

Pyrolytic Conversion of Cellulosic Pulps from “Lignin-First” Biomass Fractionation


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Lignin-First In Situ Depolymerization
2.3. Pyrolysis-GC-Polyarc-FID
3. Results and Discussion
3.1. Lignin-First Fractionation and Depolymerization
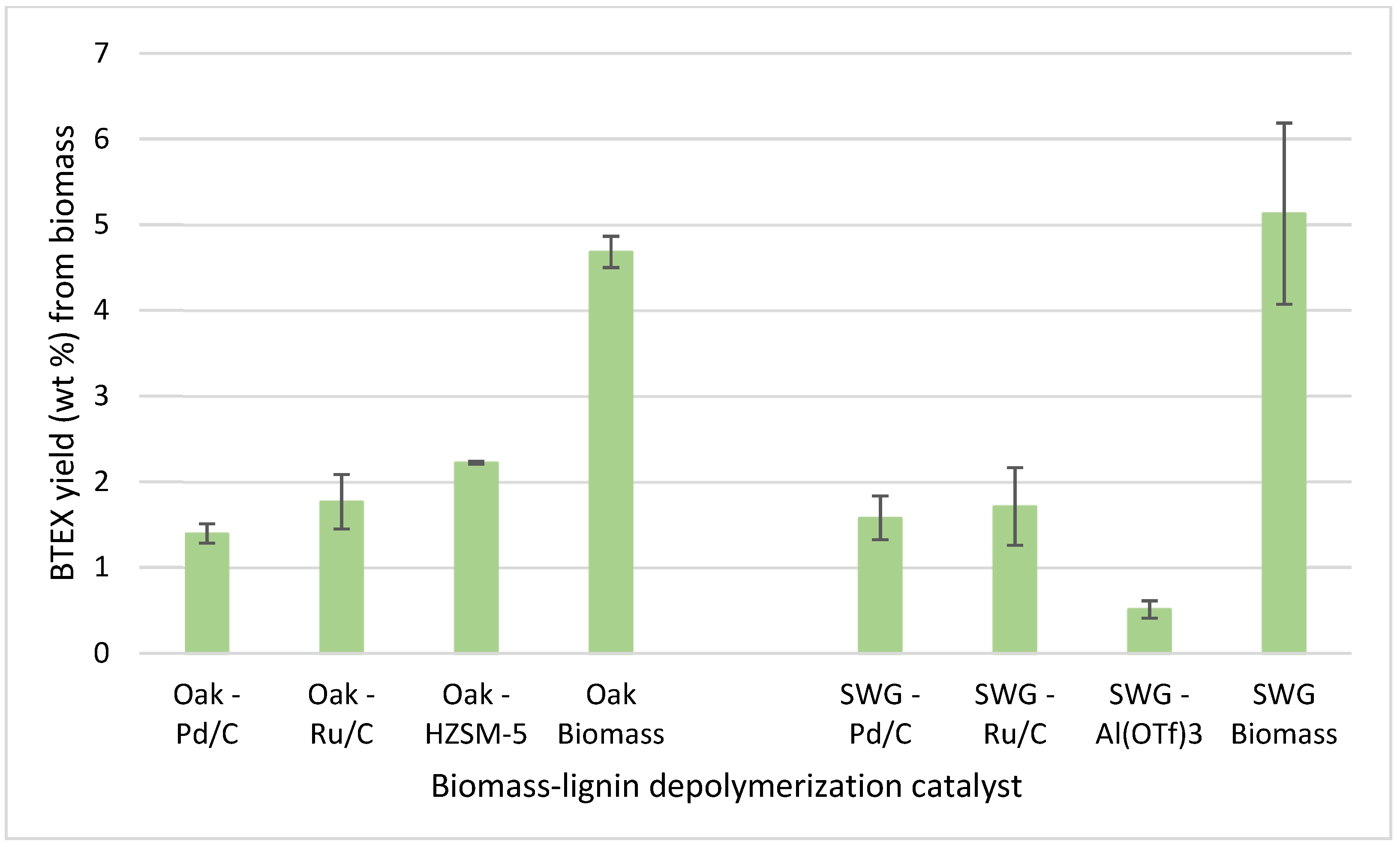
3.2. Pyrolysis of the Cellulosic Pulps
3.3. Catalytic Pyrolysis over HZSM-5
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Langholtz, M.H.; Stokes, B.J.; Eaton, L.M. 2016 Billion-Ton Report: Advancing Domestic Resources for a Thriving Bioeconomy, Volume 1: Economic Availability of Feedstocks; U.S. Department of Energy Office of Scientific and Technical Information: Oak Ridge, TN, USA, 2016.
