
Improvement of Process Conditions for H2 Production by Chemical Looping Reforming

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
- 1.
- Partial oxidation step:
- 2.
- Carrier regeneration (REG) step:
2. Materials and Methods
2.1. Material Preparation
2.2. Thermogravimetric Analysis (TGA)
- Heating in the Ar atmosphere at 30 °C/min;
- Reduction for 40 min in 4 vol.% CH4/Ar at a flow rate of 180 mL/min;
- Oxidation for 15 min with an air flow rate of 100 mL/min.
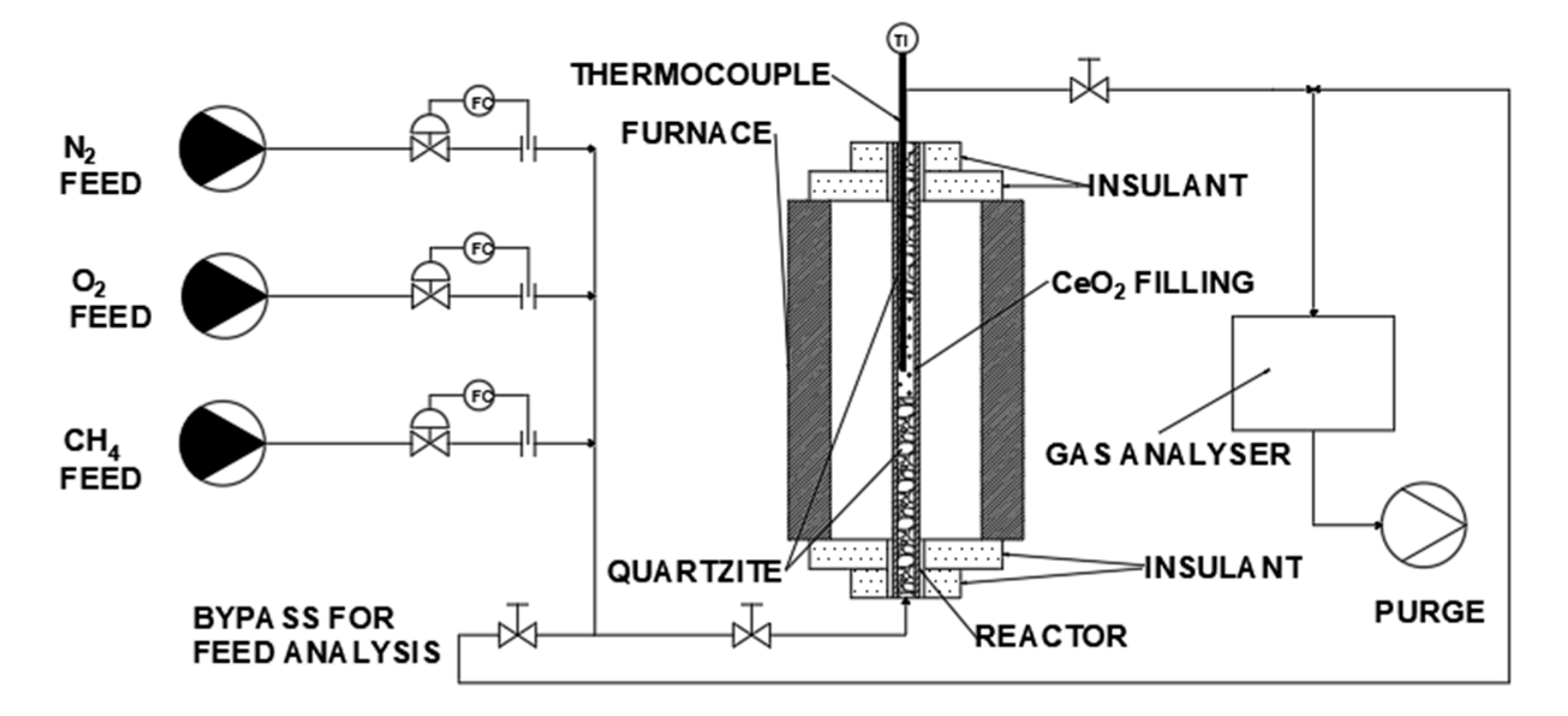
2.3. Chemical Looping Tests
2.4. Data Analysis
3. Results and Discussion
3.1. TGA
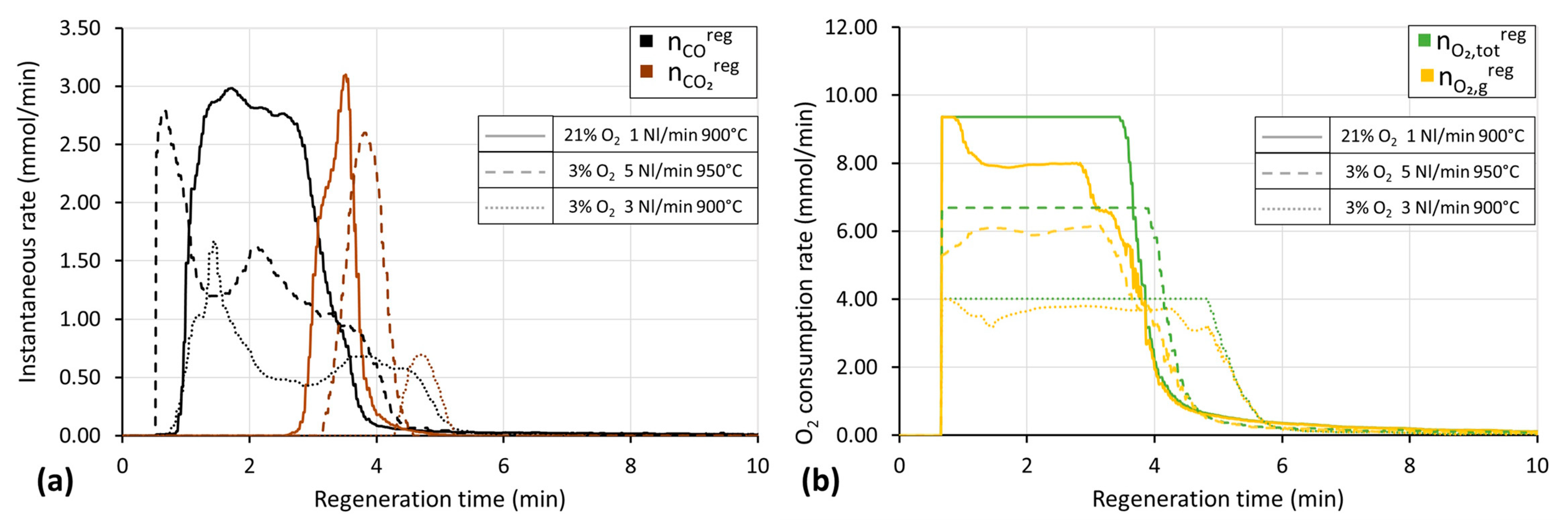
3.2. Chemical Looping Tests
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Nomenclature
| Abs | Absolute value, [-] |
| ID | Internal diameter, mm |
| m | Mass, g |
| Molar flowrate, mmol/min | |
| to | Initial time, min |
| t | Time, min |
| T | Temperature, C or K |
| τreact | Characteristic reactor time (min) |
| Y | Molar fraction |
| Subscript | |
| 0 | Initial value |
| Acc | Accumulation |
| C | Carbon formed during partial oxidation |
| F | Final |
| G | Generative term |
| i | Species i |
| in | Inlet value |
| M | Mass, g |
| R | Reactant |
| St | Stoichiometric |
| tot | Total |
| Superscripts | |
| Acc | Accumulation |
| C | Cracking contribution |
| CO2 | Regeneration step in CO2 |
| PO | Partial oxidation |
| Reg | Regeneration |
| Tot | Total |
| Greek symbols | |
| δ | Bed-average non-stoichiometric coefficient |
| Ƞ | Selectivity of the partial oxidation, [-] |
| Conversion of species i | |
| Oxygen mass fraction | |
| Acronyms | |
| SMR | Steam methane reforming |
| ASU | Air separation unit |
| TPO | Temperature-programmed oxidation |
| TGA | Thermogravimetric analysis |
| OC | Oxygen carrier |
| DR | Dry reforming |
References
- Lunsford, J.H. Catalytic Conversion of Methane to More Useful Chemicals and Fuels: A Challenge for the 21st Century. Catal. Today 2000, 63, 165–174. [Google Scholar] [CrossRef]
- Ozbilen, A.; Dincer, I.; Rosen, M.A. Comparative Environmental Impact and Efficiency Assessment of Selected Hydrogen Production Methods. Environ. Impact Assess. Rev. 2013, 42, 1–9. [Google Scholar] [CrossRef]
- Noh, Y.G.; Lee, Y.J.; Kim, J.; Kim, Y.K.; Ha, J.S.; Kalanur, S.S.; Seo, H. Enhanced Efficiency in CO2-Free Hydrogen Production from Methane in a Molten Liquid Alloy Bubble Column Reactor with Zirconia Beads. Chem. Eng. J. 2021, 428, 131095. [Google Scholar] [CrossRef]
- Abdin, Z.; Zafaranloo, A.; Rafiee, A.; Mérida, W.; Lipiński, W.; Khalilpour, K.R. Hydrogen as an Energy Vector. Renew. Sustain. Energy Rev. 2020, 120, 109620. [Google Scholar] [CrossRef]
