Abstract
The shelf-life of catalyst ink for fabricating polymer electrolyte fuel cells (PEFCs) is relevant for large-scale manufacturing with unforeseen production stops. In this study, the storage effects on the physicochemical characteristics of catalyst ink (Pt/C, Nafion, 2-propanol, water) and subsequently manufactured catalyst layers are investigated. Sedimentation analysis showed that catalyst particles are not fully stabilized by charge interaction induced by Nafion. Acetone was found to be an oxidation product, even in freshly prepared ink with platinum catalyzing the reaction. Rotating disk electrode analysis revealed that the electrochemically active surface area is, overall, minimally increased by storage, and the selectivity towards water formation (4-electron pathway) is unharmed within the first 48 h of storage. MEAs prepared from stored ink reach almost the same current density level after conditioning via potential cycling. The open-circuit voltage (OCV) increases due to increased catalyst availability. Scanning electron microscopy and mercury intrusion porosimetry showed that with increasing acetone content, the pore structure becomes finer, with a higher specific surface area. Electrochemical impedance spectroscopy revealed that this results in a more hindered mass transfer but lowered charge transfer resistance. The MEA with the highest OCV and power output and the lowest overall cell resistance was fabricated from catalyst ink stored for a duration of four weeks.
1. Introduction
To mitigate the effects of the anthropogenic climate change caused by carbon dioxide (CO2) emissions, rapid decarbonization is necessary. In 2020, 73.2% of the global CO2 emissions, equivalent to 25.8 billion tons, were caused by the energy and transport sector, where over 90% of the energy demand is covered by fossil fuels [1,2]. Despite current costs and technological readiness level restrictions, polymer electrolyte fuel cells (PEFCs) powered by renewable hydrogen could be a viable alternative for a locally CO2 emission-free energy supply [3,4,5,6,7,8].
In a PEFC, hydrogen is electrochemically converted into water, electricity and heat. The performance-determining core of a PEFC is the membrane electrode assembly (MEA). The MEA involves a polymer electrolyte membrane (PEM), anodic and cathodic catalyst layers (CLs) as well as anodic and cathodic gas diffusion layers (GDLs) [9]. The PEM is a thin film of proton-conductive perflourosulfonate ionomer [10], which is coated with the CLs to form a catalyst-coated membrane (CCM) [11,12]. This CCM is sandwiched between two GDLs to form an MEA [9].
The hydrogen oxidation reaction (HOR, anode) and the oxygen reduction reaction (ORR, cathode) are located at the CLs [9]. The CLs are formed by the application of catalyst ink (CI), e.g., via ultrasonic spray coating, slot-die coating or electrospinning, onto both sides of the PEM [9]. Therefore, the proper fabrication of the catalyst ink is decisive for the performance and durability of a PEFC. The formulation variety of catalyst ink is manifold [13,14], but it commonly consists of three components:
- Catalyst on supportive material, mostly platinum on carbon (Pt/C), for the HOR and ORR;
- Ionomer as binder to stabilize the dispersion state of the CI and for proton conduction within the CL [15,16];
- A mixture of water and an organic solvent to disperse the catalyst particles and the ionomer as well as to control the pore structure and morphology of the CL [17].
The ink is mixed and dispersed using ultrasonic methods (tip, bath) or high-shear mechanic stirrer [14] before further use or storage. The dispersion stability via charge stabilization can be characterized by the Zeta potential (mV) and is highly dependent on the ionomer content and choice of solvent, as shown in the literature [16,18,19,20,21,22,23].
The effect of storage and aging of catalyst ink has been scarcely covered in the literature. Biddinger et al. [24] investigated the effect of storage for commercially available platinum on Vulcan carbon (Pt/VC) and self-prepared nanostructured carbon with nitrogen (CNx) on the selectivity to water formation using a rotating ring disk electrode. Ethanol and Nafion were used as the solvent and ionomer. After eleven days of storage, both catalysts showed a higher number of transferred electrons and, thus, increased ORR selectivity towards water (Pt/VC: + 1%, CNx: + 10%). The authors state that the reasons behind this effect are not fully clear but could involve changes in the hydrophilicity of the catalyst surface. Koh and Strasser [25] studied the effect of ageing for dealloyed platinum nanoparticles using a rotating disk electrode (RDE). The ink consisted of 2-propanol and Nafion and was degassed before mixing. Catalyst layers on the glassy carbon electrode were stored under a nitrogen atmosphere before characterization. The storage of dry catalyst powder for up to 26 days under an air atmosphere showed no performance degradation compared to powder used immediately. Inks were stored for up to 6 days. The optimal performance (highest ORR activity and electrochemically active surface area, ECSA (m2/g)) was achieved after 24–48 h of storage. The reason stated for this behavior was an increased wetting of catalyst pores. They stated that ink should be discarded after the fourth day. Aging catalyst films demonstrate a drastic decrease in performance, which is derived from morphological changes in the CL such as cracking. Uemura et al. [26,27] explored the effects of the oxidation of 2-propanol as an organic solvent in the ink due to the catalyst (platinum on carbon, Pt/C). A pseudo ink was prepared where Pt/C was replaced by Vulcan carbon. Inks were stored for up to 20 days. In contrast to pseudo ink, catalyst ink showed an increasing amount of oxidation products of the used organic solvents (e.g., CO2, propionic acid, propionaldehyde acetaldehyde). In follow-up studies, Uemura et al. [28,29,30] investigated a mixture of ethanol/water as the solvent and determined according degradation products (e.g., acetic acid, ethyl acetate, acetaldehyde, 1,1-diethoxyethene). Hydrophobic products (e.g., acetaldehyde, ethyl acetate) are causal for an increase in the ink viscosity and crack formation of the catalyst layer. Liu et al. [31] investigated the effect of degassing catalyst ink using nitrogen. Degassed ink showed a higher (absolute) Zeta potential and contained at least 50% fewer degradation products, with a higher boiling point than the pure solvent (e.g., propionic acid, propyl propionate) than non-degassed catalyst ink. MEAs fabricated from degassed ink achieved an increase in the peak power density by approx. 35%, which can be explained by a lower cathodic resistance. The effect of added propionic acid was studied through RDE measurements. Catalyst layers from ink, where propionic acid was added, demonstrated a smoother surface due to decreased surface tension, a higher ECSA as well as enhanced ORR activity. Ren et al. [32] investigated the effect of ink storage for two days on the MEA performance. They compared inks containing a long-side-chain ionomer (Nafion) and a short-side-chain ionomer (Aquivion) in a 1:1 wt./wt. mixture of isopropanol water with an ionomer-to-carbon-weight ratio, I/C [-], of 0.65 and a solids content of Pt/C of 1 wt.%. For Nafion-based ink, the physical properties (Zeta potential, viscosity) as well as the respective MEA performance are not influenced by storage. For Aquivion-based ink, both the viscosity and (negative) Zeta potential decreased. The MEAs fabricated with this ink showed a decreasing performance at various relative humidity, especially at high current densities. The reason stated for this behavior is an altered CL microstructure (increased macroporous volume) as well as enhanced ionomer coverage of the Pt/C particles, which promotes proton conductance but hinders oxygen transport, which is especially crucial at high current densities. The authors, therefore, advise not to store Aquivion-based ink or to do so for as short a time as possible.
