
Reaction Mechanism of Pyrolysis and Combustion of Methyl Oleate: A ReaxFF-MD Analysis


Abstract
1. Introduction
2. Models and Methods
3. Results and Discussion
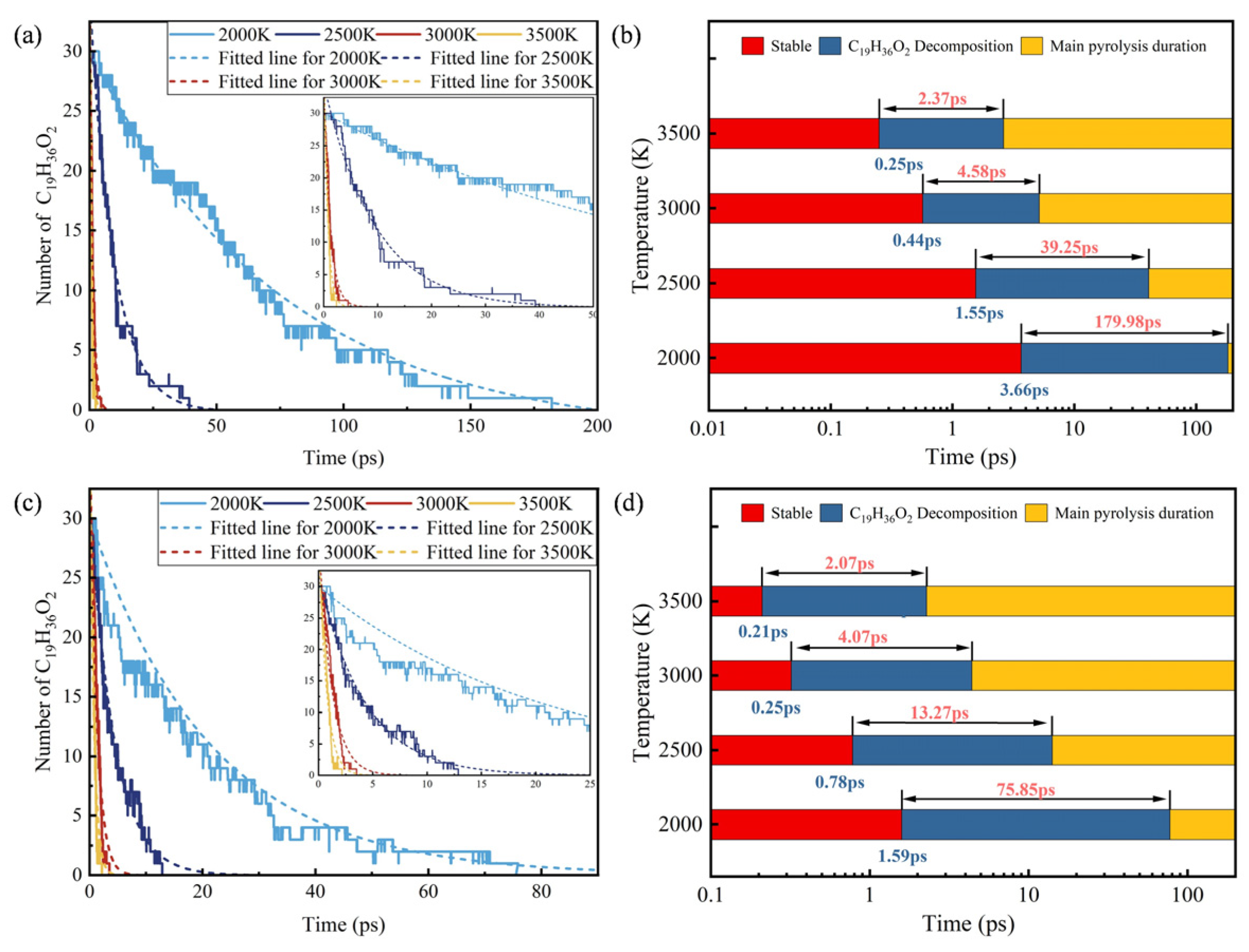