- Boscherini, M.; Storione, A.; Minelli, M.; Miccio, F.; Doghieri, F. New Perspectives on Catalytic Hydrogen Production by the Reforming, Partial Oxidation and Decomposition of Methane and Biogas. Energies 2023, 16, 6375. [Google Scholar] [CrossRef]
- Iminabo, J.T.; Iminabo, M.; Yip, A.C.K.; Pang, S. Hydrogen-Rich Syngas Production via Dry and Steam Reforming of Methane in Simulated Producer Gas over ZSM-5-Supported Trimetallic Catalysts. Energies 2023, 16, 7518. [Google Scholar] [CrossRef]
- Luberti, M.; Brown, A.; Balsamo, M.; Capocelli, M. Numerical Analysis of VPSA Technology Retrofitted to Steam Reforming Hydrogen Plants to Capture CO2 and Produce Blue H2. Energies 2022, 15, 1091. [Google Scholar] [CrossRef]
- González Palencia, J.C.; Itoi, Y.; Araki, M. Design of a Hydrogen Production System Considering Energy Consumption, Water Consumption, CO2 Emissions and Cost. Energies 2022, 15, 7938. [Google Scholar] [CrossRef]
- Cherif, A.; Atwair, M.; Atsbha, T.A.; Zarei, M.; Duncan, I.J.; Nebbali, R.; Sen, F.; Lee, C.J. Enabling Low-Carbon Membrane Steam Methane Reforming: Comparative Analysis and Multi-Objective NSGA-II-Integrated Bayesian Optimization. Energy Convers. Manag. 2023, 297, 117718. [Google Scholar] [CrossRef]
- Lu, Y.; Lee, T. Influence of the Feed Gas Composition on the Fischer-Tropsch Synthesis in Commercial Operations. J. Nat. Gas. Chem. 2007, 16, 329–341. [Google Scholar] [CrossRef]
- Bozzano, G.; Manenti, F. Efficient Methanol Synthesis: Perspectives, Technologies and Optimization Strategies. Prog. Energy Combust. Sci. 2016, 56, 71–105. [Google Scholar] [CrossRef]
- Baltrusaitis, J.; Luyben, W.L. Methane Conversion to Syngas for Gas-to-Liquids (GTL): Is Sustainable CO2 Reuse via Dry Methane Reforming (DMR) Cost Competitive with SMR and ATR Processes? ACS Sustain. Chem. Eng. 2015, 3, 2100–2111. [Google Scholar] [CrossRef]
- Osman, A.I. Catalytic Hydrogen Production from Methane Partial Oxidation: Mechanism and Kinetic Study. Chem. Eng. Technol. 2020, 43, 641–648. [Google Scholar] [CrossRef]
- Honnery, D.; Moriarty, P.J.P. Syngas Production By Short Contact Time Catalytic Partial Oxidation of Methane. In Hydrogen Production: Prospects and Processes; Honnery, D.R., Moriarty, P., Eds.; Nova Science Publishers Inc.: Hauppauge, NY, USA, 2011; pp. 95–139. ISBN 9781621002468. [Google Scholar]
- Gao, M.; Xiao, Y.; Ding, L.; Gao, Y.; Dai, Z.; Yu, G.; Yang, W.; Wang, F. Understanding of the Effect of Oxygen on Soot Formation during Non-Catalytic Partial Oxidation Process. Chem. Eng. Sci. 2024, 284, 119499. [Google Scholar] [CrossRef]