The aim of this work was to investigate the physical, chemical and electrochemical aging effects of catalyst ink due to storage. The target was to determine the shelf-life of catalyst ink before it should be discarded. Physical aging is quantified by sedimentation and dispersion stability analysis. For chemical aging, degradation products in the liquid phase of the ink are determined and quantified. The effect of storage on the electrochemical characteristics of catalyst layers is investigated via RDE and MEA measurements. The results show that catalyst ink can be stored for days without major performance losses.
2. Materials and Methods
For all experiments, the catalyst ink composition was kept constant and based on the work by Grandi et al. [33]. The used catalyst powder was Pt/C 20 (platinum, nominally 20 w.% on carbon black, Thermo Fisher Scientific, Schwerte, Germany). For pseudo inks, the Vulcan XC72-R (CABOT Corporation, Boston, MA, USA) was used instead of the catalyst. The particle concentration in the ink was 4 mg/mL in pure solvent. As ionomer, Nafion (Nafion 117, 5% mixture in alcohol and water, Sigma Aldrich, St. Louis, MO, USA) was added with an I/C ratio of 0.55. The solvent for the inks consisted of a premixed 3:7 v/v mixture of ultrapure water (resistance > 17 MΩ, Barnstead NANOpureWater Purification system, Thermo Fisher Scientific) and 2-propanol (>99.9%, Carl Roth, Karlsruhe, Germany). The ink was dispersed in an ultrasonic bath (Sonotex Super RK 31 H, Bandelin Sonorex, Berlin, Germany) for 30 min. No degassing of the ink was performed. After this dispersion time, the time count was started. Samples were stored at 6 °C. Vials had a filling ratio of approx. 50%.
2.1. Physical Analysis—UV-Vis Spectroscopy, Zeta Potential
The dispersion stability of the catalyst ink was determined by the Zeta potential. The ink was diluted with a ratio of 1:200 v/v using ultrapure water [34] and analyzed at 25 °C using a Litesizer 500 (Anton Paar GmbH, Graz, Austria). The analysis of the Zeta potential was executed by the application of the Smoluchowski approximation and a Henry factor of 1.5. Reproducibility was checked through trifold measurement.
Sedimentation analysis was performed using UV-Vis spectroscopy and two sets of catalyst ink. The total height of the ink in the vials was 30 mm. From the first set, samples from the top of the catalyst ink (immersion depth approx. 3 mm) were taken, while from the second set, samples from the bottom (immersion depth approx. 27 mm) were obtained. The samples were diluted with a ratio of 1:100 v./v. using ultrapure water [35]. Measurements were executed at 20 °C using a UV-1800 (Shimadzu, Kyoto, Japan) with ultrapure water as a blank. Non-interfering semi micro quartz glass cuvettes (ROTILABO, Carl Roth, Karlsruhe, Germany) were used for analysis. As results in the region of maximum absorbance were too noisy, the absorbance at a wavelength 600 nm was taken as representative value [36].
2.2. Chemical Analysis—GC/MS, GC/FID
To determine degradation products of the catalyst ink, gas chromatography coupled with mass spectroscopy (GC/MS) was used. Samples were taken from the gas phase (headspace). For analysis, a GCMS-QP2010 SE (Shimadzu, Kyoto, Japan) with a semi-polar column (Agilent J&W GC Columns, Agilent Technologies, Santa Klara, CA, USA) was used. The injection volume was 50 µL. The temperature profile started at 40 °C and rose with a rate of 5 K/min up to 240 °C.
For quantification of the degradation products in the liquid phase of the ink, gas chromatography with flame ionization detector (GC/FID, Agilent 6890N, Agilent Technologies, Santa Klara, CA, USA) was used to avoid oversaturation of the mass spectroscopy detector. The chromatogram of the pure solvent was used as baseline for peak integration to determine degradation products solely formed in the ink. One single ink sample per set storage time was used. Pseudo-samples, where Pt/C was replaced by Vulcan XC-72R, for all investigated time steps were prepared to isolate the formation of degradation products to the presence of platinum. Before analysis, particles were removed via filtration (hydrophobic PTFE syringe filters, pore size of 0.45 µm). Filters were cooled to avoid product loss. The column was of type Agilent DB-624 UI (Agilent Technologies, Santa Klara, CA, USA). A similar temperature profile was applied. Based on the findings from GC/MS, a calibration line for each substance with concentrations from 100 to 10,000 ppm, w. was established.
2.3. Electrochemical Analysis—RDE, MEA
Samples are denoted as “RDE” or “MEA” with the ink storage duration in hours added (e.g., MEA-0 represents an MEA fabricated from fresh ink). In addition to purely stored ink, an extreme example with added degradation product (notation EX) is characterized, as discussed in Section 3.2. To explore the effect of storage on the catalyst activity, the ink was electrochemically investigated using a rotating disk electrode (RDE) setup from Gamry (Gamry Instruments Reference 600, RDE710, Gamry Instruments, Warminster, PA, USA). A reversible hydrogen electrode (RHE, Gaskatel GmbH, Kassel, Germany) and a platinized titanium rod (Bank Elektronik–Intelligent Controls GmbH, Pohlheim, Germany) were used as reference and counter electrode, respectively. The working electrode consisted of a glassy carbon substrate with an active area of 0.1963 cm2. For each storage time, a separate ink sample was prepared. After storage, inks were redispersed for 15 min. Two layers each of 5 µL ink were applied and dried at ambient temperature at a rotational speed N of 700 rpm for 10 min. The catalyst loading, lPt (g/cm2), was 40 µg/cm2. Measurements were conducted at 30 °C in 0.1 M HClO4 (Carl Roth, Karlsruhe, Germany) as electrolyte. Before measurements, the electrolyte was deaerated using N2 for a minimum of 30 min. Cyclic voltammetry was performed as conditioning method. The ECSA of the 1st, 9th and 25th cycle was evaluated through determination of the hydrogen desorption charge, QHUPD (C/cm2) via integration of the hydrogen desorption peak (HUPD, Figure S1) and Equation (1) to verify the conditioning progress [37].