- Lynd, L.R.; Beckham, G.T.; Guss, A.M.; Jayakody, L.N.; Karp, E.M.; Maranas, C.; McCormick, R.L.; Amador-Noguez, D.; Bomble, Y.J.; Davison, B.H.; et al. Toward low-cost biological and hybrid biological/catalytic conversion of cellulosic biomass to fuels. Energy Environ. Sci. 2022, 15, 938–990. [Google Scholar] [CrossRef]
- Devi, A.; Bajar, S.; Kour, H.; Kothari, R.; Pant, D.; Singh, A. Lignocellulosic Biomass Valorization for Bioethanol Production: A Circular Bioeconomy Approach. Bioenergy Res. 2022, 15, 1820–1841. [Google Scholar] [CrossRef]
- Huang, C.; Jiang, X.; Shen, X.; Hu, J.; Tang, W.; Wu, X.; Ragauskas, A.; Jameel, H.; Meng, X.; Yong, Q. Lignin-enzyme interaction: A roadblock for efficient enzymatic hydrolysis of lignocellulosics. Renew. Sustain. Energy Rev. 2022, 154, 111822. [Google Scholar] [CrossRef]
- Sun, S.; Sun, S.; Cao, X.; Sun, R. The role of pretreatment in improving the enzymatic hydrolysis of lignocellulosic materials. Bioresour. Technol. 2016, 199, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, P.R.; Brown, R.C.; Shanks, B.H. Understanding the fast pyrolysis of lignin. ChemSusChem 2011, 4, 1629–1636. [Google Scholar] [CrossRef]
- Sharma, V.; Tsai, M.L.; Nargotra, P.; Chen, C.W.; Sun, P.P.; Singhania, R.R.; Patel, A.K.; Dong, C.D. Journey of lignin from a roadblock to bridge for lignocellulose biorefineries: A comprehensive review. Sci. Total Environ. 2023, 861, 160560. [Google Scholar] [CrossRef]
- Mullen, C.A. Thermochemical and Catalytic Conversion of Lignin. In Biomass Utilization: Conversion Strategies; Springer International Publishing: Cham, Switzerland, 2022; pp. 133–200. [Google Scholar]
- Patil, V.; Adhikari, S.; Cross, P.; Jahromi, H. Progress in the solvent depolymerization of lignin. Renew. Sustain. Energy Rev. 2020, 133, 110359. [Google Scholar] [CrossRef]
- Mullen, C.A.; Strahan, G.D.; Elkasabi, Y. A comparison of the solvent liquefaction of lignin in ethanol and 1,4-butanediol. J. Anal. Appl. Pyrolysis 2022, 164, 105522. [Google Scholar] [CrossRef]
- Abu-Omar, M.M.; Barta, K.; Beckham, G.T.; Luterbacher, J.S.; Ralph, J.; Rinaldi, R.; Román-Leshkov, Y.; Samec, J.S.M.; Sels, B.F.; Wang, F. Guidelines for performing lignin-first biorefining. Energy Environ. Sci. 2021, 14, 262–292. [Google Scholar] [CrossRef]
- De Santi, A.; Galkin, M.V.; Lahive, C.W.; Deuss, P.J.; Barta, K. Lignin-First Fractionation of Softwood Lignocellulose Using a Mild Dimethyl Carbonate and Ethylene Glycol Organosolv Process. ChemSusChem 2020, 13, 4468–4477. [Google Scholar] [CrossRef]