- Makaryan, I.A.; Salgansky, E.A.; Arutyunov, V.S.; Sedov, I.V. Non-Catalytic Partial Oxidation of Hydrocarbon Gases to Syngas and Hydrogen: A Systematic Review. Energies 2023, 16, 2916. [Google Scholar] [CrossRef]
- Basini, L.; Guarinoni, A.; Aragno, A. Molecular and Temperature Aspects in Catalytic Partial Oxidation of Methane. J. Catal. 2000, 190, 284–295. [Google Scholar] [CrossRef]
- Wen, C.; Liu, Y.; Guo, Y.; Wang, Y.; Lu, G. Strategy to Eliminate Catalyst Hot-Spots in the Partial Oxidation of Methane: Enhancing Its Activity for Direct Hydrogen Production by Reducing the Reactivity of Lattice Oxygen. Chem. Commun. 2010, 46, 880–882. [Google Scholar] [CrossRef]
- Zhu, Z.; Guo, W.; Zhang, Y.; Pan, C.; Xu, J.; Zhu, Y.; Lou, Y. Research Progress on Methane Conversion Coupling Photocatalysis and Thermocatalysis. Carbon. Energy 2021, 3, 519–540. [Google Scholar] [CrossRef]
- Siang, T.J.; Jalil, A.A.; Liew, S.Y.; Owgi, A.H.K.; Rahman, A.F.A. A Review on State-of-the-Art Catalysts for Methane Partial Oxidation to Syngas Production. Catal. Rev. Sci. Eng. 2024, 66, 343–399. [Google Scholar] [CrossRef]
- Bharadwaj, S.S.; Schmidt, L.D. Catalytic Partial Oxidation of Natural Gas to Syngas. Fuel Process. Technol. 1995, 42, 109–127. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Efstathiou, A.M. Hydrogen Production Technologies: Current State and Future Developments. Conf. Pap. Energy 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Li, D.; Xu, R.; Gu, Z.; Zhu, X.; Qing, S.; Li, K. Chemical-Looping Conversion of Methane: A Review. Energy Technol. 2020, 8, 1900925. [Google Scholar] [CrossRef]
- Haeussler, A.; Abanades, S.; Julbe, A.; Jouannaux, J.; Cartoixa, B. Two-Step CO2 and H2O Splitting Using Perovskite-Coated Ceria Foam for Enhanced Green Fuel Production in a Porous Volumetric Solar Reactor. J. CO2 Util. 2020, 41, 101257. [Google Scholar] [CrossRef]
- Khan, M.N.; Cloete, S.; Amini, S. Efficient Production of Clean Power and Hydrogen through Synergistic Integration of Chemical Looping Combustion and Reforming. Energies 2020, 13, 3443. [Google Scholar] [CrossRef]
- Padula, S.; Tregambi, C.; Troiano, M.; Di Benedetto, A.; Salatino, P.; Landi, G.; Solimene, R. Chemical Looping Reforming with Perovskite-Based Catalysts for Thermochemical Energy Storage. Energies 2022, 15, 8556. [Google Scholar] [CrossRef]
- Czakiert, T.; Krzywanski, J.; Zylka, A.; Nowak, W. Chemical Looping Combustion: A Brief Overview. Energies 2022, 15, 1563. [Google Scholar] [CrossRef]
- Tian, M.; Wang, C.; Han, Y.; Wang, X. Recent Advances of Oxygen Carriers for Chemical Looping Reforming of Methane. ChemCatChem 2021, 13, 1615–1637. [Google Scholar] [CrossRef]