To correct the oxygen reduction reaction (ORR) measurements, a cyclic voltammogram (CV) was recorded at similar conditions. Prior to the investigation of the ORR, the electrolyte was saturated with O2, which was constantly supplied during the measurements. The diffusion limited current density, id (mA/cm2), was evaluated as arithmetic mean between 350 and 450 mV vs. RHE of the anodic scan (see Figure S2a). The number of transferred electrons was calculated by applying Koutecky–Levich analysis [38], as shown in Equation (2), where id is connected to the square root of the rotational speed of the working electrode, ω (rad/s), by the direct proportionality factor, k (mA/cm2 rad0.5s0.5). This factor k can be determined graphically from the Koutecky–Levich plot as the slope of the straight line through the origin obtained by plotting id over ω1/2 (see Figure S2b).
This factor k can be further defined according to Equation (3)
with F as the Faraday constant (96,485 C/mol), DO2,elec and cO2,elec as the diffusion coefficient (1.90 ∙ 10−9 m2/s) and bulk concentration (1.18 mol/m3) of oxygen in the electrolyte, νelec as the kinematic viscosity of the electrolyte (8.93 ∙ 10−7 m2/s) and n as the number of transferred electrons [-], respectively [39]. The measurement protocol is summarized in Table 1.

Table 1.
Measurement protocol for the investigation of ink storage on the catalytic performance using rotating disk electrode analysis.
For the investigation of the effect of aged catalyst ink on the performance of fuel cells, MEAs were fabricated. Reproducibility was checked and confirmed for MEA-0 and -168. For each storage time, a separate ink sample was prepared, and inks were redispersed for 15 min. A catalyst-coated membrane (CCM) was made by application of the catalyst ink on a Nafion membrane (Nafion N211, 25.4 µm, Fuel Cell Store, Bryan, TX, USA) via ultrasonic spray coating (Sonotech ExactaCoat OP3, SonoTek Corporation, Milton, NY, USA). The active area of the CCM was 25 cm2 with a Pt loading of 0.1 mg/cm2 on both the anode and cathode. Full MEAs were assembled with gas diffusion layers (Sigracet 22BB, SGL Carbon SE, Wiesbaden, Germany).
Measurements were conducted in a commercial test cell setup (balticFuelCells GmBH, Schwerin, Germany) at 80 °C cell temperature and 100% relative humidity. The gas inlet temperature was 85 °C to prevent condensation. All gases used (AirLiquide Alphagas, Paris, France) had a purity higher than 99.99%. Measurements were conducted using a PP242 power potentiostat coupled with a Zennium Pro workstation (Zahner-elektrik GmbH & Co. KG, Kronach, Germany). The measurement protocol is summarized in Table 2. First, the ECSA was determined similarly to the RDE before conditioning. Afterwards, the MEA was conditioned based on the protocol by Grandi et al. [33] via potential cycling, as demonstrated in Figure S3. Each potential step was held for 60 s. After conditioning, CVs were performed again to determine the improvement in the ECSA from conditioning. The individual cell resistances were determined via electrochemical impedance spectroscopy (EIS). At each recorded current density step, an amplitude, a (mA/cm2), of 10% of i and frequencies, f (Hz), from 1 kHz to 300 mHz were applied. An equivalent circuit based on the work of Santana et al. [40] as well as Ferreira et al. [41], shown in Figure 1, was simulated by Zahner Analysis 3.2.0. This equivalent circuit consists of an inductive coil for the cables from the cell to the potentiostat (Lcable), a membrane resistance (RPEM) and two parallel circuits in series with each a resistance (R) and constant phase element (CPE) for charge transfer (CT) and mass transfer (MT) impedance. Lastly, a polarization curve was recorded under similar conditions as during the EIS measurement in galvanostatic mode and with stoichiometric gas flows (stoichiometry λ [-]).
Table 2.
Measurement protocol for the investigation of ink storage on the single fuel cell performance.
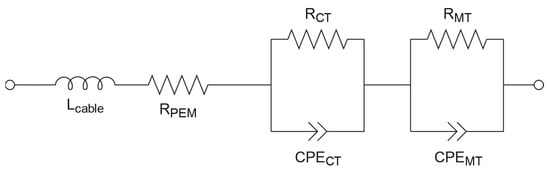
Figure 1.
Equivalent circuit for the determination of individual cell resistances [40,41] via electrochemical impedance spectroscopy.
The morphology and structure of the CL before characterization were investigated using scanning electron microscopy (SEM) and mercury (Hg) intrusion porosimetry. For SEM, the CCMs were cut into small pieces of approx. 0.5 × 0.5 cm with a scalpel and fixed with conductive adhesive tape on a specimen holder. The CL morphology was scrutinized with an FEI-XL20 SEM by Philips (Amsterdam, Netherlands) and the element distribution with an energy dispersive spectroscopy (EDX) detector manufactured by remX GmbH (Bruchsal, Germany).
Hg intrusion was performed using a PASCAL 140 and 440 Series Mercury Porosimetry equipped with a CD3 dilatometer (Thermo Fisher Scientific, Waltham, MA, USA). The sample amount of CCMs was approx. 20 mg. Blank measurement was performed with the empty dilatometers. A measurement started with outgassing, followed by an air pulse and a filling run in which the dilatometer was filled with mercury. After the low-pressure measurement (PASCAL 140 device), the sample was carefully transferred into the PASCAL 440 porosimeter where a high-pressure measurement (400 MPa) was performed. Calculations were made based on the Washburn equation in SOL.I.D software (Version 1.6.6, Thermo Fisher Scientific).
3. Results and Discussion
The following section shows and discusses results obtained from the above-mentioned methods to investigate the storage effects of catalyst ink.
3.1. Physical Analysis—UV-Vis Spectroscopy, Zeta Potential
The sedimentation behavior of the catalyst ink can be seen in Figure 2. In Figure 2a, the recorded spectrum of samples extracted from the top of the ink is shown. The acquired absorbance at 600 nm over a storage time of two weeks (336 h) for samples taken from the top and bottom of the ink is demonstrated in Figure 2b. The particle concentration decreased at the top of the ink, where it more than halved, while it increased at the bottom with a doubling over the experimental time. A low standard deviation indicates the reproducibility of the measurements.
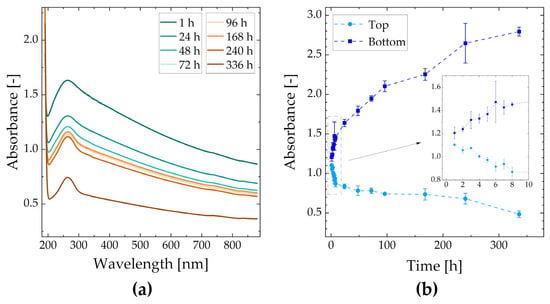
Figure 2.