- Huang, X.; Zhu, J.; Koranyi, T.I.; Boot, M.D.; Hensen, E.J. Effective Release of Lignin Fragments from Lignocellulose by Lewis Acid Metal Triflates in the Lignin-First Approach. ChemSusChem 2016, 9, 3262–3267. [Google Scholar] [CrossRef]
- Kramarenko, A.; Etit, D.; Laudadio, G.; D’Angelo, F.N. β-Zeolite-Assisted Lignin-First Fractionation in a Flow-Through Reactor. ChemSusChem 2021, 14, 3838–3849. [Google Scholar] [CrossRef]
- Renders, T.; Van den Bosch, S.; Koelewijn, S.F.; Schutyser, W.; Sels, B.F. Lignin-first biomass fractionation: The advent of active stabilisation strategies. Energy Environ. Sci. 2017, 10, 1551–1557. [Google Scholar] [CrossRef]
- Renders, T.; Van den Bossche, G.; Vangeel, T.; Van Aelst, K.; Sels, B. Reductive catalytic fractionation: State of the art of the lignin-first biorefinery. Curr. Opin. Biotechnol. 2019, 56, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Galkin, M.V.; Smit, A.T.; Subbotina, E.; Artemenko, K.A.; Bergquist, J.; Huijgen, W.J.; Samec, J.S. Hydrogen-free catalytic fractionation of woody biomass. ChemSusChem 2016, 9, 3280–3287. [Google Scholar] [CrossRef]
- Liu, X.; Bouxin, F.P.; Fan, J.; Budarin, V.L.; Hu, C.; Clark, J.H. Recent Advances in the Catalytic Depolymerization of Lignin towards Phenolic Chemicals: A Review. ChemSusChem 2020, 13, 4296–4317. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, S.; Schutyser, W.; Vanholme, R.; Driessen, T.; Koelewijn, S.F.; Renders, T.; De Meester, B.; Huijgen, W.J.J.; Dehaen, W.; Courtin, C.M.; et al. Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps. Energy Environ. Sci. 2015, 8, 1748–1763. [Google Scholar] [CrossRef]
- Huang, X.; Ouyang, X.; Hendriks, B.M.S.; Gonzalez, O.M.M.; Zhu, J.; Koranyi, T.I.; Boot, M.D.; Hensen, E.J.M. Selective production of mono-aromatics from lignocellulose over Pd/C catalyst: The influence of acid co-catalysts. Faraday Discuss. 2017, 202, 141–156. [Google Scholar] [CrossRef]
- Deuss, P.J.; Lahive, C.W.; Lancefield, C.S.; Westwood, N.J.; Kamer, P.C.; Barta, K.; de Vries, J.G. Metal Triflates for the Production of Aromatics from Lignin. ChemSusChem 2016, 9, 2974–2981. [Google Scholar] [CrossRef]
- Ahmed, R.; Liu, G.; Yousaf, B.; Abbas, Q.; Ullah, H.; Ali, M.U. Recent advances in carbon-based renewable adsorbent for selective carbon dioxide capture and separation-A review. J. Clean. Prod. 2020, 242, 118409. [Google Scholar] [CrossRef]
