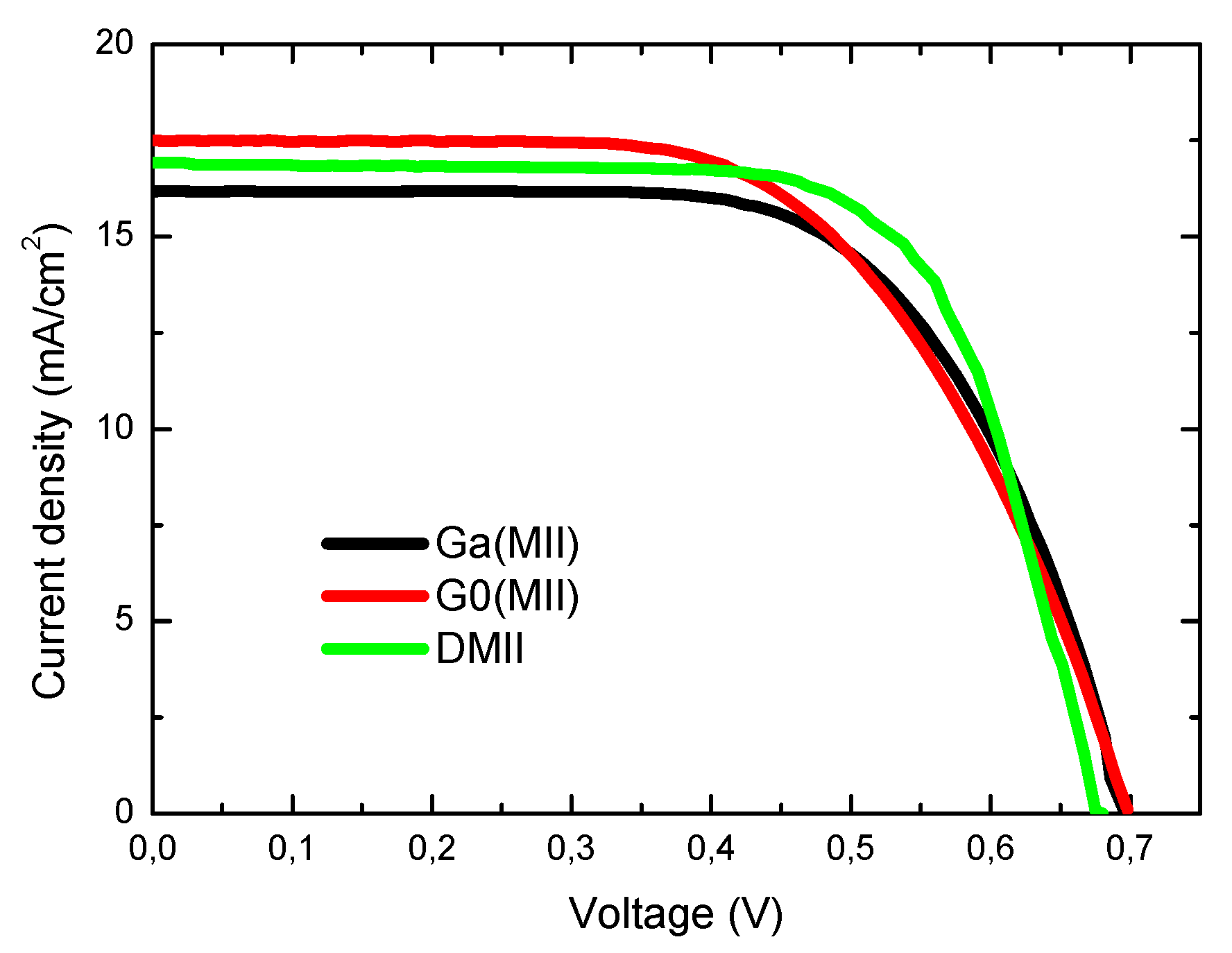
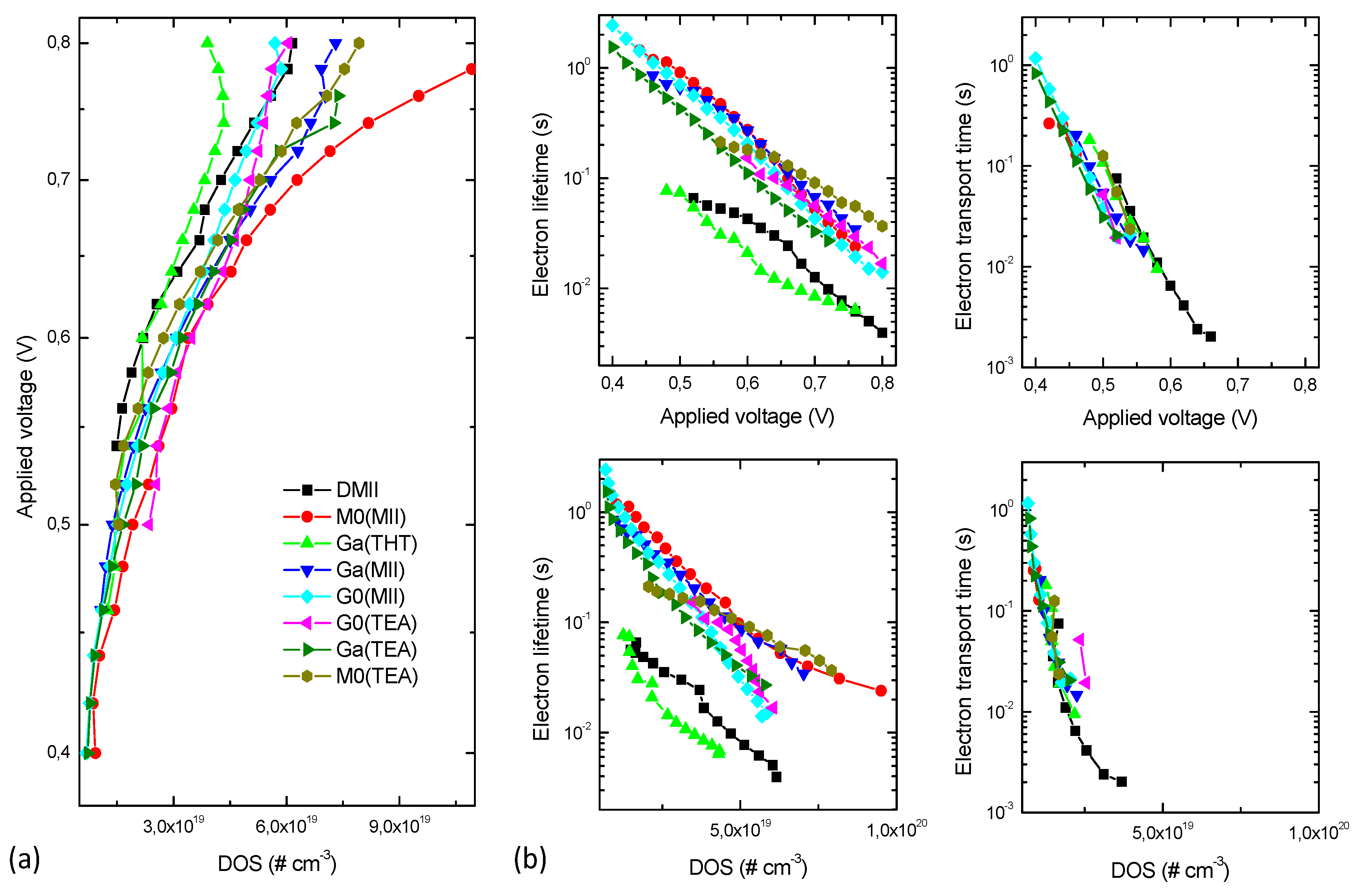
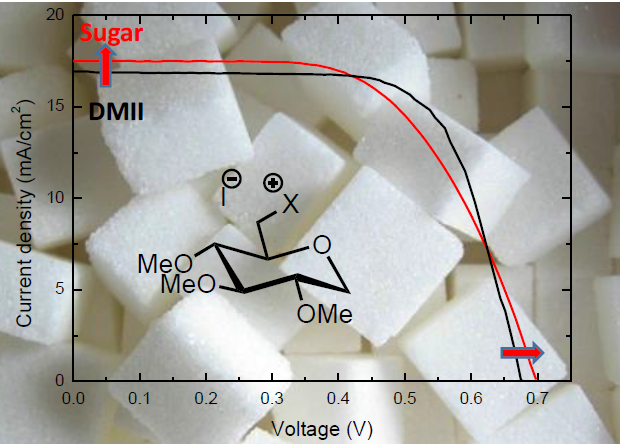
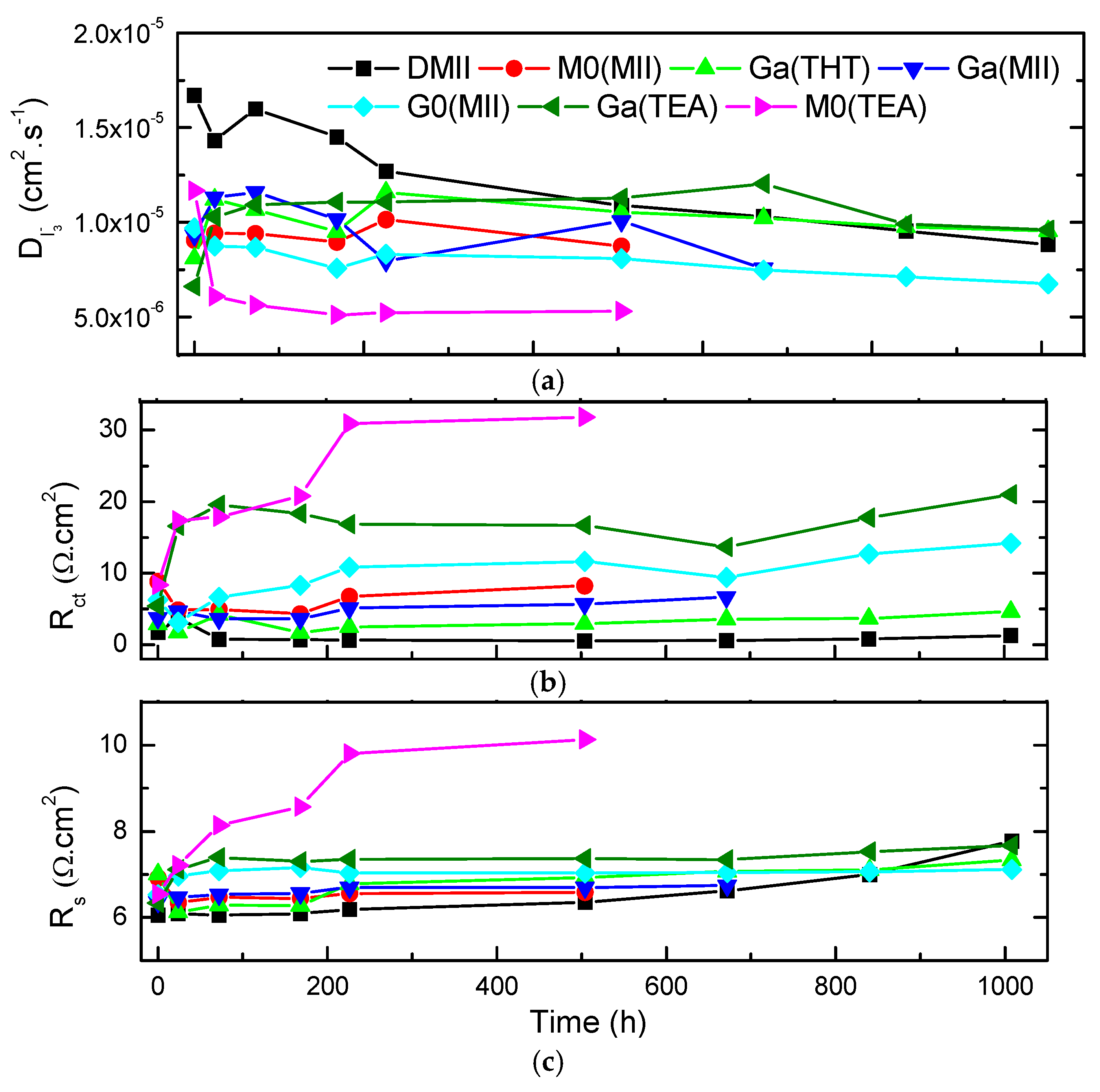
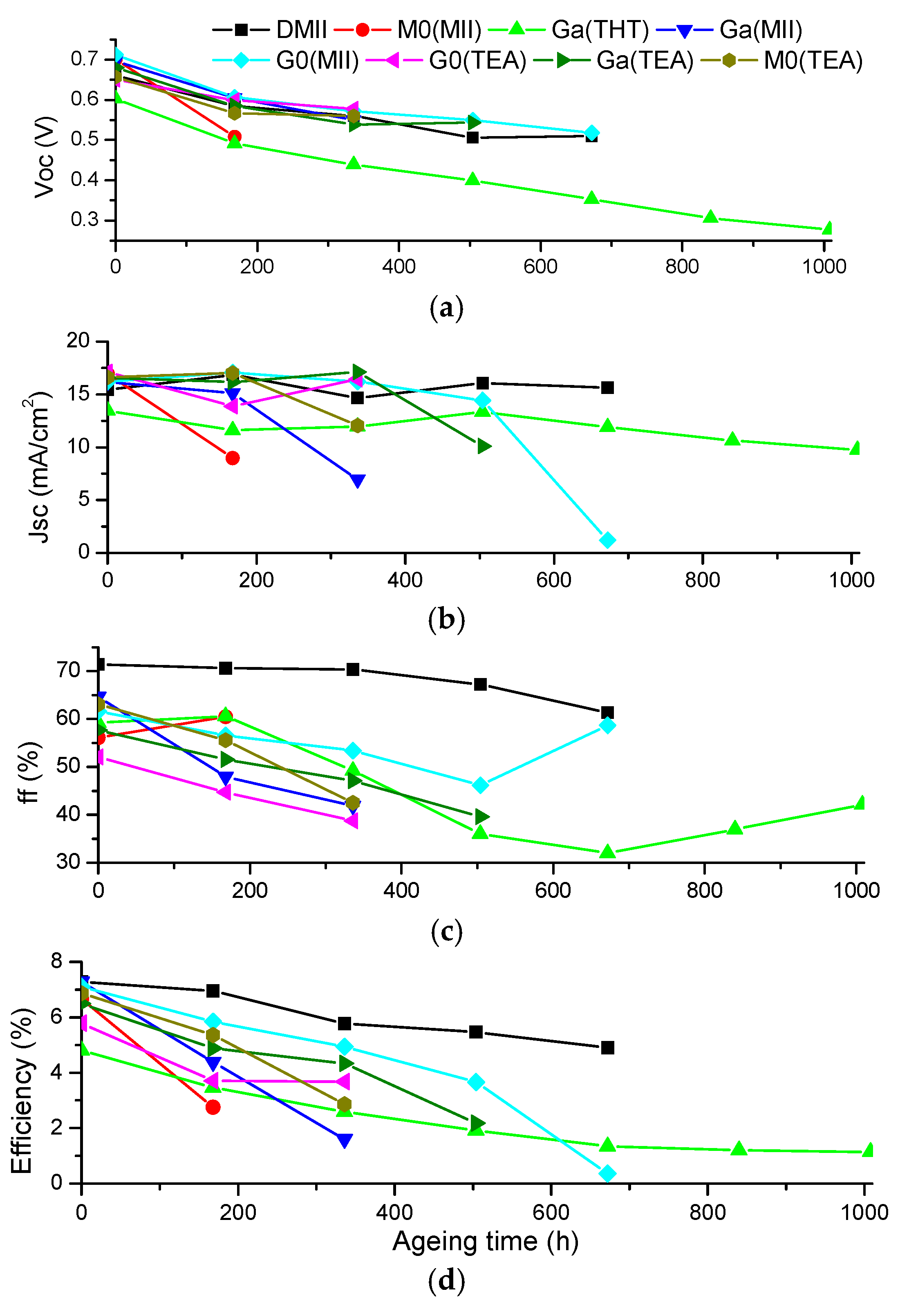