Absorbance spectra of sedimenting catalyst ink for varying storage times (a); absorbance of catalyst ink (top and bottom samples) at a wavelength of 600 nm (b).
The recorded Zeta potential was −45.32 ± 0.49 mV, which indicates that the catalyst ink was a very stable dispersion due to repulsive interaction [42,43]. The recorded value is comparable to literature values where Nafion was used as the ionomer [13,32,43]. Thus, the sedimentation behavior observed via UV-Vis spectroscopy is unexpected, especially the strong deviation rate from the initial particle concentration in the first few hours. This could be explained by the absence of an inner structure of the catalyst ink because a very small amount of Nafion as stabilizing polymer is present.
The results of the physical aging indicate that catalyst ink should be used immediately after mixing. A lower particle concentration, as expected, might not be visible to the naked eye and, hence, a mistake in further fuel cell manufacturing could be the result. Moreover, the Zeta potential should not be taken solely into account when developing a new catalyst ink formulation when evaluating the dispersion stability, but UV-Vis spectroscopy should be considered as well.
3.2. Chemical Analysis—GC/MC, GC/FID
The headspace analysis of the catalyst inks revealed the presence of acetone as the main degradation product, which is formed according to Equation (4). Further products that are formed via the oxidation and condensation of ethanol (acetaldehyde, diethylether), solvent of the used Nafion dispersion, were determined in traces. As the amount of these two products was below the detection limit of the GC/FID, they were not further treated.
The liquid phase analysis of the catalyst ink over four weeks (672 h) using GC/FID is shown in Figure 3. Low error bars indicate the high reproducibility of the conducted measurements. An acetone content of 21.94 ± 0.5 ppm, w. was found in the pure solvent. The first three points were recorded after storage times of 0, 3 and 6 h. It is demonstrated that acetone is not only present in fresh ink in non-negligible amounts (1233 ppm, w.) but its concentration rises rapidly within the first week (2433 ppm, w.). Afterwards, the reaction rate seems to decrease and stay approx. linear. This indicates that, even after 4 weeks, an equilibrium concentration of acetone in the catalyst ink was not reached, and there was enough oxygen in the gas phase above the liquid ink within the sample vile to diffuse in the ink and react with the solvent. The acetone concentration in pseudo ink can be explained by Nafion dispersion impurities and non-catalyzed oxidation. As the concentration of acetone in the pseudo ink does not rise significantly over the whole experimental period, it can be concluded that the oxidation of 2-propanol was catalyzed by platinum.
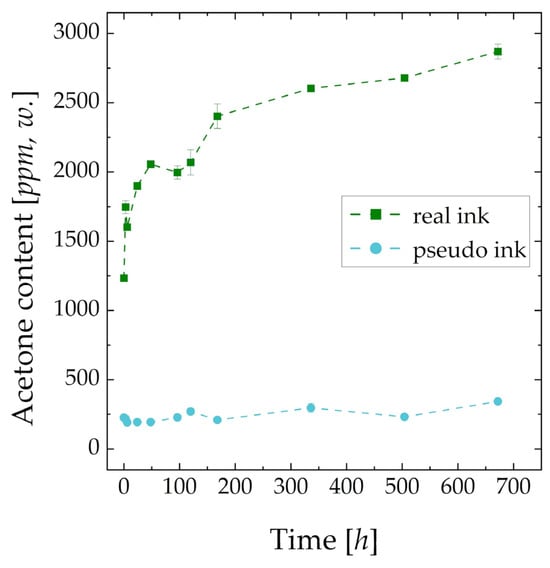
Figure 3.
Measured concentration of acetone as degradation product in real and pseudo catalyst ink over a storage time of 4 weeks (672 h); pure solvent: 21.94 ± 0.5 ppm, w.
As mentioned in Section 2.3, an extreme example of degraded catalyst ink was through the addition of acetone as the degradation product to more directly investigate the effect of acetone in the catalyst ink. The chosen representative amount was 10 wt.%, whereas the amounts of 2-propanol and water were adjusted, respectively, according to Equation (4).
3.3. Electrochemical Analysis—RDE, MEA
3.3.1. RDE
ECSA—Figure 4a demonstrates the CVs of the first cycle from fresh, stored and EX-ink. The height of the HUPD and hydrogen evolution peak increase with increasing acetone content, which indicates that acetone in the ink increases the initial availability of Pt for protons [44,45]. Despite being visually dry, the presence of acetone in the CL can be seen by a slight voltage shift in the Pt reduction peak [46]. This indicates that the initial availability of Pt, characterized by the HUPD, is harmed by neither storage nor an increased acetone content. Compared to Figure 4b, where the cyclic voltammogram of the last conditioning cycle is shown, the Pt oxidation peak is lowered. Moreover, peaks in the CV (e.g., HUPD, Pt-oxidation peak) become more distinct after storage or with added acetone. This can be explained by the formation of secondary pores within the CL due to a comparable higher evaporation rate of acetone [46,47]. These pores are reached by the electrolyte during the conditioning. It cannot be stated whether or not Nafion is dissolved, as demonstrated in Section 3.3. Figure 4c compares the ECSA of the 1st, 9th and 25th (last) cleaning cycle obtained from catalyst inks after various storage times and RDE-EX. Generally, values do not differ strongly from each other. Considering the first conditioning cycle, the ECSA appears to be approximately constant after a storage time of 168 h. Within the first 72 h of storage, an increased ECSA for the 9th and 25th cycle compared to fresh ink is visible. A comparably lower mean ECSA of RDE-96 can be explained through CL fabrication issues, as observable due to the size of the corresponding error bars. After the 25th cycle, RDE-168 to RDE-672 provide a similar to slightly higher ECSA than RDE-0. This indicates that storage for up to four weeks and the formation of acetone do not negatively affect the ECSA. For RDE-EX, the ECSA is similar for the first cycle but notably higher for both the 9th and 25th cycle compared to RDE-0 and RDE-672. It can, therefore, be said that an amount of acetone higher than that generated via solvent oxidation increases the availability of the catalyst after conditioning using CVs.
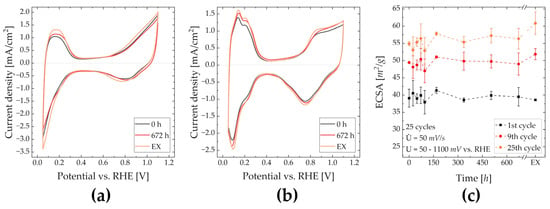
Figure 4.