3.1. Influence of Density on the Reaction Rrocess
3.2. Based on the ReaxFF-MD Method for Calculating Ea
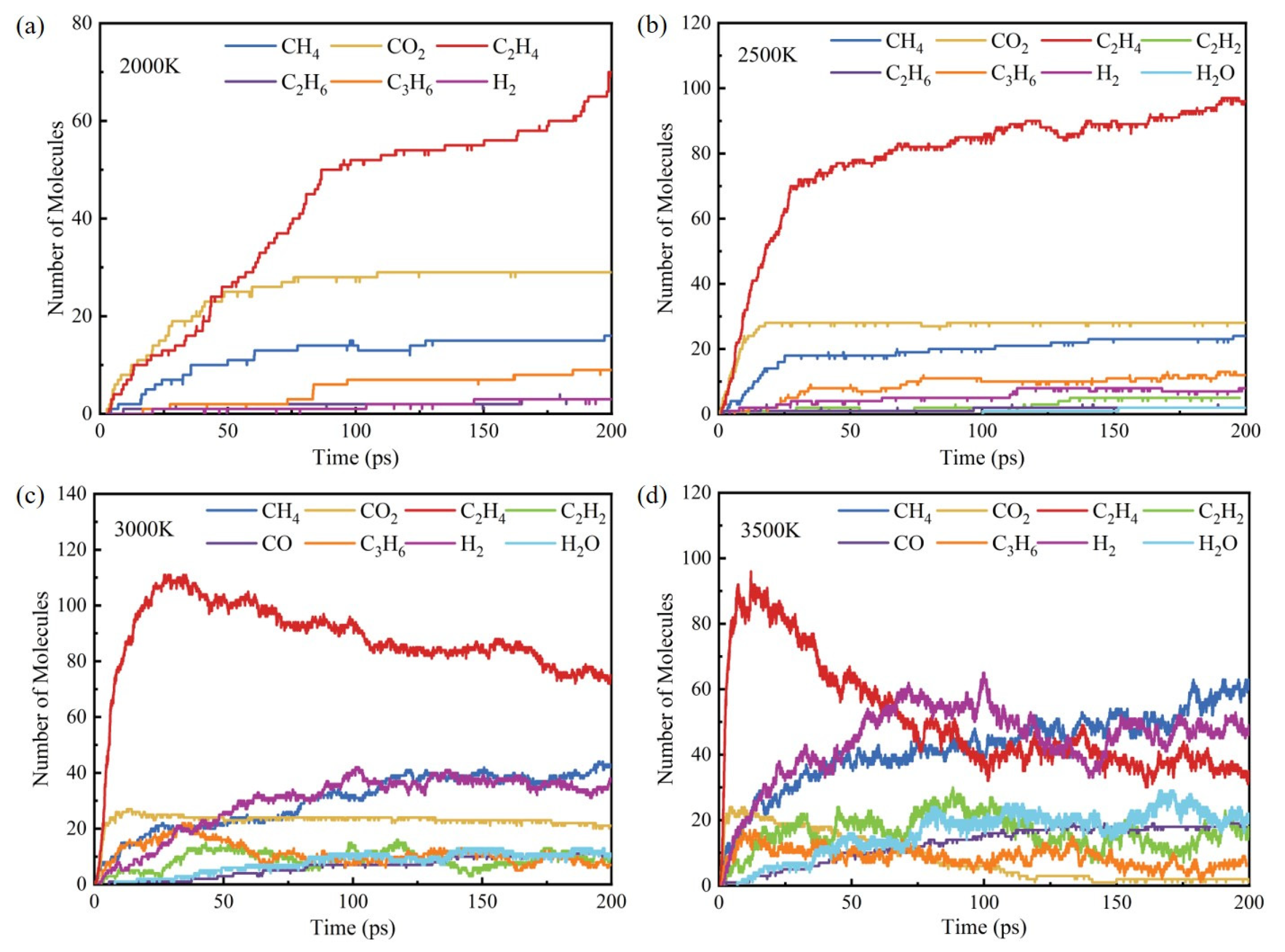
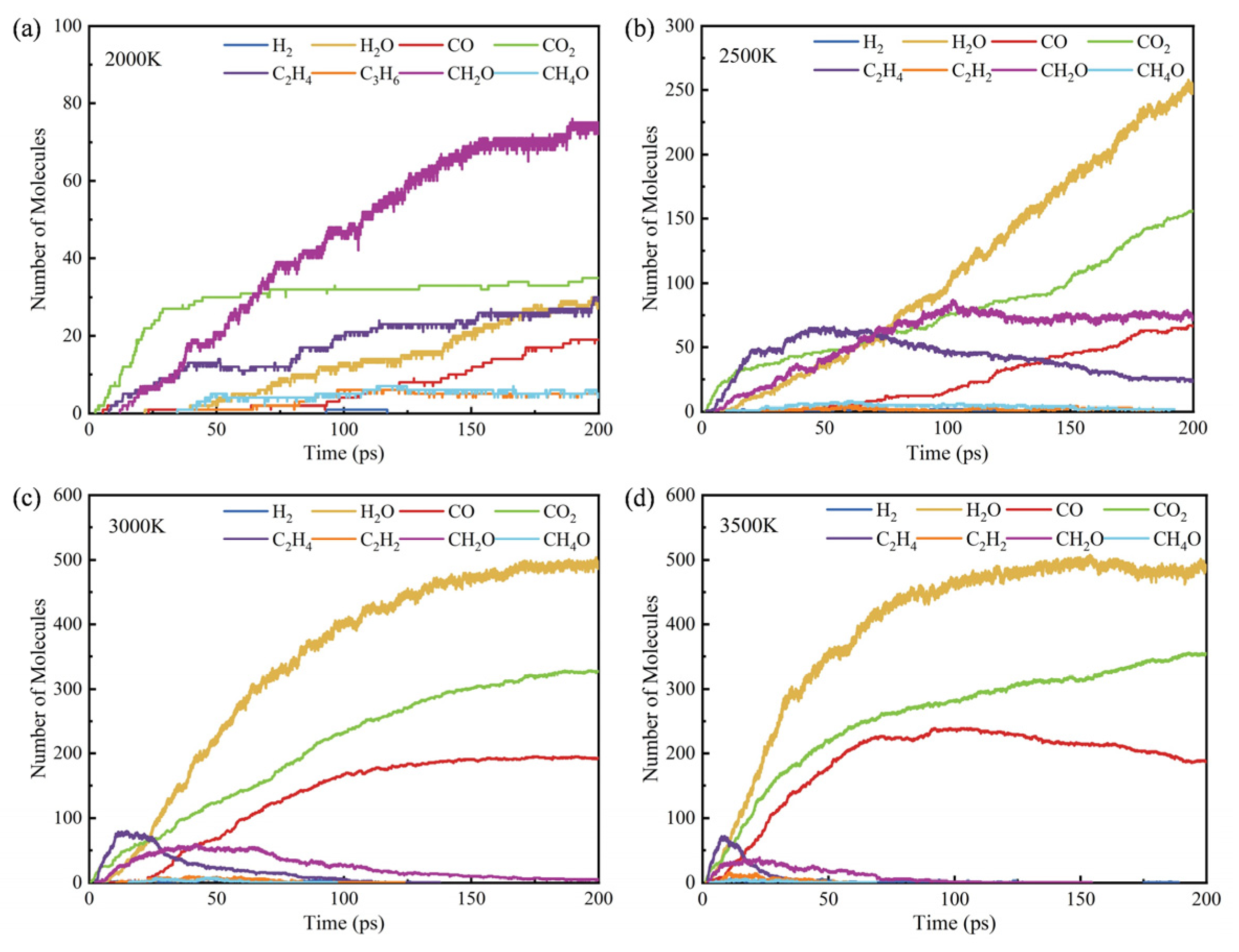
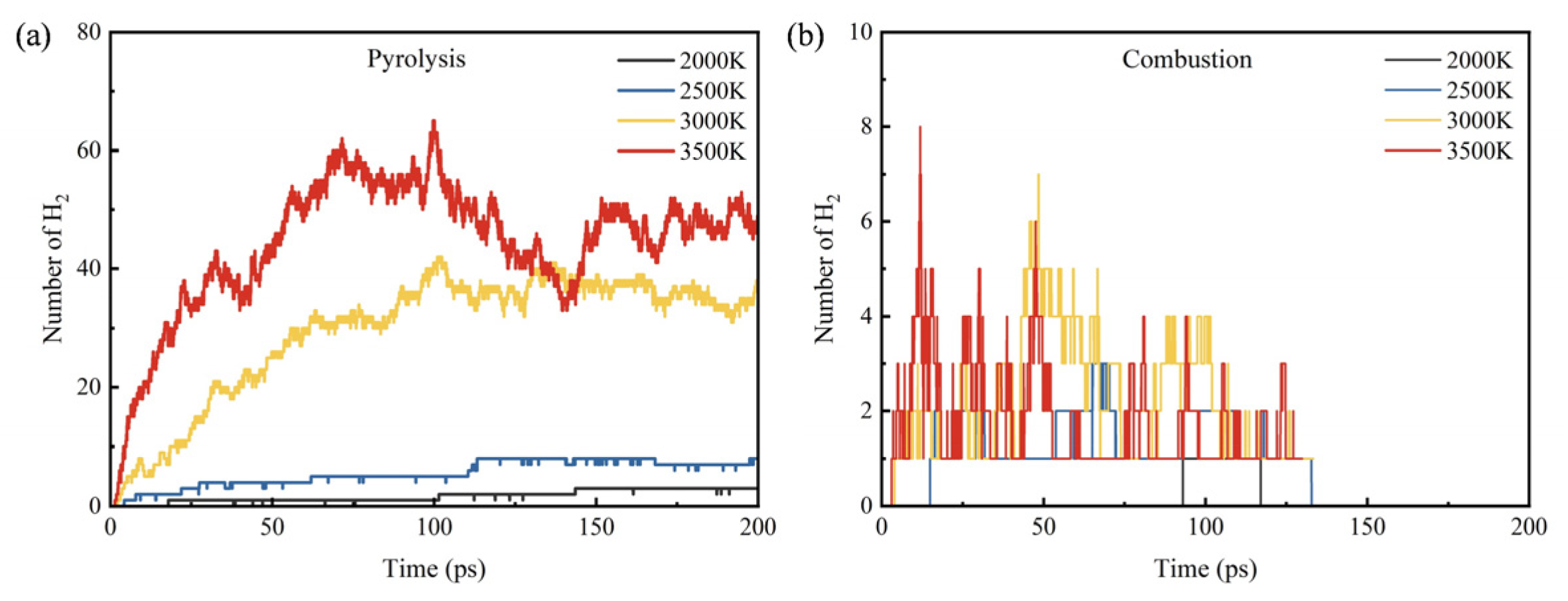
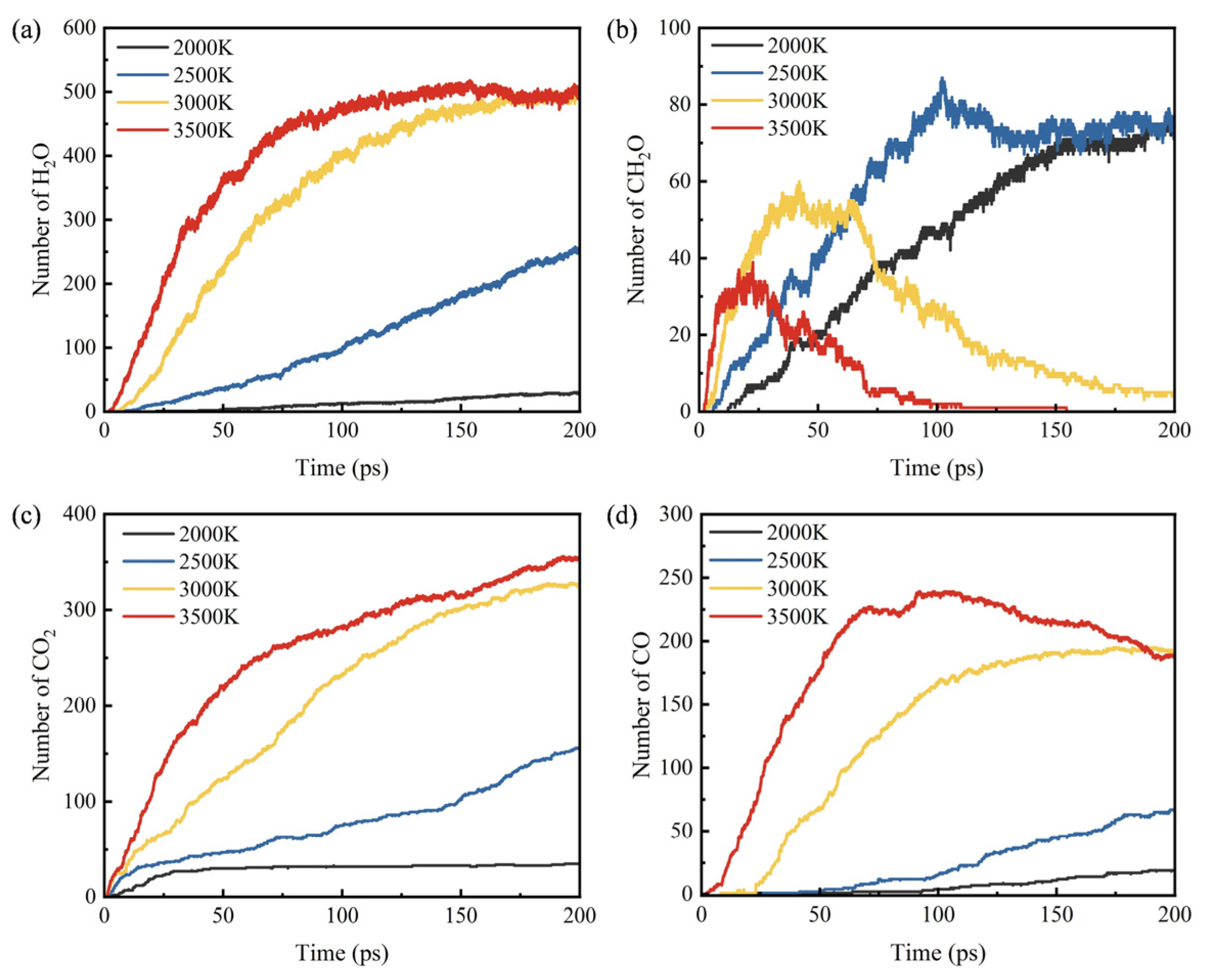