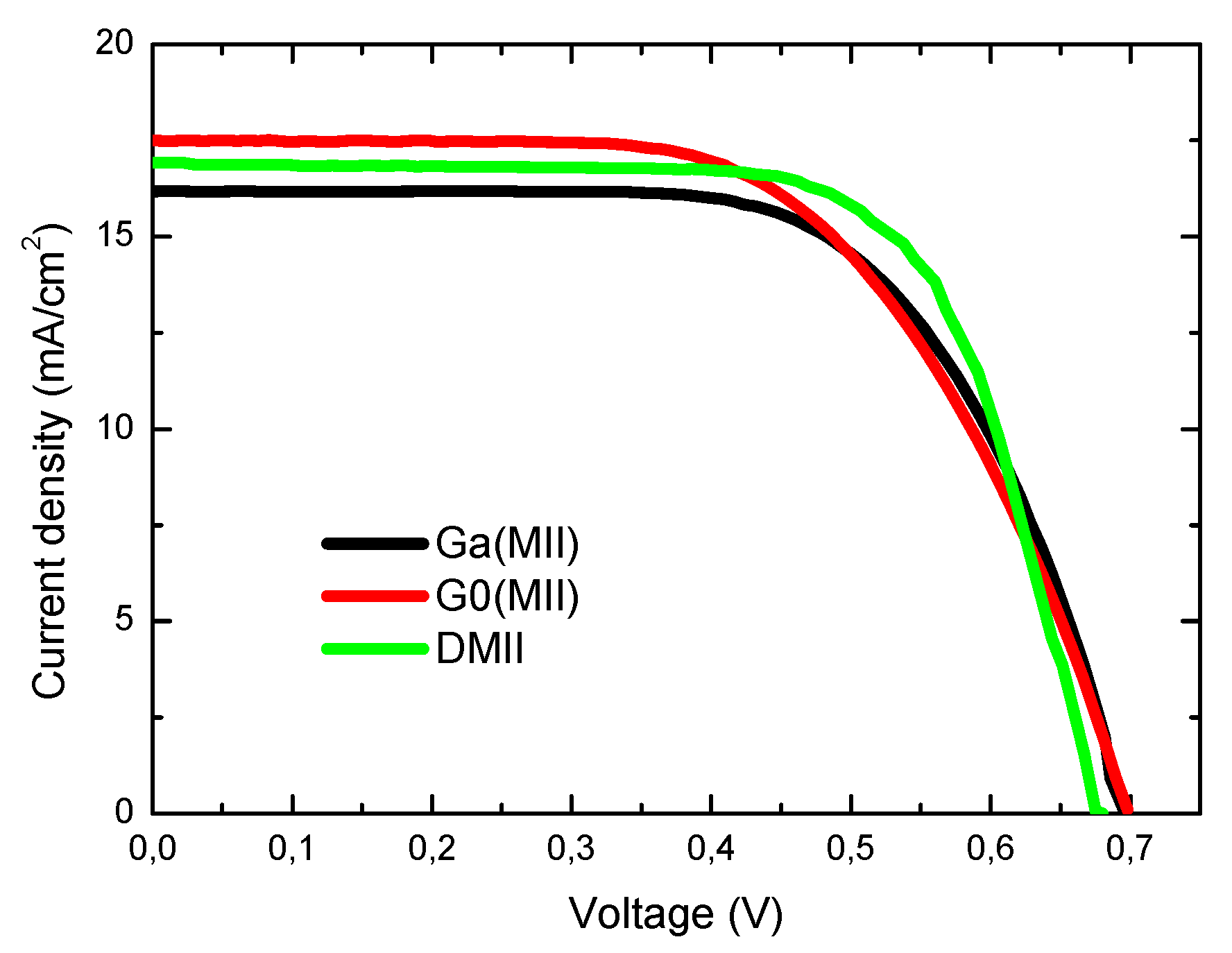
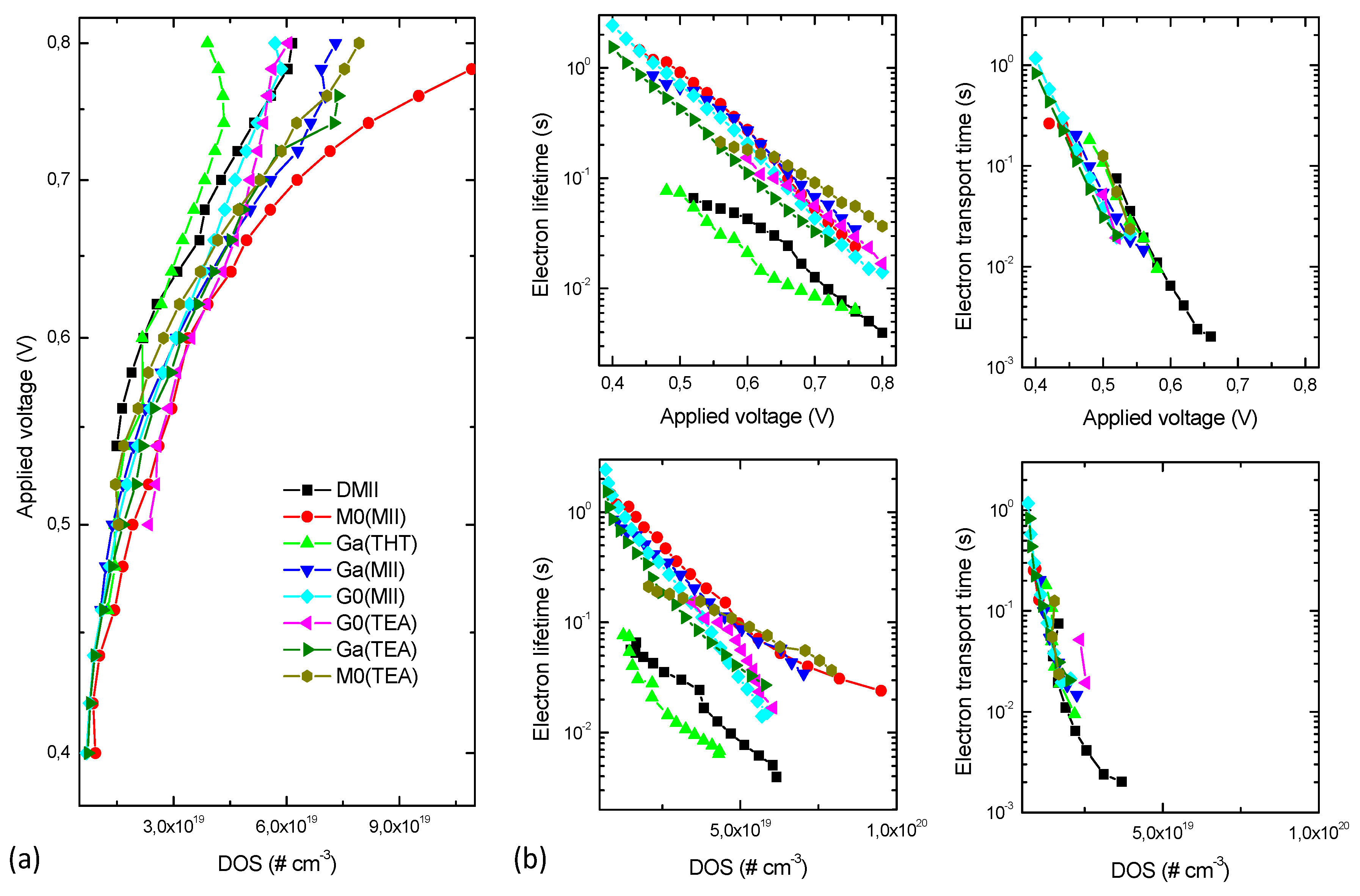
2. Results and Discussion
3. Conclusions
4. Experimental Section
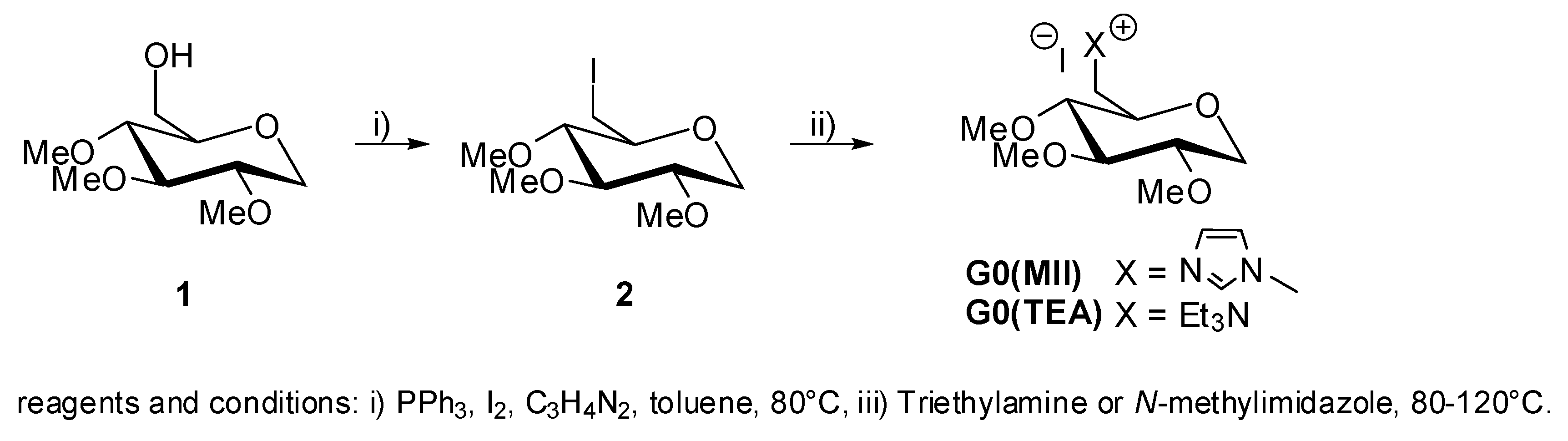