CVs for conditioning during RDE measurements; first cycle for RDE-0, -672 and –EX (a); last cycle for RDE-0, -672, and –EX (b); ECSA of first, 9th and 25th (last) conditioning cycle over the catalyst ink storage time including EX (c).
ORR-activity—In Figure 5, the evolution of n, calculated from the Koutecky–Levich plot, over the ink storage time is shown. Within the first 48 h of storage, the selectivity towards water formation is not negatively influenced, which is comparable to results from Koh and Strasser [25]. For RDE-96, the first minimum in selectivity is reached with an n equaling 3.39 or a selectivity of 69.5% towards water formation. A similar local minimum is visible in the ECSA, albeit significantly less pronounced. Comparably large error bars for both the n and ECSA may indicate that the performance of RDE-96 is very sensitive to statistical errors, e.g., application of ink on the glassy carbon electrode. It appears that acetone is present on the surface of Pt, even though the electrode history indicates the oxidation of acetone. For RDE-168 to RDE-504, n recovers and, ultimately, RDE-504 performs similarly to RDE-0. This behavior can be explained by the increasing ECSA and cleaning effect in Figure 4c. The sharp drop in n for RDE-672 would contradict this theory; however, it cannot be excluded that this value is an outlier [48,49]. A decreasing n for RDE-EX compared to RDE-504 indicates that relatively high amounts of acetone in the ink negatively influence the selectivity towards water formation.
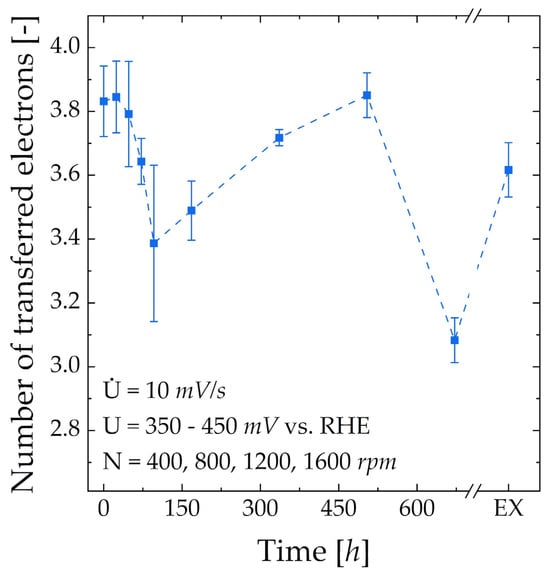
Figure 5.
Evolution of number of transferred electrons from Koutecky–Levich analysis (RDE) over catalyst ink storage time.
To summarize, the catalyst ink prepared with the ingredients used in this work and investigated via RDE should be used within 48 h or after storage between two and three weeks. Conditioning does not fully remove acetone from the Pt surface. While the ECSA after conditioning increases with increasing acetone content, a clear connection between the acetone content and n cannot be made. The ECSA does not fully relate to n, especially at high acetone contents.
3.3.2. MEA
Morphology—In Figure 6, the morphology of the CL of the three extremum examples MEA-0, MEA-672 and MEA-EX (from left to right) before conditioning is shown. At 120x magnification (top row), all three CLs appear similarly homogeneous, which indicates that neither ink storage nor the addition of acetone leads to macroscopic agglomerations during ink application and drying via the ultrasonic spray coating process. Compared to 2-propanol, acetone shows a slightly higher polarity index (5.1 vs 3.9 [50]) as well as dielectric constant (19.4 vs. 20.6 [51,52]), which indicates that the CL should appear more even with the presence of acetone [53,54]. This is indeed the case at a higher magnification (650×, bottom row), where the CL becomes smoother with increasing acetone content. As demonstrated in Figure S4 through EDX, the homogeneous dispersion of the catalyst and ionomer is not affected by storage or the presence of acetone.

Figure 6.
Scanning electron microscopy images of catalyst layers of MEA-0 (a), MEA-672 (b) and MEA-EX (c) at low (1) and high (2) magnification.
Another explanation for this could be that the boiling point of the catalyst ink is lowered due to the addition of acetone [55]. This results in a higher evaporation rate, where solvent vapor has to migrate through the drying CL, resulting in the formation of a finer pore structure with increased porosity [56] as solid particles do not have enough time to form large agglomerates [53,56].
The results from Hg porosimetry show that with increasing acetone content, the average pore size decreases and the specific surface area increases (Table 3). Adsorption/desorption isotherms in Figure 7 show that the pore shape is altered. For MEA-0, pores appear to have a prism-like shape (e.g., cylindrical) because the adsorption and desorption isotherm show a similar path. For MEA-672, a hysteresis effect is visible, which indicates that pores have a bottle-like shape. This effect is further intensified for MEA-EX, which implies that with increasing acetone content, the bottle neck of pores in the CL becomes smaller.
Table 3.
Average pore diameter and specific surface area for MEA-0, MEA-672 and MEA-EX, determined through Hg porosimetry.
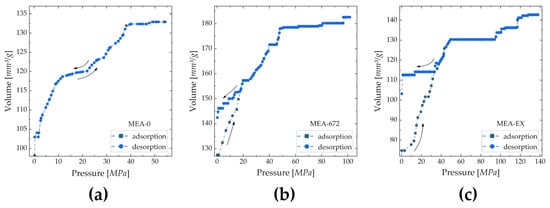
Figure 7.
Adsorption and desorption isotherms of MEA-0 (a), MEA-672 (b) and MEA-EX (c) recorded via Hg intrusion porosimetry.
Conditioning—The mean current densities during conditioning at 0.6 V and 0.4 V for four representative MEAs (MEA-0, -336, -672, -EX) are shown in Figure 8a,b, respectively. The results for all MEAs can be found in Figure S5. As expected, the current densities for all MEAs increase during the conditioning procedure, and a plateau is reached by its end. However, the relative improvements between the MEAs differ significantly, as shown in Table 4. The lowest and highest relative current increase is observed for MEA-0 and MEA-672, respectively. At 0.6 V, a steady increase in the relative improvement and decrease in the current density within the first conditioning loop with increasing ink storage duration are visible. After the last cycle, MEA-336 and MEA-672 perform similarly to MEA-0, whereas MEA-672 appears not to have reached its maximum yet. For MEA-EX, the initial current density is similar to MEA-0, but the final current density is approx. 11% higher. At 0.4 V, MEA-0 has almost reached its final performance within the first six cycles, while all other MEAs show a sharp performance increase, especially MEA-672, within the first ten to eleven cycles. The highest performance was reached by MEA-0, whereas it should be noted that all cells show a comparable performance after the last cycle.