- Chang, W.; Hu, Y.; Xu, W.; Huang, C.; Chen, H.; He, J.; Han, Y.; Zhu, Y.; Ma, X.; Wang, X. Recent Advances of Oxygen Carriers for Hydrogen Production via Chemical Looping Water-Splitting. Catalysts 2023, 13, 279. [Google Scholar] [CrossRef]
- Storione, A.; Minelli, M.; Doghieri, F.; Landi, E.; Miccio, F. Thermodynamic Study on the Feasibility of a New Combined Chemical Looping Process for Syngas Production. Chem. Eng. Trans. 2021, 86, 1267–1272. [Google Scholar] [CrossRef]
- Kang, D.; Lee, M.; Lim, H.S.; Lee, J.W. Chemical Looping Partial Oxidation of Methane with CO2 Utilization on the Ceria-Enhanced Mesoporous Fe2O3 Oxygen Carrier. Fuel 2018, 215, 787–798. [Google Scholar] [CrossRef]
- García-García, F.R.; Metcalfe, I.S. Chemical Looping Dry Reforming of Methane Using Mixed Oxides of Iron and Cerium: Operation Window. Catal. Commun. 2021, 160, 106356. [Google Scholar] [CrossRef]
- Cao, Z.; Zhu, X.; Li, K.; Wei, Y.; He, F.; Wang, H. Moderate-Temperature Chemical Looping Splitting of CO2 and H2O for Syngas Generation. Chem. Eng. J. 2020, 397, 125393. [Google Scholar] [CrossRef]
- Pant, K.K.; Jain, R.; Jain, S. Renewable Hydrogen Production by Steam Reforming of Glycerol over Ni/CeO2 Catalyst Prepared by Precipitation Deposition Method. Korean J. Chem. Eng. 2011, 28, 1859–1866. [Google Scholar] [CrossRef]
- Löfberg, A.; Guerrero-Caballero, J.; Kane, T.; Rubbens, A.; Jalowiecki-Duhamel, L. Ni/CeO2 Based Catalysts as Oxygen Vectors for the Chemical Looping Dry Reforming of Methane for Syngas Production. Appl. Catal. B 2017, 212, 159–174. [Google Scholar] [CrossRef]
- Guerrero-Caballero, J.; Kane, T.; Haidar, N.; Jalowiecki-Duhamel, L.; Löfberg, A. Ni, Co, Fe Supported on Ceria and Zr Doped Ceria as Oxygen Carriers for Chemical Looping Dry Reforming of Methane. Catal. Today 2019, 333, 251–258. [Google Scholar] [CrossRef]
- Haxel, G.B.; Hedrick, J.B.; Orris, G.J. Rare Earth Elements—Critical Resources for High Technology. Available online: https://pubs.usgs.gov/fs/2002/fs087-02/ (accessed on 25 May 2022).
- Li, K.; Chen, J.; Zou, D. Extraction and Recovery of Cerium from Rare Earth Ore by Solvent Extraction. In Cerium Oxide—Applications and Attributes; IntechOpen: London, UK, 2019. [Google Scholar]
- Allahkarami, E.; Rezai, B. A Literature Review of Cerium Recovery from Different Aqueous Solutions. J. Environ. Chem. Eng. 2021, 9, 104956. [Google Scholar] [CrossRef]
- Li, P.; Chen, X.; Li, Y.; Schwank, J.W. A Review on Oxygen Storage Capacity of CeO2-Based Materials: Influence Factors, Measurement Techniques, and Applications in Reactions Related to Catalytic Automotive Emissions Control. Catal. Today 2019, 327, 90–115. [Google Scholar] [CrossRef]