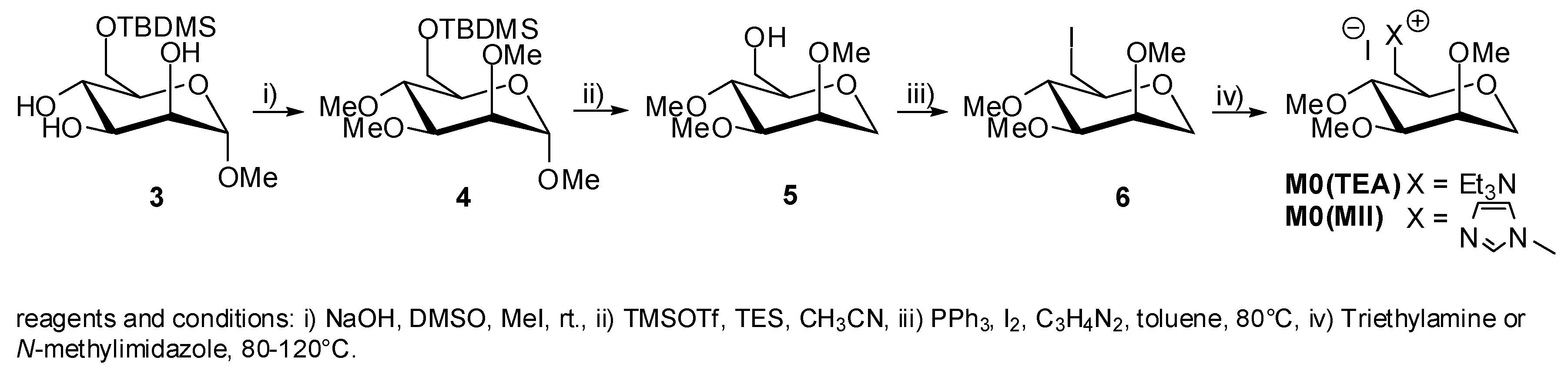
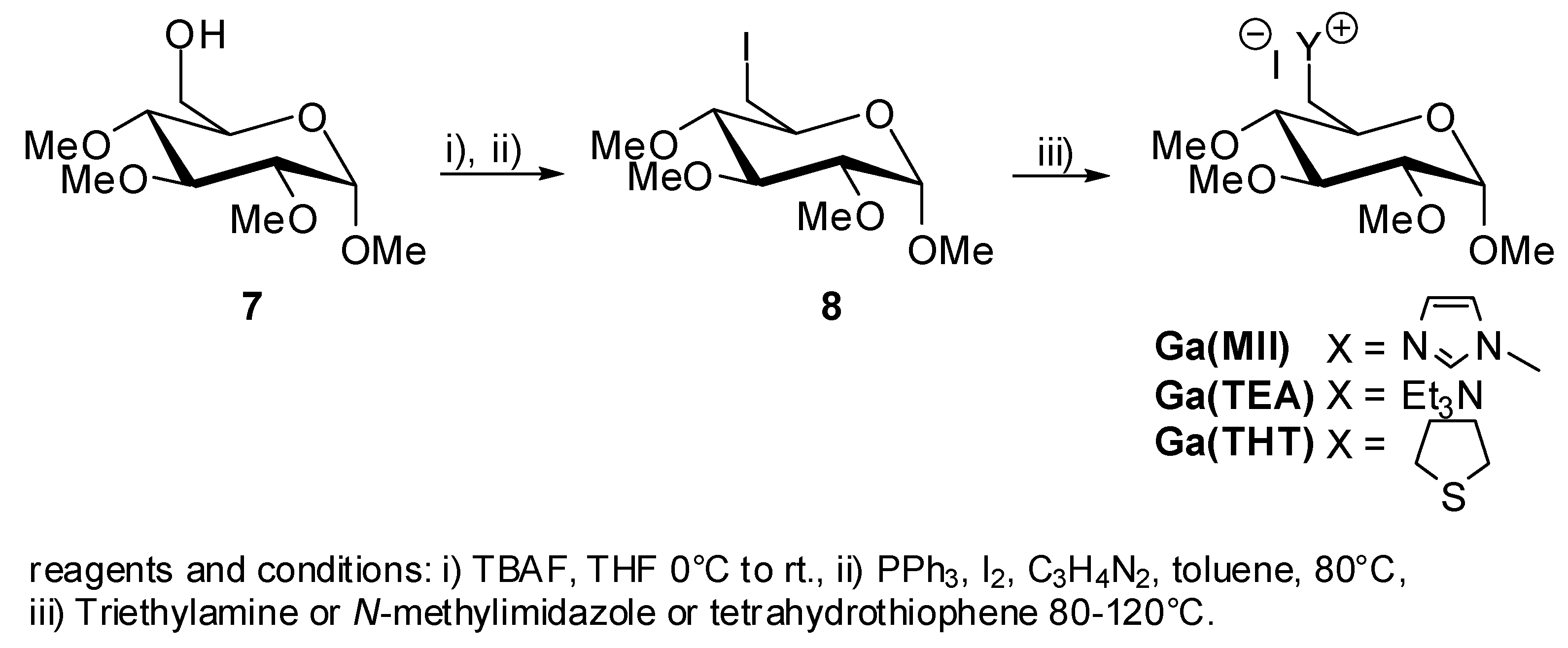
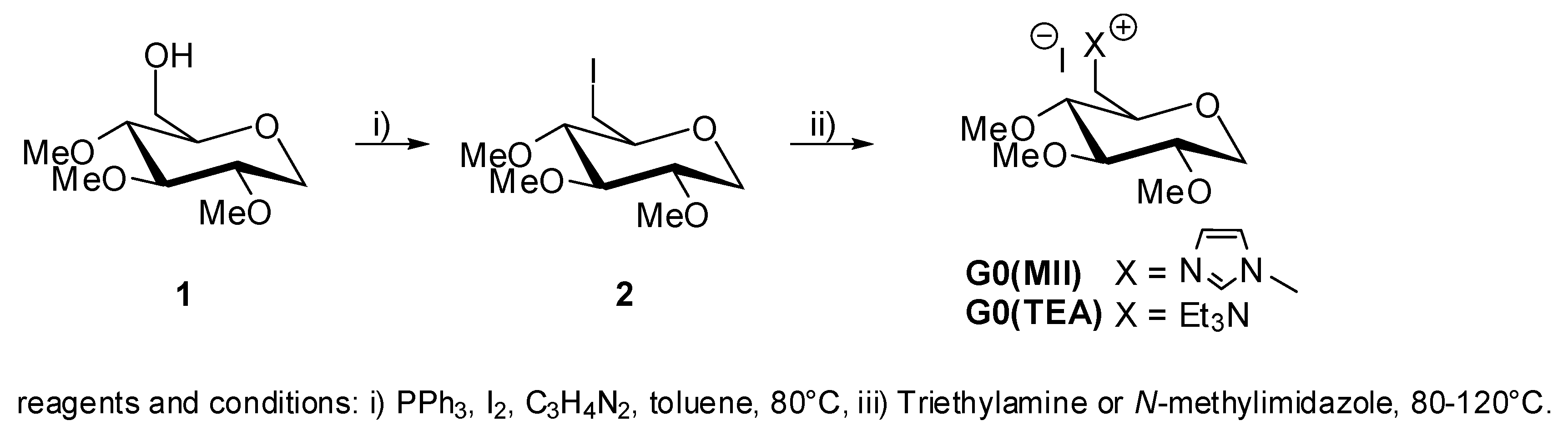
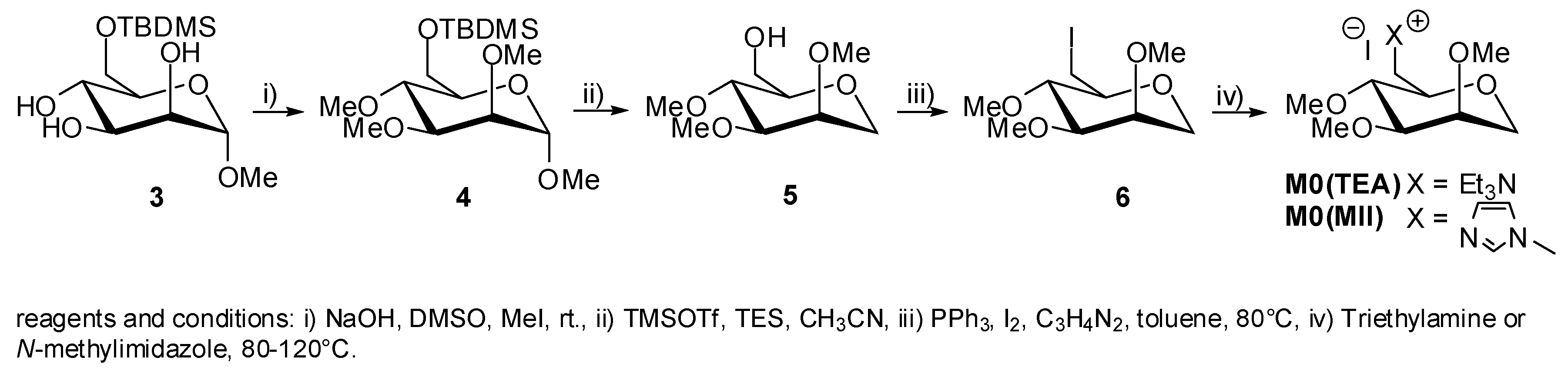
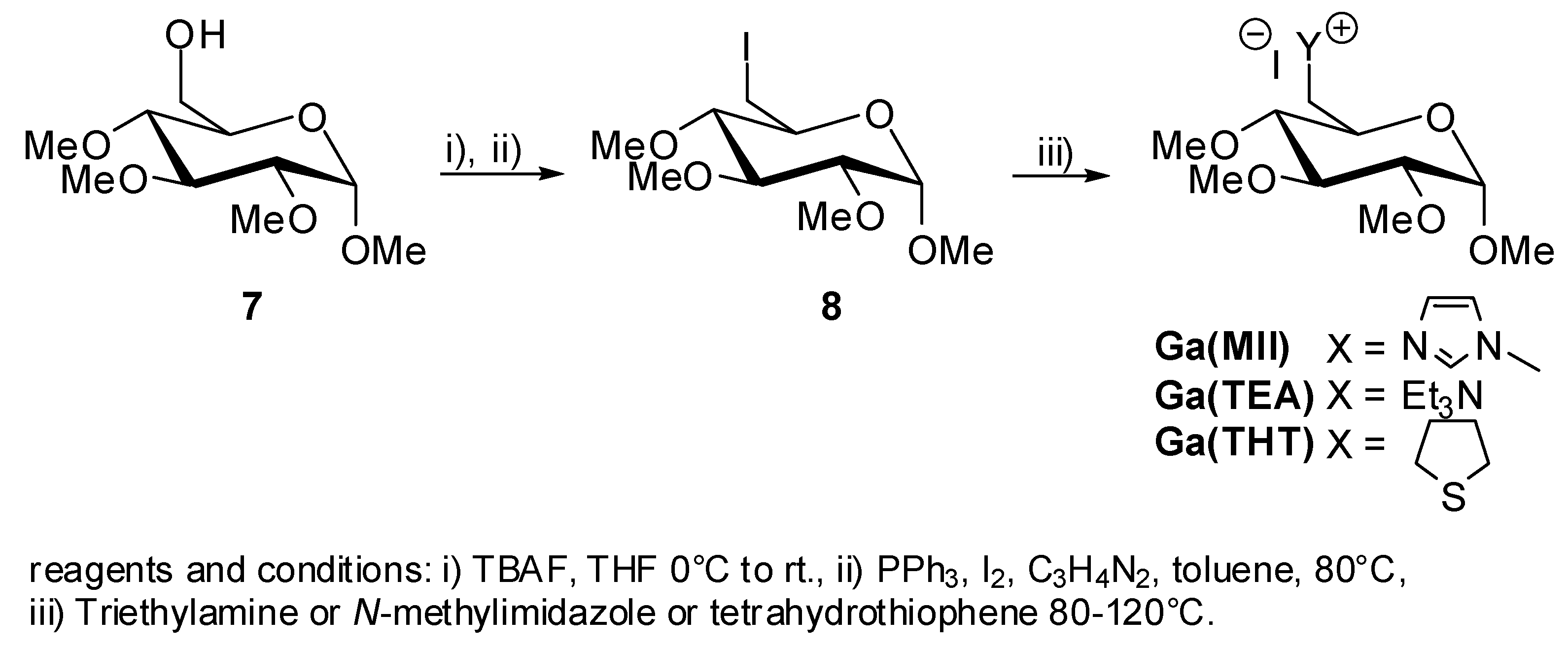
5. Synthesis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Green, M.A. Third Generation Photovoltaics: Advanced Solar Energy Conversion; Springer: Berlin, Germany, 2006; Volume 12. [Google Scholar]
- Lewis, N.S. Toward cost-effective solar energy use. Science 2007, 315, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Schiermeier, Q.; Tollefson, J.; Scully, T.; Witze, A.; Morton, O. Energy alternatives: Electricity without carbon. Nat. News 2008, 454, 816–823. [Google Scholar] [CrossRef] [PubMed]
- World Energy Outlook; International Energy Agency: Paris, France, 2013.