3.3. Influence of Temperature on Products
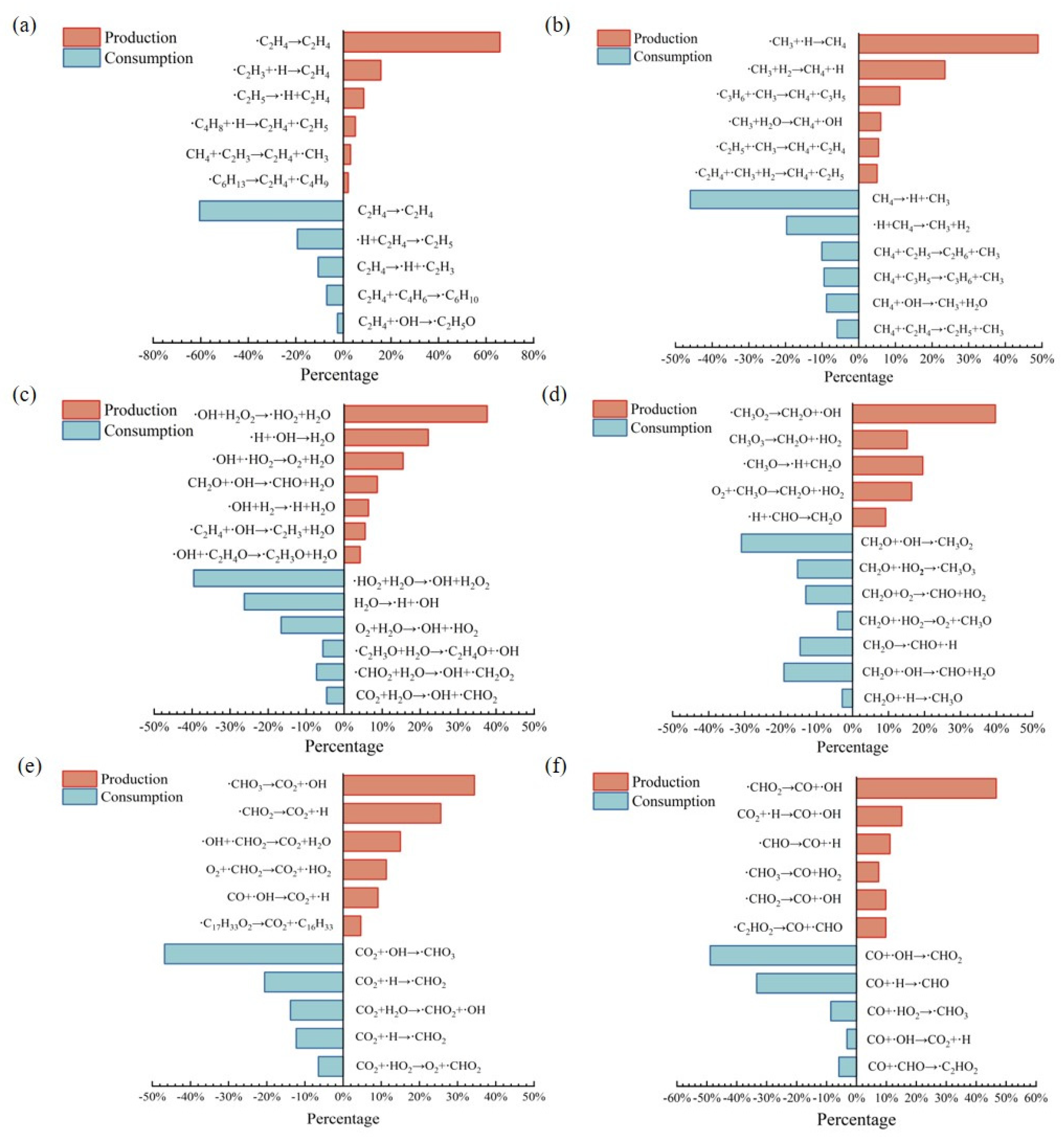
3.4. Main Product Reaction Pathways
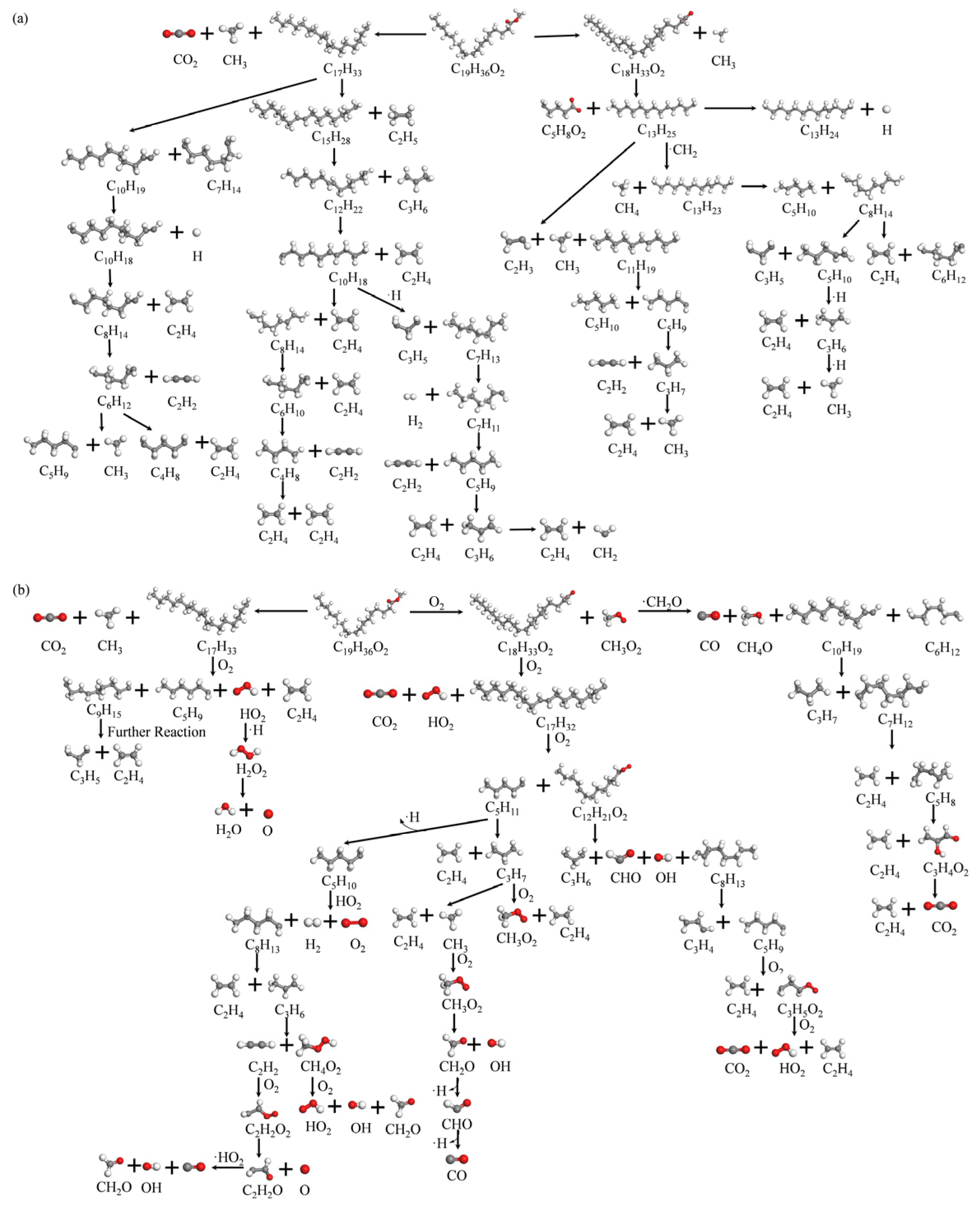
3.5. C19H36O2 Reaction Mechanism
4. Conclusions
- (1)
- The initial pyrolysis path of methyl oleate is the breaking of the C-C bond, generating large molecular intermediates and methane. Subsequently, long-chain species continue to decompose into small molecule gaseous substances, and finally evolve to produce species such as methane and ethylene. In contrast, the intermediate products generated during combustion are mainly short carbon chain gases, which further combust to generate stable CO2 and H2O.