- Schmitt, R.; Nenning, A.; Kraynis, O.; Korobko, R.; Frenkel, A.I.; Lubomirsky, I.; Haile, S.M.; Rupp, J.L.M. A Review of Defect Structure and Chemistry in Ceria and Its Solid Solutions. Chem. Soc. Rev. 2020, 49, 554–592. [Google Scholar] [CrossRef]
- Rotaru, C.G.; Postole, G.; Florea, M.; Matei-Rutkovska, F.; Pârvulescu, V.I.; Gelin, P. Dry Reforming of Methane on Ceria Prepared by Modified Precipitation Route. Appl. Catal. A Gen. 2015, 494, 29–40. [Google Scholar] [CrossRef]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and Catalytic Applications of CeO2-Based Materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef]
- Ackermann, S.; Scheffe, J.R.; Steinfeld, A. Diffusion of Oxygen in Ceria at Elevated Temperatures and Its Application to H2O/CO2 Splitting Thermochemical Redox Cycles. J. Phys. Chem. C 2014, 118, 5216–5225. [Google Scholar] [CrossRef]
- Ramírez-Cabrera, E.; Atkinson, A.; Chadwick, D. Reactivity of Ceria, Gd- and Nb-Doped Ceria to Methane. Appl. Catal. B 2002, 36, 193–206. [Google Scholar] [CrossRef]
- Teh, L.P.; Setiabudi, H.D.; Timmiati, S.N.; Aziz, M.A.A.; Annuar, N.H.R.; Ruslan, N.N. Recent Progress in Ceria-Based Catalysts for the Dry Reforming of Methane: A Review. Chem. Eng. Sci. 2021, 242, 116606. [Google Scholar] [CrossRef]
- Miccio, F.; Landi, E.; Murri, A.N.; Minelli, M.; Doghieri, F.; Storione, A. Fluidized Bed Reforming of Methane by Chemical Looping with Cerium Oxide Oxygen Carriers. Chem. Eng. Res. Des. 2023, 191, 568–577. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S.; Rodat, S. Stepwise Solar Methane Reforming and Water-Splitting via Lattice Oxygen Transfer in Iron and Cerium Oxides. Energy Technol. 2020, 8, 1–12. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S.; Rodat, S. Solar Chemical Looping Reforming of Methane Combined with Isothermal H2O/CO2 Splitting Using Ceria Oxygen Carrier for Syngas Production. J. Energy Chem. 2020, 41, 60–72. [Google Scholar] [CrossRef]
- Gao, X.; Vidal, A.; Bayon, A.; Bader, R.; Hinkley, J.; Lipinski, W.; Tricoli, A. Efficient Ceria Nanostructures for Enhanced Solar Fuel Production via High-Temperature Thermochemical Redox Cycles. J. Mater. Chem. A Mater. 2016, 4, 9614–9624. [Google Scholar] [CrossRef]
- Nair, M.M.; Abanades, S. Tailoring Hybrid Nonstoichiometric Ceria Redox Cycle for Combined Solar Methane Reforming and Thermochemical Conversion of H2O/CO2. Energy Fuels 2016, 30, 6050–6058. [Google Scholar] [CrossRef]
- Mantripragada, H.C.; Veser, G. Hydrogen Production via Chemical Looping Dry Reforming of Methane: Process Modeling and Systems Analysis. AIChE J. 2022, 68, e17612. [Google Scholar] [CrossRef]