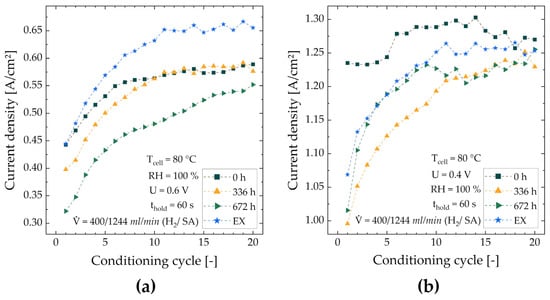
Figure 8.
Current density evolution during the conditioning process of MEA-0, -336, -672 and -EX at 0.6 V (a) and 0.4 V (b) (mean over the holding time of each voltage step).
Table 4.
Current density at 0.6 V and 0.4 V of first and last conditioning cycle including the relative increase of the current density for MEAs fabricated from fresh ink (MEA-0), stored ink (MEA-336, MEA-672) and ink with added acetone (MEA-EX).
The results from the conditioning cycles show that for MEAs fabricated from stored ink, storage has a negative effect on the initial performance. However, their performance approx. equalizes to MEA-0 during the conditioning phase, which implies that ink for MEA fabrication can be stored for at least up to four weeks, with final performance losses of less than 7% if conditioned according to the used protocol. With a comparably higher amount of acetone in the catalyst ink (MEA-EX), the final performance is higher at low current densities (0.6 V) but similar to MEAs from stored ink at high current densities. This indicates that the addition of acetone to the catalyst ink has a positive effect on the cell performance at lower current densities. Furthermore, it is shown that the presence of acetone has no delaying effect on the activation of MEAs.
ECSA—The ECSA before and after conditioning for all MEAs is shown in Table 5. In addition to MEA-336, the ECSA before conditioning shows a slightly decreasing trend over the storage time, which can be explained by the pore structure observed and potential catalyst poisoning. While MEA-0 has larger pores, the initial humidification of the CL by the gas flows is less hindered and, therefore, more catalyst is available within the three-phase boundary (catalyst/ionomer/gas). As the acetone content increases, especially for MEA-EX, pores in the CL become smaller, and more catalyst is initially unavailable, decreasing the ECSA. After conditioning, when the CL is penetrated with water via self-humidification, the results are opposite, similar to the results obtained from the RDE. In addition to MEA-168, the ECSA for MEAs from inks with an increased acetone content is higher. The relative increase improves with increasing acetone content and is approx. constant for MEA-168 to -672. This behavior can be also explained by the change in morphology of the CL observed in Figure 6 as well as the increased specific surface area. Throughout the conditioning process, the pores in the CL, including finer ones at increasing acetone content, are humidified, which enhances the proton conductivity of the ionomer and, hence, leads to an increased availability of the catalyst. Similar to CVs obtained from RDE analysis, acetone as a contaminant is present (see Figure S6). However, the effects are less pronounced, which can be explained by the ultrasonic spray-coating process for ink deposition, during which acetone can evaporate more easily due to induced heat.
Table 5.
ECSA before and after conditioning including its relative increase for all MEAs characterized.
Polarization curve—The polarization and power density curves for all cells investigated are shown in Figure 9. In Table 6, representative values (OCV, current densities in linear region, voltage at maximum current density) are summarized. Generally, performance results between the MEAs are not significantly different. The OCV increases with ink storage time, which, again, indicates that the catalyst availability is increased, and the MEA stays hydrated at low current densities due to the fine pore structure of the CL. A deteriorated OCV for MEA-EX shows that the membrane can be attacked by acetone at concentrations ≥ 10 w.%, which triggers fuel crossover [44,45,57]. The cell performance in the ohmic region of MEAs from stored ink initially worsens compared to MEA-0 but recovers later for MEA-504 and further improves. The decreased performance of MEA-168 is explainable by the also comparably low ECSA, which indicates that the catalyst might be partially poisoned by acetone, as demonstrated by Bondue et al. [58]. The reason for an equally low current density below 0.7 V of MEA-336 is not evident from the measured data. In addition to membrane effects (shown in the next subsection), differences between the MEAs in the ohmic part of the polarization curve may be explained by mass transfer modifications induced by morphological changes in the CL, shown in Figure 7 and Table 3. At low current densities, i.e., away from the limiting current regime, the mass transfer overpotential is considered pseudo-ohmic. The high performance of MEA-672 can be explained by both the ECSA and the morphology. The fine pore structure partially retains water and, hence, keeps both the CL and PEM humidified, which increases the performance. The eventual poisoning of the catalyst is overwhelmed by these effects. The current density of MEA-EX is similar to MEA-0 above 0.7 V and lies between MEA-0 and -672 at lower voltage. In addition to potential catalyst poisoning, the pore structure of MEA-EX might be too fine for water removal and gas supply, which leads to increased mass transfer resistance. It can, therefore, be said that an optimal pore structure is obtained after 672 h of ink storage. The effects shown in Figure 9 can be explained by EIS in the next subsection.
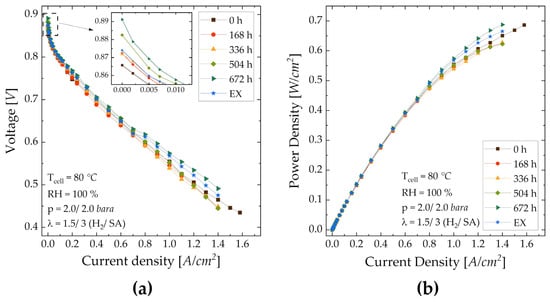
Figure 9.
Polarization curve of all MEAs tested (fresh ink, storage of 168 h to 672 h, with added acetone) (a); resulting power density over the current density (b).
Table 6.
Polarization data (OCV, current density in ohmic region, voltage at maximum current density) for all MEAs characterized.