- Grätzel, M. Solar energy conversion by dye-sensitized photovoltaic cells. Inorg. Chem. 2005, 44, 6841–6851. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Kay, A.; Rodicio, I.; Humphry-Baker, R.; Müller, E.; Liska, P.; Vlachopoulos, N.; Grätzel, M. Conversion of light to electricity by cis-X2bis (2,2′-bipyridyl-4, 4′-dicarboxylate)ruthenium(II) charge-transfer sensitizers (X = Cl−, Br−, I−, CN−, and SCN−) on nanocrystalline titanium dioxide electrodes. J. Am. Chem. Soc. 1993, 115, 6382–6390. [Google Scholar] [CrossRef]
- Kakiage, K.; Aoyama, Y.; Yano, T.; Oya, K.; Fujisawa, J.; Hanaya, M. Highly-efficient dye-sensitized solar cells with collaborative sensitization by silyl-anchor and carboxy-anchor dyes. Chem. Commun. 2015, 51, 15894–15897. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, F.; Chhor, S.; Marchioro, A.; Moser, J.-E.; Graetzel, M. Butyronitrile-based electrolyte for dye-sensitized solar cells. J. Am. Chem. Soc. 2011, 133, 13103–13109. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhou, D.; Shi, Y.; Si, X.; Wang, Y.; Wang, P. Stable and efficient dye-sensitized solar cells: Photophysical and electrical characterizations. Energy Environ. Sci. 2010, 3, 1722–1725. [Google Scholar] [CrossRef]
- Marszalek, M.; Arendse, F.D.; Decoppet, J.-D.; Babkair, S.S.; Ansari, A.A.; Habib, S.S.; Wang, M.; Zakeeruddin, S.M.; Grätzel, M. Ionic Liquid-Sulfolane Composite Electrolytes for High-Performance and Stable Dye-Sensitized Solar Cells. Adv. Energy Mater. 2014, 4, 1301235–1301242. [Google Scholar] [CrossRef]
- Stergiopoulos, T.; Kontos, A.G.; Jiang, N.; Milliken, D.; Desilvestro, H.; Likodimos, V.; Falaras, P. High boiling point solvent-based dye solar cells pass a harsh thermal ageing test. Sol. Energy Mater. Sol. Cells 2016, 144, 457–466. [Google Scholar] [CrossRef]
- Zakeeruddin, S.M.; Graetzel, M. Solvent-Free Ionic Liquid Electrolytes for Mesoscopic Dye-Sensitized Solar Cells. Adv. Funct. Mater. 2009, 19, 2187–2202. [Google Scholar] [CrossRef]
- Lee, C.-P.; Chen, P.-Y.; Ho, K.-C. Ionic Liquid Based Electrolytes for Dye-Sensitized Solar Cells, Ionic Liquids: Theory, Properties, New Approaches; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- Wyss, P.; Moehl, T.; Zakeeruddin, S.M.; Grätzel, M. Influence of cations of the electrolyte on the performance and stability of dye sensitized solar cells. J. Mater. Chem. 2012, 22, 24424–24429. [Google Scholar] [CrossRef]
- Strappaveccia, G.; Ismalaj, E.; Petrucci, C.; Lanari, D.; Marrocchi, A.; Drees, M.; Facchetti, A.; Vaccaro, L. A biomass-derived safe medium to replace toxic dipolar solvents and access cleaner Heck coupling reactions. Green Chem. 2015, 17, 365–372. [Google Scholar] [CrossRef]
- Ghavre, M.; Byrne, O.; Altes, L.; Surolia, P.K.; Spulak, M.; Quilty, B.; Thampi, K.R.; Gathergood, N. Low toxicity functionalised imidazolium salts for task specific ionic liquid electrolytes in dye-sensitised solar cells: a step towards less hazardous energy production. Green Chem. 2014, 16, 2252–2265. [Google Scholar] [CrossRef]
- Kuang, D.; Klein, C.; Ito, S.; Moser, J.-E.; Humphry-Baker, R.; Evans, N.; Duriaux, F.; Graetzel, C.; Zakeeruddin, S.M.; Grätzel, M. High-Efficiency and Stable Mesoscopic Dye-Sensitized Solar Cells Based on a High Molar Extinction Coefficient Ruthenium Sensitizer and Nonvolatile Electrolyte. Adv. Mater. 2007, 19, 1133–1137. [Google Scholar] [CrossRef]
- Wilkes, J.S. A short history of ionic liquids—From molten salts to neoteric solvents. Green Chem. 2002, 4, 73–80. [Google Scholar] [CrossRef]
- Mudring, A.-V. Solidification of ionic liquids: Theory and techniques. Aust. J. Chem. 2010, 63, 544–564. [Google Scholar] [CrossRef]
- Wu, J.; Lan, Z.; Lin, J.; Huang, M.; Huang, Y.; Fan, L.; Luo, G. Electrolytes in Dye-Sensitized Solar Cells. Chem. Rev. 2015, 115, 2136–2173. [Google Scholar] [CrossRef] [PubMed]
- Ue, M.; Ida, K.; Mori, S. Electrochemical Properties of Organic Liquid Electrolytes Based on Quaternary Onium Salts for Electrical Double-Layer Capacitors. J. Electrochem. Soc. 1994, 141, 2989–2996. [Google Scholar] [CrossRef]
- Kambe, S.; Nakade, S.; Kitamura, T.; Wada, Y.; Yanagida, S. Influence of the electrolytes on electron transport in mesoporous TiO2-electrolyte systems. J. Phys. Chem. B 2002, 106, 2967–2972. [Google Scholar] [CrossRef]
- Son, K.M.; Kang, M.G.; Vittal, R.; Lee, J.; Kim, K.-J. Effects of substituents of imidazolium cations on the performance of dye-sensitized TiO2 solar cells. J. Appl. Electrochem. 2008, 38, 1647–1652. [Google Scholar] [CrossRef]
- Wang, M.; Chen, P.; Humphry-Baker, R.; Zakeeruddin, S.M.; Grätzel, M. The Influence of Charge Transport and Recombination on the Performance of Dye-Sensitized Solar Cells. ChemPhysChem 2009, 10, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Flasque, M.; Van Nhien, A.N.; Swiatowska, J.; Seyeux, A.; Davoisne, C.; Sauvage, F. Interface Stability of a TiO2/3-Methoxypropionitrile-Based Electrolyte: First Evidence for Solid Electrolyte Interphase Formation and Implications. ChemPhysChem 2014, 15, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Bai, Y.; Yu, Q.; Cheng, Y.; Liu, S.; Shi, D.; Gao, F.; Wang, P. Dye-sensitized solar cells with a high absorptivity ruthenium sensitizer featuring a 2-(hexylthio) thiophene conjugated bipyridine. J. Phys. Chem. C 2009, 113, 6290–6297. [Google Scholar] [CrossRef]