- Aramouni, N.A.K.; Touma, J.G.; Tarboush, B.A.; Zeaiter, J.; Ahmad, M.N. Catalyst Design for Dry Reforming of Methane: Analysis Review. Renew. Sustain. Energy Rev. 2018, 82, 2570–2585. [Google Scholar] [CrossRef]
- Warren, K.J.; Carrillo, R.J.; Greek, B.; Hill, C.M.; Scheffe, J.R. Solar Reactor Demonstration of Efficient and Selective Syngas Production via Chemical-Looping Dry Reforming of Methane over Ceria. Energy Technol. 2020, 8, 1–13. [Google Scholar] [CrossRef]
- Farooqui, A.; Bose, A.; Boaro, M.; Llorca, J.; Santarelli, M. Assessment of Integration of Methane-Reduced Ceria Chemical Looping CO2/H2O Splitting Cycle to an Oxy-Fired Power Plant. Int. J. Hydrogen Energy 2020, 45, 6184–6206. [Google Scholar] [CrossRef]
- Li, K.; Wang, H.; Wei, Y.; Yan, D. Syngas Production from Methane and Air via a Redox Process Using Ce-Fe Mixed Oxides as Oxygen Carriers. Appl. Catal. B 2010, 97, 361–372. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, K.; Wang, H.; Zhu, X.; Wei, Y.; Zheng, M.; Wang, Y. Enhanced Activity of CeO2-ZrO2 Solid Solutions for Chemical-Looping Reforming of Methane via Tuning the Macroporous Structure. Energy Fuels 2016, 30, 638–647. [Google Scholar] [CrossRef]
- Laqdiem, M.; Carrillo, A.J.; Dimitrakopoulos, G.; Balaguer, M.; Garcia-Fayos, J.; Ghoniem, A.F.; Serra, J.M. Impact of Lattice Properties on the CO2 Splitting Kinetics of Lanthanide-Doped Cerium Oxides for Chemical Looping Syngas Production. Solid. State Ion. 2023, 394, 116192. [Google Scholar] [CrossRef]
- Li, S.; Wang, X.; Cao, M.; Lu, J.; Qiu, L.; Yan, X. Engineering the Interface and Interaction Structure on Highly Coke-Resistant Ni/CeO2-Al2O3 Catalyst for Dry Reforming of Methane. Jiegou Huaxue 2022, 41, 2212007–2212014. [Google Scholar] [CrossRef]
- Khalid, M.A.A.; Abdullah, N.; Ibrahim, M.N.M.; Taib, R.M.; Rosid, S.J.M.; Shukri, N.M.; Yahaya, N.F.; Abdullah, W.N.B.W. Catalytic Pyrolysis of Waste Oil into Hydrocarbon Fuel Utilizing Cerium Oxide Catalyst. Korean J. Chem. Eng. 2022, 39, 1487–1495. [Google Scholar] [CrossRef]
- Kim, J.-K.; Kim, Y.; Park, J.-W.; Bae, J.-S.; Yoon, D.-Y.; Lee, J.-G.; Kim, J.-H.; Han, C. Preparation of CuO-CeO2-Al2O3 Catalyst with Mesopore Structure for Water Gas Shift Reaction. Korean J. Chem. Eng. 2009, 26, 32–35. [Google Scholar] [CrossRef]
- Damyanova, S.; Perez, C.A.; Schmal, M.; Bueno, J.M.C. Characterization of Ceria-Coated Alumina Carrier. Appl. Catal. A Gen. 2002, 234, 271–282. [Google Scholar] [CrossRef]