EIS—In Figure 10, the impedance spectra for four representative cells (MEA-0, MEA-336, MEA-672, MEA-EX) as well as the fitted membrane, charge transfer and mass transfer resistances for all cells at low, medium and high current density are shown. All recorded impedance spectra of all MEAs are shown in the Supplementary Material (Figure S7). MEA-168 shows a spectrum identical to MEA-0 for most current densities; hence, the comparably lower performance can be explained by the lower ECSA. At all current densities, the total cell resistance has a maximum for MEA-336 and a minimum for MEA-672, which explains its comparably high power output. This indicates that ink storage beyond two weeks is beneficial for cell resistance. During storage, RPEM increases marginally. Related to MEA-0 and -EX, RPEM for MEA-336 and -672 is slightly higher, which indicates a lower crossover and short-circuit current and explains the increasing OCV of MEAs fabricated from stored ink. For almost all cells and current densities, RCT represents the highest resistance. At low current density (i = 0.4 A/cm2), RCT decreases with increasing ink storage time but increases for MEA-EX. The reason for this is a lowered activation loss due to water storage and a smoother CL structure [40], which further explains the comparably high performance of MEA-672. Additionally, as demonstrated by Ren et al. [32] for Aquivion, the ionomer coverage of catalyst particles is increased over storage time, which promotes proton conduction. For RMT, an increase with increasing current density is observed, which is explainable by a growing water formation rate that blocks pores in the CL and GDL. The maximum for MEAs prepared from stored ink for MEA-336 could be explained by the pore structure. Here, pores are smaller than for MEA-0 but start showing a bottle-like shape, as for MEA-672, which appears to be the worst case for water removal and gas access within the MEA. With increasing acetone content (i.e., finer pore structure), the difference between RCT and RMT becomes smaller. The negative effect of a very fine pore structure on mass transfer is also visible at MEA-EX, where RMT becomes the dominant cell resistance at medium current density. As impedance spectra were recorded from high to low current density, it appears that water produced at higher current density was retained within the finer pore structure. A lower RMT for MEA-0, especially at high current density, verifies that through the pore structure, gas can permeate without major hindrance by water.

Figure 10.
Impedance spectra recorded by EIS (1) and resistances according to the used equivalence circuit (2) for MEA-0, -336, -672 and -EX at 0.4 A/cm2 (a), 0.8 A/cm2 (b) and 1.2 A/cm2 (c).
4. Conclusions
In this work, the effect of the storage of catalyst ink for PEFCs was investigated. UV-Vis spectroscopy revealed that catalyst particles show a strong sedimentation tendency, despite being stabilized by charge interaction. Hence, catalyst ink should be used as soon as possible after dispersion or continuously dispersed during application. Acetone as an oxidation product of the used organic solvent (2-propanol) was found via GC analysis, even in freshly prepared ink. Compared to samples with pseudo ink, it was found that the oxidation reaction is catalyzed by platinum. An equilibrium concentration was not reached after a storage time of four weeks. Based on GC results, an extreme example with 10 wt.% acetone in the catalyst (notation EX) was additionally characterized electrochemically.
The ECSA recorded using RDE showed no negative influence of storage but increased with higher acetone content after conditioning. A slight shift in the Pt reduction peak in the first CV due to residual acetone is visible. The number of transferred electrons, calculated from Koutecky–Levich analysis, illustrated that the selectivity towards water formation is constant for the first 48 h of storage but decreases rapidly until a storage time of 96 h to 168 h. A recovering trend is visible, with ink stored for three weeks performing similarly to fresh ink. Another selectivity drop for ink stored for four weeks requires further studies. In addition to statistical errors, an altered structural change in the CL on the glassy carbon electrode and residual acetone impurities are plausible. It can be concluded that catalyst ink with ingredients used within this work should be used within the first 48 h for RDE analysis to ensure a performance similar to fresh ink.
SEM and Hg porosimetry of MEA CLs before conditioning proved that CLs show a finer pore structure and more bottle-shaped pores as the catalyst ink age increases. With extra acetone added, an even finer pore structure is obtained, which attributes the morphology change observed to the acetone content. The homogeneous distribution of the catalyst and ionomer within the CL is negatively influenced neither by ink storage nor by ink acetone content. MEAs prepared from stored catalyst ink show an initially deteriorated performance compared to MEA-0, which is almost recovered throughout the applied conditioning process. The ECSA before conditioning behaves similarly but increasingly improves with increasing acetone content in the ink, which implies that, as well as changes in the pore structure, the catalyst might be partially poisoned by acetone at first but is even better recovered after conditioning compared to MEA-0. Considering performance, the OCV increases with increasing ink age. In relation to MEA-0, less activation and charge transfer losses but higher mass transfer losses, especially for MEA-EX, are observed. Ink stored for up to three weeks leads to a slightly weakened power output, while ink stored for four weeks and ink with added acetone demonstrate a particularly higher power output. It is, therefore, proposed that catalyst ink for MEAs can be stored for at least up to four weeks at 6 °C without major performance losses. The addition of 10 wt.% acetone slightly increases the overall performance compared to fresh ink, which indicates that acetone as an additional organic solvent, at least in small amounts in the catalyst ink, should be further considered. These findings are especially relevant for large-scale MEA production in the case of unexpected long-time production shutdowns as, eventually, the produced catalyst does not have to be discarded immediately. It should be noted that this is only the case for the ink composition used and if this is properly conditioned, as shown by the used conditioning protocol. Future studies will consider different organic solvents (e.g., 1-propanol), a varying organic solvent to water ratio, other catalysts, different ionomers, I/C ratios, altered conditioning protocols (e.g., cathode starvation) and the structural change in catalyst ink during storage (e.g., particle agglomeration through Ostwald ripening) to potentially further improve the cell performance. With respect to long-term MEA stability, accelerated stress tests and various operating conditions (e.g., high current, low relative humidity, low stoichiometry) will be investigated.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/en16197011/s1. Figure S1: Cyclic voltammogram with integrated hydrogen desorption peak for ECSA evaluation. Figure S2: ORR activity (CV) recorded with RDE at varying rotational speeds (a); mean diffusion limited current density over the square root of the rotational speed for with linear fit for Koutecky-Levich analysis (b). Figure S3: Potential profile used as conditioning method for MEAs. Figure S4: SEM-EDX images of CLs of MEA-0 (a), -672 (b) and –EX (c) for catalyst (Pt La) (1) and Nafion (F Ka) (2). Figure S5: Current density evolution (mean over the holding time of each voltage step) during the conditioning process for all MEAs characterized at 0.6 V (a) and 0.4 V (b). Figure S6: CVs of MEA-0, -672 and -EX before conditioning (a) and after conditioning (b). Figure S7: Impedance spectra recorded by EIS for all MEAs characterized at 0.4 A/cm2 (a), 0.6 A/cm2 (b), 0.8 A/cm2 (c), 1.0 A/cm2 (d) and 1.2 A/cm2 (e).