- Lourenço, A.; Pereira, H. Compositional Variability of Lignin in Biomass. In Lignin—Trends and Applications; IntechOpen: London, UK, 2018. [Google Scholar]
- Choi, H.S.; Meier, D. Fast pyrolysis of Kraft lignin—Vapor cracking over various fixed-bed catalysts. J. Anal. Appl. Pyrolysis 2013, 100, 207–212. [Google Scholar] [CrossRef]
- Zhou, S.; Xue, Y.; Sharma, A.; Bai, X. Lignin Valorization through Thermochemical Conversion: Comparison of Hardwood, Softwood and Herbaceous Lignin. ACS Sustain. Chem. Eng. 2016, 4, 6608–6617. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, S.; Pecha, B.; Westerhof, R.J.M.; Garcia-Perez, M. Effect of Pyrolysis Temperature and Sulfuric Acid During the Fast Pyrolysis of Cellulose and Douglas Fir in an Atmospheric Pressure Wire Mesh Reactor. Energy Fuels 2014, 28, 5167–5177. [Google Scholar] [CrossRef]
- Hilbers, T.J.; Wang, Z.; Pecha, B.; Westerhof, R.J.M.; Kersten, S.R.A.; Pelaez-Samaniego, M.R.; Garcia-Perez, M. Cellulose-Lignin interactions during slow and fast pyrolysis. J. Anal. Appl. Pyrolysis 2015, 114, 197–207. [Google Scholar] [CrossRef]
- Mullen, C.A. Upgrading of biomass via catalytic fast pyrolysis (CFP). In Chemical Catalysts for Biomass Upgrading; Wiley-VCH: Weinheim, Germany, 2020; pp. 1–33. [Google Scholar]
- Du, S.; Valla, J.A.; Bollas, G.M. Characteristics and origin of char and coke from fast and slow, catalytic and thermal pyrolysis of biomass and relevant model compounds. Green Chem. 2013, 15, 3214–3229. [Google Scholar] [CrossRef]
- Wang, K.; Kim, K.H.; Brown, R.C. Catalytic pyrolysis of individual components of lignocellulosic biomass. Green Chem. 2014, 16, 727–735. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oak | Switchgrass | |
|---|---|---|
| Moisture | 6.6 | 6.4 |
| VM, d.b. | 81.0 | 87.7 |
| FC, d.b. | 17.4 | 9.3 |
| Ash, d.b. | 1.6 | 3.1 |
| C | 50.52 | 43.8 |
| H 1 | 6.24 | 4.7 |
| N | 0.12 | 0.5 |
| O 1 | 35.04 | 41.6 |
| Cellulose | 29.71 | 36.77 |
| Hemicellulose | 21.62 | 33.07 |
| Lignin | 37.08 | 27.42 |
| Biomass—Catalyst | Phenolic Oil Yield (from Biomass) | Phenolic Oil Yield (from Lignin) | Pulp Yield (from Biomass) | Pulp Yield (from Cellulose) |
|---|---|---|---|---|
| Oak—Pd/C | 28.0 | 75.6 | 27.7 | 93.2 |
| Oak—Ru/C | 34.2 | 92.2 | 38.5 | 129.5 |
| Oak—HZSM-5 | 39.5 | 106.6 | 36.3 | 122.3 |
| SWG—Pd/C | 27.1 | 98.9 | 31.1 | 84.6 |
| SWG—Ru/C | 41.6 | 151.9 | 33.4 | 90.8 |
| SWG—Al(OTf)3 | 37.0 | 135.0 | 24.5 | 66.7 |
| Mn (g/mol) | Mw (g/mol) | |
|---|---|---|
| Oak—Pd/C | 484 | 953 |
| Oak—Ru/C | 556 | 1128 |