- Prakash, A.S.; Shivakumara, C.; Hegde, M.S. Single Step Preparation of CeO2/CeAlO3/γ-Al2O3 by Solution Combustion Method: Phase Evolution, Thermal Stability and Surface Modification. Mater. Sci. Eng. B 2007, 139, 55–61. [Google Scholar] [CrossRef]
- Warren, K.J.; Scheffe, J.R. Kinetic Insights into the Reduction of Ceria Facilitated via the Partial Oxidation of Methane. Mater. Today Energy 2018, 9, 39–48. [Google Scholar] [CrossRef]
- Wheeler, D.W. Kinetics and Mechanism of the Oxidation of Cerium in Air at Ambient Temperature. Corros. Sci. 2016, 111, 52–60. [Google Scholar] [CrossRef]
- Bulfin, B.; Call, F.; Vieten, J.; Roeb, M.; Sattler, C.; Shvets, I.V. Oxidation and Reduction Reaction Kinetics of Mixed Cerium Zirconium Oxides. J. Phys. Chem. C 2016, 120, 2027–2035. [Google Scholar] [CrossRef]
- Mullins, D.R. The Surface Chemistry of Cerium Oxide. Surf. Sci. Rep. 2015, 70, 42–85. [Google Scholar] [CrossRef]
- Schweke, D.; Rafailov, G.; Zalkind, S.; Azulai, O.; Rabinovitch, L.; Hayun, S. Elucidating the Role of Hydrogen Species Originating from Water Vapor in the Oxidation Mechanism of Cerium. Corros. Sci. 2022, 196, 110030. [Google Scholar] [CrossRef]
- Bulfin, B.; Lowe, A.J.; Keogh, K.A.; Murphy, B.E.; Lübben, O.; Krasnikov, S.A.; Shvets, I.V. Analytical Model of CeO2 Oxidation and Reduction. J. Phys. Chem. C 2013, 117, 24129–24137. [Google Scholar] [CrossRef]
- Zhao, Z.; Uddi, M.; Tsvetkov, N.; Yildiz, B.; Ghoniem, A.F. Redox Kinetics Study of Fuel Reduced Ceria for Chemical-Looping Water Splitting. J. Phys. Chem. C 2016, 120, 16271–16289. [Google Scholar] [CrossRef]
- Zhao, Z.; Uddi, M.; Tsvetkov, N.; Yildiz, B.; Ghoniem, A.F. Enhanced Intermediate-Temperature CO2 Splitting Using Nonstoichiometric Ceria and Ceria-Zirconia. Phys. Chem. Chem. Phys. 2017, 19, 25774–25785. [Google Scholar] [CrossRef] [PubMed]
- Grünbacher, M.; Klötzer, B.; Penner, S. CO2 Reduction by Hydrogen Pre-Reduced Acceptor-Doped Ceria. ChemPhysChem 2019, 20, 1706–1718. [Google Scholar] [CrossRef]
- Zhou, Q.; Akber, H.; Zhao, A.; Yang, F.; Liu, Z. Interaction of Water with Ceria Thin Film. ChemCatChem 2023, 15, e202300318. [Google Scholar] [CrossRef]
- Otsuka, K.; Ushiyama, T.; Yamanaka, I. Partial Oxidation of Methane Using the Redox of Cerium Oxide. Chem. Lett. 1993, 22, 1517–1520. [Google Scholar] [CrossRef]
- Haeussler, A.; Chuayboon, S.; Abanades, S. Solar Redox Cycling of Ceria in a Monolithic Reactor for Two-Step H2O/CO2 Splitting: Isothermal Methane-Induced Reduction versus Temperature-Swing Cycle. AIP Conf. Proc. 2020, 2303, 170009. [Google Scholar] [CrossRef]
- Fosheim, J.R.; Hathaway, B.J.; Davidson, J.H. High Efficiency Solar Chemical-Looping Methane Reforming with Ceria in a Fixed-Bed Reactor. Energy 2019, 169, 597–612. [Google Scholar] [CrossRef]