- Peter, L.M. Dye-sensitized nanocrystalline solar cells. Phys. Chem. Chem. Phys. 2007, 9, 2630–2642. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, B.; Durrant, J.R.; Sommeling, P.M.; Bakker, N.J. Influence of the TiCl4 treatment on nanocrystalline TiO2 films in dye-sensitized solar cells. 2. Charge density, band edge shifts, and quantification of recombination losses at short circuit. J. Phys. Chem. C 2007, 111, 14001–14010. [Google Scholar]
- Bisquert, J.; Fabregat-Santiago, F.; Mora-Seró, I.; Garcia-Belmonte, G.; Giménez, S. Electron lifetime in dye-sensitized solar cells: Theory and interpretation of measurements. J. Phys. Chem. C 2009, 113, 17278–17290. [Google Scholar] [CrossRef]
- Ito, S.; Murakami, T.N.; Comte, P.; Liska, P.; Grätzel, C.; Nazeeruddin, M.K.; Grätzel, M. Fabrication of thin film dye sensitized solar cells with solar to electric power conversion efficiency over 10%. Thin Solid Films 2008, 516, 4613–4619. [Google Scholar] [CrossRef]
- Hauch, A.; Georg, A. Diffusion in the electrolyte and charge-transfer reaction at the platinum electrode in dye-sensitized solar cells. Electrochim. Acta 2001, 46, 3457–3466. [Google Scholar] [CrossRef]
- Fabregat-Santiago, F.; Bisquert, J.; Garcia-Belmonte, G.; Boschloo, G.; Hagfeldt, A. Influence of electrolyte in transport and recombination in dye-sensitized solar cells studied by impedance spectroscopy. Sol. Energy Mater. Sol. Cells 2005, 87, 117–131. [Google Scholar]
- Fabregat-Santiago, F.; Bisquert, J. Dye-Sensitized Solar Cells; EPFL Press: Lausanne, Switzerland, 2010. [Google Scholar]
- Poletti, L.; Chiappe, C.; Lay, L.; Pieraccini, D.; Polito, L.; Russo, G. Glucose-derived ionic liquids: Exploring low-cost sources for novel chiral solvents. Green Chem. 2007, 9, 337–341. [Google Scholar] [CrossRef]
- El-Badri, M.H.; Willenbring, D.; Tantillo, D.J.; Gervay-Hague, J. Mechanistic studies on the stereoselective formation of β-mannosides from mannosyl iodides using α-deuterium kinetic isotope effects. J. Org. Chem. 2007, 72, 4663–4672. [Google Scholar] [CrossRef] [PubMed]
- Boultadakis-Arapinis, M.; Prost, E.; Gandon, V.; Lemoine, P.; Turcaud, S.; Micouin, L.; Lecourt, T. Carbene-Mediated Functionalization of the Anomeric C-H Bond of Carbohydrates: Scope and Limitations. Chem. Eur. J. 2013, 19, 6052–6066. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Melting Point (°C) | Viscosity at 25 °C (cP) | Conductivity (S/cm) | I3− Diffusion Coefficient (cm2/s) | I− Diffusion Coefficient (cm2/s) | Rct (Ω·cm2) | |
|---|---|---|---|---|---|---|---|
| CV | EIS | ||||||
DMII | 93.6 | 2.1 | 7.1 × 10−3 | 3.3 × 10−4 | 1.6 × 10−5 | 9.4 × 10−4 | 1.70 |
M0(MII)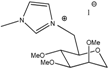 | Liquid | 3.8 | 5.0 × 10−3 | 1.2 × 10−4 | 9.1 × 10−6 | 3.3 × 10−4 | 8.80 |
M0(TEA)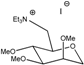 | 140 | 3.3 | 2.3 × 10−3 | 1.3 × 10−4 | 1.2 × 10−5 | 9.4 × 10−4 | 8.30 |
Ga(MII) | Liquid | 2.6 | 3.5 × 10−3 | 1.6 × 10−4 | 9.3 × 10−6 | 6.0 × 10−4 | 3.80 |
G0(MII) | 134 | 3.2 | 3.5 × 10−3 | 1.4 × 10−4 | 9.7 × 10−6 | 7.1 × 10−4 | 6.30 |
Ga(THT)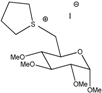 | 136 | - | - | - | 8.1 × 10−6 | - | 2.90 |
Ga(TEA)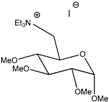 | 118 | 3.5 | 4.1 × 10−3 | 1.2 × 10−4 | 6.6 × 10−6 | 7.9 × 10−4 | 5.50 |
G0(TEA)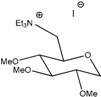 | 138 | 3.2 | 3.1 × 10−3 | 1.5 × 10−4 | 1.2 × 10−5 | 8.8 × 10−4 | 8.30 |
| Characteristic | Ga(MII) | G0(MII) | M0(TEA) | M0(MII) | Ga(TEA) | G0(TEA) | Ga(THT) | DMII |
|---|---|---|---|---|---|---|---|---|
| Voc (V) | 0.696 | 0.698 | 0.656 | 0.701 | 0.680 | 0.650 | 0.581 | 0.680 |
| Jsc (mA/cm2) | 16.2 | 17.5 | 16.6 | 16.9 | 16.5 | 17.1 | 16.1 | 16.9 |
| ff (%) | 64.7 | 60.0 | 63.0 | 56.1 | 57.7 | 52.1 | 53.2 | 69.3 |
| η (%) | 7.3 | 7.3 | 6.9 | 6.7 | 6.5 | 5.8 | 5.0 | 8.0 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sagaidak, I.; Huertas, G.; Nguyen Van Nhien, A.; Sauvage, F. Towards Renewable Iodide Sources for Electrolytes in Dye-Sensitized Solar Cells. Energies 2016, 9, 241. https://doi.org/10.3390/en9040241
Sagaidak I, Huertas G, Nguyen Van Nhien A, Sauvage F. Towards Renewable Iodide Sources for Electrolytes in Dye-Sensitized Solar Cells. Energies. 2016; 9(4):241. https://doi.org/10.3390/en9040241
Chicago/Turabian StyleSagaidak, Iryna, Guillaume Huertas, Albert Nguyen Van Nhien, and Frédéric Sauvage. 2016. "Towards Renewable Iodide Sources for Electrolytes in Dye-Sensitized Solar Cells" Energies 9, no. 4: 241. https://doi.org/10.3390/en9040241
APA StyleSagaidak, I., Huertas, G., Nguyen Van Nhien, A., & Sauvage, F. (2016). Towards Renewable Iodide Sources for Electrolytes in Dye-Sensitized Solar Cells. Energies, 9(4), 241. https://doi.org/10.3390/en9040241