- (2)
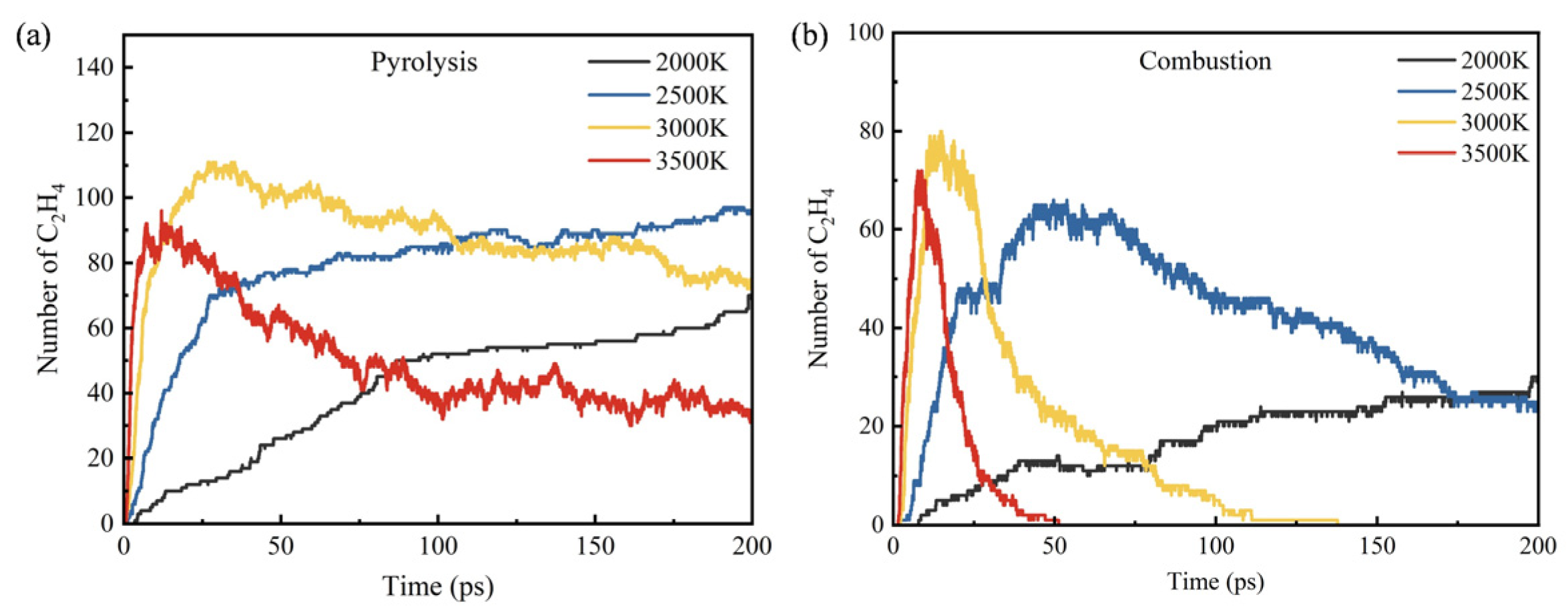
- During the pyrolysis and combustion processes, ethylene, water, and carbon dioxide are the main products with the highest cumulative amounts. As the reaction process progresses, the amount of ethylene first increases rapidly, and then rapidly decreases, while the amounts of water and carbon dioxide keep increasing.
- (3)
- The pyrolysis and combustion processes are simulated at temperatures of 2000 K, 2500 K, 3000 K, and 3500 K. The results show that the chemical reactivity of methyl oleate is stronger in the combustion process. The activation energy of the pyrolysis process is 190.02 kJ/mol, and the combustion activation energy is 144.89 kJ/mol.