- Warren, K.J.; Scheffe, J.R. Role of Surface Oxygen Vacancy Concentration on the Dissociation of Methane over Nonstoichiometric Ceria. J. Phys. Chem. C 2019, 123, 13208–13218. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Case i | Case ii | Case iii | Case iiii |
|---|---|---|---|---|
| TPO (°C) | 900 | 900 | 950 | 1000 |
| TREG (°C) | 400 | 600 | 900 | 900 |
| PO rate (min−1) | 0.01 | 0.36 ± 0.01 | 0.01 | 0.08 |
| Parameter | Case i | Case ii | Case iii |
|---|---|---|---|
| TREG (°C) | 400 | 600 | 900 |
| TPO (°C) | 900 | 900 | 950–1000 |
| tREG (min) | 2.5 0.1 | 2.3 0.1 | 3.0 0.1 |
| REG rate (min−1) | 1.0 ± 0.1 | 1.2 ± 0.2 | 1.44 ± 0.05 |
| Sample | Gas | tPO (min) | ηCO2 | ηCO | ηC | CO Yield (mmol/gcarrier) | H2 Yield (mmol/gcarrier) | χCH4 [%] |
|---|---|---|---|---|---|---|---|---|
| CeO2 | O2 | 30 | 0.05 ± 0.01 | 0.68 ± 0.04 | 0.26 ± 0.04 | 1.80 ± 0.07 | 5.01 ± 0.11 | 30.4 ± 0.9 |
| CeO2 | O2 | 20 | 0.08 ± 0.02 | 0.82 ± 0.05 | 0.10 ± 0.04 | 1.24 ± 0.25 | 2.88 ± 0.50 | 25.4 ± 4.1 |
| CeO2 | CO2 | 20 | 0.08 ± 0.01 | 0.76 ± 0.03 | 0.16 ± 0.03 | 1.52 ± 0.01 | 3.61 ± 0.11 | 34.5 ± 1.2 |
| CeO2-Al2O3 | O2 | 20 | 0.27 ± 0.02 | 0.63 ± 0.01 | 0.10 ± 0.01 | 1.38 ± 0.17 | 2.71 ± 0.35 | 38.4 ± 3.8 |
| CeO2-Al2O3 | CO2 | 20 | 0.18 ± 0.02 | 0.75 ± 0.01 | 0.07 ± 0.02 | 1.33 ± 0.05 | 2.76 ± 0.12 | 31.3 ± 1.0 |
| Cycle | Accumulated C (mmol/gCeO2) | |
|---|---|---|
| O2 Regen | CO2 Regen | |
| 1 | 0 | 0.275 |
| 2 | 0.020 | 0.241 |
| 3 | 0 | 0.252 |
| 4 | 0.025 | 0.278 |
| 5 | 0.030 | 0.312 |
| 6 | 0.026 | 0.340 |
| 7 | 0.030 | 0.373 |
| 8 | (-) | 0.405 |
| 9 | (-) | 0.440 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Storione, A.; Boscherini, M.; Miccio, F.; Landi, E.; Minelli, M.; Doghieri, F. Improvement of Process Conditions for H2 Production by Chemical Looping Reforming. Energies 2024, 17, 1544. https://doi.org/10.3390/en17071544
Storione A, Boscherini M, Miccio F, Landi E, Minelli M, Doghieri F. Improvement of Process Conditions for H2 Production by Chemical Looping Reforming. Energies. 2024; 17(7):1544. https://doi.org/10.3390/en17071544
Chicago/Turabian StyleStorione, Alba, Mattia Boscherini, Francesco Miccio, Elena Landi, Matteo Minelli, and Ferruccio Doghieri. 2024. "Improvement of Process Conditions for H2 Production by Chemical Looping Reforming" Energies 17, no. 7: 1544. https://doi.org/10.3390/en17071544
APA StyleStorione, A., Boscherini, M., Miccio, F., Landi, E., Minelli, M., & Doghieri, F. (2024). Improvement of Process Conditions for H2 Production by Chemical Looping Reforming. Energies, 17(7), 1544. https://doi.org/10.3390/en17071544