| Oak—HZSM-5 | 533 | 994 |
| SWG—Pd/C | 400 | 802 |
| SWG—Ru/C | 384 | 741 |
| SWG—Al(OTf)3 | 1141 | 1659 |
| Oak Pd/C | Oak Ru/C | Oak HZSM-5 | SWG Pd/C | SWG Ru/C | SWG Al(OTf)3 | |
|---|---|---|---|---|---|---|
| Moisture | 3.2 | 3.2 | 5.6 | 2.6 | 2.4 | 5.3 |
| VM, d.b. | 80.9 | 76.9 | 70.98 | 66.8 | 76.5 | 42.9 |
| FC, d.b. | 16.6 | 19.4 | 28.0 | 28.0 | 17.9 | 47.4 |
| Ash, d.b. | 2.5 | 3.7 | 4.1 | 5.2 | 5.6 | 9.7 |
| C | 55.79 | 52.50 | 40.52 | 55.79 | 50.05 | 58.95 |
| H 1 | 5.72 | 3.82 | 4.03 | 3.88 | 3.58 | 3.84 |
| N | 0.63 | 0.55 | 0.25 | 0.62 | 0.20 | 0.20 |
| O 1 | 32.15 | 36.23 | 45.5 | 31.91 | 38.17 | 22.01 |
| C/O (mol) | 2.33 | 1.92 | 1.19 | 2.33 | 1.74 | 3.57 |
| H/C (mol) | 1.23 | 0.87 | 1.06 | 0.83 | 0.85 | 0.78 |
| (wt % Yield) | Oak-Pd/C | Oak-Ru/C | Oak—HZSM-5 | Oak—Biomass | SWG—Pd/C | SWG—Ru/C | SWG—Al(OTf)3 | SWG—Biomass | Cellulose |
|---|---|---|---|---|---|---|---|---|---|
| Hydroxy-acetaldehyde | 2.95 | 1.29 | 5.28 | 2.41 | 0.55 | 1.51 | 0.08 | 1.63 | 1.98 |
| Acetic Acid | 0.47 | 0.37 | 1.10 | 6.76 | 0.69 | 0.54 | 0.17 | 3.60 | 0.32 |
| Acetol | 0.81 | 0.28 | 1.63 | 2.20 | 0.28 | 1.08 | 0.21 | 3.22 | 0.88 |
| 2-Cyclopenten-1-one | 0.41 | 0.08 | 0.18 | 0.15 | 0.00 | 0.18 | 0.04 | 0.20 | 0.13 |
| Furfural | 0.82 | 0.38 | 0.86 | 0.76 | 0.35 | 0.86 | 0.13 | 0.75 | 0.52 |
| Hydroxymethyl-furfural (HMF) | 0.63 | 0.17 | 0.46 | 0.13 | 0.09 | 0.29 | 0.15 | 0.11 | 0.34 |
| Phenol | 0.03 | 0.08 | 0.08 | 0.06 | 0.00 | 0.07 | 0.20 | 0.14 | 0.01 |
| Guaiacol | 0.18 | 0.09 | 0.10 | 0.22 | 0.00 | 0.11 | 0.13 | 0.18 | nd |
| Syringol | 0.14 | 0.07 | 0.15 | 0.38 | 0.00 | 0.11 | 0.07 | 0.00 | nd |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mullen, C.A.; Ellison, C.; Elkasabi, Y. Pyrolytic Conversion of Cellulosic Pulps from “Lignin-First” Biomass Fractionation. Energies 2023, 16, 3236. https://doi.org/10.3390/en16073236
Mullen CA, Ellison C, Elkasabi Y. Pyrolytic Conversion of Cellulosic Pulps from “Lignin-First” Biomass Fractionation. Energies. 2023; 16(7):3236. https://doi.org/10.3390/en16073236
Chicago/Turabian StyleMullen, Charles A., Candice Ellison, and Yaseen Elkasabi. 2023. "Pyrolytic Conversion of Cellulosic Pulps from “Lignin-First” Biomass Fractionation" Energies 16, no. 7: 3236. https://doi.org/10.3390/en16073236
APA StyleMullen, C. A., Ellison, C., & Elkasabi, Y. (2023). Pyrolytic Conversion of Cellulosic Pulps from “Lignin-First” Biomass Fractionation. Energies, 16(7), 3236. https://doi.org/10.3390/en16073236