- (4)
- This study is dedicated to the development of methyl oleate. Through in-depth research, a kinetic model for the high-temperature conversion of methyl oleate has been initially established, which will enhance our understanding of the thermochemical conversion of biodiesel fuels.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knothe, G.; Razon, L.F. Biodiesel fuels. Prog. Energy Combust. Sci. 2017, 58, 36–59. [Google Scholar] [CrossRef]
- Glisic, S.B.; Pajnik, J.M.; Orlović, A.M. Process and techno-economic analysis of green diesel production from waste vegetable oil and the comparison with ester type biodiesel production. Appl. Energy 2016, 170, 176–185. [Google Scholar] [CrossRef]
- Ando, S.; Wu, Y.; Nakaya, S.; Tsue, M. Droplet combustion behavior of oxidatively degraded methyl laurate and methyl oleate in microgravity. Combust. Flame 2020, 214, 199–210. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, Z.; Liang, Y.; Zhang, X.; Yu, L.; Lu, X. The effect of the unsaturation degree on the gas-phase autoignition of methyl oleate and methyl linoleate: Experimental and modeling study. Combust. Flame 2024, 263, 113381. [Google Scholar] [CrossRef]
- Ahmad, T.; Danish, M.; Kale, P.; Geremew, B.; Adeloju, S.B.; Nizami, M.; Ayoub, M. Optimization of process variables for biodiesel production by transesterification of flaxseed oil and produced biodiesel characterizations. Renew. Energy 2019, 139, 1272–1280. [Google Scholar] [CrossRef]
- Bhatia, S.K.; Bhatia, R.K.; Jeon, J.; Pugazhendhi, A.; Awasthi, M.K.; Kumar, D.; Kumar, G.; Yoon, J.; Yang, G. An overview on advancements in biobased transesterification methods for biodiesel production: Oil resources, extraction, biocatalysts, and process intensification technologies. Fuel 2021, 285, 119117. [Google Scholar] [CrossRef]
- Herbinet, O.; Pitz, W.J.; Westbrook, C. Detailed chemical kinetic mechanism for the oxidation of biodiesel fuels blend surrogate. Combust. Flame 2010, 157, 893–908. [Google Scholar] [CrossRef]
- Sui, M.; Li, F.; Wang, S.; Wang, H. Molecular dynamics simulation and experimental research on the oxidation reaction of methyl linoleate at low oxygen and high temperature. Fuel 2021, 305, 121478. [Google Scholar] [CrossRef]
- Soloiu, V.; Moncada, J.D.; Gaubert, R.; Knowles, A.; Molina, G.; Ilie, M.; Harp, S.; Wiley, J.T. Reactivity Controlled Compression Ignition combustion and emissions using n-butanol and methyl oleate. Energy 2018, 165, 911–924. [Google Scholar] [CrossRef]
- Zhou, W.; Liang, Y.; Pei, X.; Zhang, Y.; Yu, L.; Lu, X. Autoignition of methyl palmitate in low to intermediate temperature: Experiments in rapid compression machine and kinetic modeling. Combust. Flame 2023, 249, 112619. [Google Scholar] [CrossRef]
- Jiaqiang, E.; Liu, T.; Yang, W.; Deng, Y.; Gong, J. A skeletal mechanism modeling on soot emission characteristics for biodiesel surrogates with varying fatty acid methyl esters proportion. Appl. Energy 2016, 181, 322–331. [Google Scholar]
- Demirbas, A. Progress and recent trends in biodiesel fuels. Energy Convers. Manag. 2009, 50, 14–34. [Google Scholar] [CrossRef]
- Knothe, G. Biodiesel and renewable diesel: A comparison. Prog. Energy Combust. Sci. 2010, 36, 364–373. [Google Scholar] [CrossRef]
- Türck, J.; Schmitt, F.; Anthofer, L.; Türck, R.; Ruck, W.; Krahl, J. Extension of Biodiesel Aging Mechanism–the Role and Influence of Methyl Oleate and the Contribution of Alcohols Through the Use of SolketaL. ChemSusChem 2023, 16, e202300263. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Zhou, Y.; Yu, H.; Lu, C.; Han, K. Investigation on thermal degradation properties of oleic acid and its methyl and ethyl esters through TG-FTIR. Energy Convers. Manag. 2017, 149, 495–504. [Google Scholar] [CrossRef]
- Campbell, M.F.; Davidson, D.F.; Hanson, R.K. Ignition delay times of methyl oleate and methyl linoleate behind reflected shock waves. Proc. Combust. Inst. 2013, 34, 419–425. [Google Scholar] [CrossRef]
- Soloiu, V.; Knowles, A.R.; Carapia, C.E.; Moncada, J.D.; Wiley, J.T.; Kilpatrick, M.; Williams, J.; Rahman, M.; Ilie, M. n-Butanol and Oleic Acid Methyl Ester, Combustion and NVH Characteristics In Reactivity Controlled Compression Ignition. Energy 2020, 207, 118183. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, F.; Hu, J.; Yang, S.; Liu, H.; Wang, H. Investigation of Pyrolysis Characteristics and Product Evolution Behavior of Methyl Oleate under the Effect of Copper Slag. Catal. Lett. 2024. [Google Scholar] [CrossRef]
- Naik, C.V.; Westbrook, C.K.; Herbinet, O.; Pitz, W.J.; Mehl, M. Detailed chemical kinetic reaction mechanism for biodiesel components methyl stearate and methyl oleate. Proc. Combust. Inst. 2011, 33, 383–389. [Google Scholar] [CrossRef]
- Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Baltin, R. One-dimensional hypervirial theorems in density functional theory. Phys. Lett. A 1985, 113, 121–125. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, W.; Pei, Q.; Zhang, X.; Zhou, Z.; Xu, S.; Lan, Y.; Jiao, F.; Shi, X.; Xu, S.; et al. Effect of stearic acid coating on the flame propagation and reaction mechanism of AlH3. Fuel 2024, 358, 130140. [Google Scholar] [CrossRef]
- Zhao, T.; Li, T.; Xin, Z.; Zou, L.; Zhang, L. A ReaxFF-Based Molecular Dynamics Simulation of the Pyrolysis Mechanism for Polycarbonate. Energy Fuels 2018, 32, 2156–2162. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, S.; Lv, Y.; Hu, P.; Huang, Y.; Kong, M.; Li, G. Atomic-scale insight into the pyrolysis of poly-car-bonate by ReaxFF-based reactive molecular dynamics simulation. Fuel 2021, 287, 119484. [Google Scholar] [CrossRef]
- Zheng, H.; Wang, Z.; Yang, T.; Yao, W.; Cai, S.; Li, X.; Liu, C.; Yang, E. Investigation on pyrolysis mechanism of palm olein and the effect of moisture on its pyrolysis. J. Mol. Liq. 2021, 339, 116824. [Google Scholar] [CrossRef]
- Zhang, H.; Li, W.; Nian, Y.; Liu, M.; Zhang, J.; Han, Y. Insights into the pyrolytic coking process of RP-3 fuel from ReaxFF molecular dynamics. Chem. Eng. Sci. 2024, 291, 119935. [Google Scholar] [CrossRef]
- Goncalves, R.; Iha, B.; Rocco, J. Reactive molecular dynamics of pyrolysis and combustion of alternative jet fuels: A ReaxFF study. Fuel 2022, 310, 122157. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Wang, J.; Li, X.; Ma, J. Effect of CO2 and methyl groups reaction kinetics on the ignition and combustion of diesel surrogate fuel: Part I. Reaction mechanisms. Fuel 2023, 351, 128984. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, X.; Liu, H.; Li, J.; Wang, H. Pyrolysis and combustion reaction mechanisms of methyl palmitate with ReaxFF-MD method. Comput. Theor. Chem. 2024, 1231, 114446. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Zhang, A.; Wang, H. ReaxFF based molecular dynamics simulation of ethyl butyrate in pyrolysis and combustion. Chem. Eng. Sci. 2024, 284, 119528. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Liu, R.; Xu, S.; Xu, S. Thermal decomposition, flame propagation, and combustion reactions behaviours of stearic acid by experiments and molecular dynamic simulation. Chem. Eng. J. 2023, 461, 141906. [Google Scholar] [CrossRef]
- Chenoweth, K.; Van, D.; Goddard, W.A. ReaxFF Reactive Force Field for Molecular Dynamics Simulations of Hydrocarbon Oxidation. J. Phys. Chem. A 2008, 112, 1040–1053. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Zeng, J.; Cao, L.; Chin, C.; Ren, H.; Zhang, H.; Zhu, T. ReacNetGenerator: An automatic reaction network generator for reactive molecular dynamics simulations. Phys. Chem. Chem. Phys. 2020, 22, 683–691. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Wang, F.; Chen, L.; Geng, D.; Lu, J.; Wu, J. Effect of density on the thermal decomposition mechanism of ε-CL-20: A ReaxFF reactive molecular dynamics simulation study. Phys. Chem. Chem. Phys. 2018, 20, 22600–22609. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.F.; Davidson, D.F.; Hanson, R.K. Ignition delay times of very-low-vapor-pressure biodiesel surrogates behind reflected shock waves. Fuel 2014, 126, 271–281. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Process | T (K) | Duration (ps) | Density (g/cm3) |
|---|---|---|---|---|
| 30 C19H36O2 | Pyrolysis | 300–3000 + 3000 | 135 + 100 | 0.47 |
| 0.67 | ||||
| 0.87 | ||||
| 1.07 | ||||
| 30 C19H36O2 + 810 O2 | Combustion | 2000 | 200 | 0.87 |
| 2500 | ||||
| 3000 | ||||
| 3500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Zhang, X.; Qing, S.; Wang, H. Reaction Mechanism of Pyrolysis and Combustion of Methyl Oleate: A ReaxFF-MD Analysis. Energies 2024, 17, 3536. https://doi.org/10.3390/en17143536
Wei Y, Zhang X, Qing S, Wang H. Reaction Mechanism of Pyrolysis and Combustion of Methyl Oleate: A ReaxFF-MD Analysis. Energies. 2024; 17(14):3536. https://doi.org/10.3390/en17143536
Chicago/Turabian StyleWei, Yu, Xiaohui Zhang, Shan Qing, and Hua Wang. 2024. "Reaction Mechanism of Pyrolysis and Combustion of Methyl Oleate: A ReaxFF-MD Analysis" Energies 17, no. 14: 3536. https://doi.org/10.3390/en17143536
APA StyleWei, Y., Zhang, X., Qing, S., & Wang, H. (2024). Reaction Mechanism of Pyrolysis and Combustion of Methyl Oleate: A ReaxFF-MD Analysis. Energies, 17(14), 3536. https://doi.org/10.3390/en17143536