Author Contributions
M.K.: conceptualization, data evaluation, experimental work, formal analysis, funding acquisition, overall methodology, supervision, visualization, writing—original draft preparation; M.R.: conceptualization, experimental work, graphical illustration, methodology, writing—original draft preparation, writing—review and editing; W.Y.K.: data evaluation, experimental work, methodology; F.B.: data evaluation, experimental work, methodology, writing—original draft preparation; M.G.: conceptualization, methodology, writing—review and editing; M.B.: conceptualization, methodology, writing—review and editing; V.H.: formal analysis, funding acquisition, supervision, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
Open Access Funding by the Graz University of Technology.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank the CEET lab team, Herta Luttenberger and Tanja Weiß, as well as Silvia Maitz, for their support during practical work, as well as Brigitte Hammer and Bettina Koch for their diligent work in the secretary of CEET. Special thanks go to the Hacker working group, Sebastian Rohde (Institute of Chemistry and Technology of Materials) as well as Adrian Baumunk and Bastian Etzold (Darmstadt University of Technology) for helpful input. Financial support from the TU Graz Open Access Publishing Fund is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ritchie, H.; Roser, M.; Rosado, P. CO2 and Greenhouse Gas Emissions. Available online: https://ourworldindata.org/co2-emissions?utm_source=tri-city%20news&utm_campaign=tricity%20news%3A%20outbound&utm_medium=referral (accessed on 25 July 2023).
- U.S. Energy Information Administration. Use of Energy Explained: Energy Use for Transportation. Available online: https://www.eia.gov/energyexplained/use-of-energy/transportation.php (accessed on 11 April 2022).
- Cullen, D.A.; Neyerlin, K.C.; Ahluwalia, R.K.; Mukundan, R.; More, K.L.; Borup, R.L.; Weber, A.Z.; Myers, D.J.; Kusoglu, A. New roads and challenges for fuel cells in heavy-duty transportation. Nat. Energy 2021, 6, 462–474. [Google Scholar] [CrossRef]
- Fan, L.; Tu, Z.; Chan, S.H. Recent development of hydrogen and fuel cell technologies: A review. Energy Rep. 2021, 7, 8421–8446. [Google Scholar] [CrossRef]
- Olabi, A.; Wilberforce, T.; Abdelkareem, M.A. Fuel cell application in the automotive industry and future perspective. Energy 2021, 214, 118955. [Google Scholar] [CrossRef]
- Trencher, G. Strategies to accelerate the production and diffusion of fuel cell electric vehicles: Experiences from California. Energy Rep. 2020, 6, 2503–2519. [Google Scholar] [CrossRef]
- Trencher, G.; Taeihagh, A.; Yarime, M. Overcoming barriers to developing and diffusing fuel-cell vehicles: Governance strategies and experiences in Japan. Energy Policy 2020, 142, 111533. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Fan, Y. Techno-Economic Challenges of Fuel Cell Commercialization. Engineering 2018, 4, 352–360. [Google Scholar] [CrossRef]
- Grandi, M.; Rohde, S.; Liu, D.; Gollas, B.; Hacker, V. Recent advancements in high performance polymer electrolyte fuel cell electrode fabrication—Novel materials and manufacturing processes. J. Power Sources 2023, 562, 232734. [Google Scholar] [CrossRef]
- Peron, J.; Mani, A.; Zhao, X.; Edwards, D.; Adachi, M.; Soboleva, T.; Shi, Z.; Xie, Z.; Navessin, T.; Holdcroft, S. Properties of Nafion® NR-211 membranes for PEMFCs. J. Membr. Sci. 2010, 356, 44–51. [Google Scholar] [CrossRef]
- Heras, A.D.L.; Vivas, F.; Segura, F.; Andújar, J. From the cell to the stack. A chronological walk through the techniques to manufacture the PEFCs core. Renew. Sustain. Energy Rev. 2018, 96, 29–45. [Google Scholar] [CrossRef]
- Mayyas, A.; Mann, M. Emerging Manufacturing Technologies for Fuel Cells and Electrolyzers. Procedia Manuf. 2019, 33, 508–515. [Google Scholar] [CrossRef]
- Berlinger, S.A.; Garg, S.; Weber, A.Z. Multicomponent, multiphase interactions in fuel-cell inks. Curr. Opin. Electrochem. 2021, 29, 100744. [Google Scholar] [CrossRef]
- Bapat, S.; Giehl, C.; Kohsakowski, S.; Peinecke, V.; Schäffler, M.; Segets, D. On the state and stability of fuel cell catalyst inks. Adv. Powder Technol. 2021, 32, 3845–3859. [Google Scholar] [CrossRef]
- Yang, F.; Xin, L.; Uzunoglu, A.; Qiu, Y.; Stanciu, L.; Ilavsky, J.; Li, W.; Xie, J. Investigation of the Interaction between Nafion Ionomer and Surface Functionalized Carbon Black Using Both Ultrasmall Angle X-ray Scattering and Cryo-TEM. ACS Appl. Mater. Interfaces 2017, 9, 6530–6538. [Google Scholar] [CrossRef]
- Shukla, S.; Bhattacharjee, S.; Weber, A.Z.; Secanell, M. Experimental and Theoretical Analysis of Ink Dispersion Stability for Polymer Electrolyte Fuel Cell Applications. J. Electrochem. Soc. 2017, 164, F600–F609. [Google Scholar] [CrossRef]
- Chisaka, M.; Matsuoka, E.; Daiguji, H. Effect of Organic Solvents on the Pore Structure of Catalyst Layers in Polymer Electrolyte Membrane Fuel Cells. J. Electrochem. Soc. 2010, 157, B1218–B1221. [Google Scholar] [CrossRef]
- Dixit, M.B.; Harkey, B.A.; Shen, F.; Hatzell, K.B. Catalyst Layer Ink Interactions That Affect Coatability. J. Electrochem. Soc. 2018, 165, F264–F271. [Google Scholar] [CrossRef]
- Berlinger, S.A.; Chowdhury, A.; Van Cleve, T.; He, A.; Dagan, N.; Neyerlin, K.C.; McCloskey, B.D.; Radke, C.J.; Weber, A.Z. Impact of Platinum Primary Particle Loading on Fuel Cell Performance: Insights from Catalyst/Ionomer Ink Interactions. ACS Appl. Mater. Interfaces 2022, 14, 36731–36740. [Google Scholar] [CrossRef]
- Guo, Y.; Pan, F.; Chen, W.; Ding, Z.; Yang, D.; Li, B.; Ming, P.; Zhang, C. The Controllable Design of Catalyst Inks to Enhance PEMFC Performance: A Review. Electrochem. Energy Rev. 2021, 4, 67–100. [Google Scholar] [CrossRef]
- Alink, R.; Singh, R.; Schneider, P.; Christmann, K.; Schall, J.; Keding, R.; Zamel, N. Full Parametric Study of the Influence of Ionomer Content, Catalyst Loading and Catalyst Type on Oxygen and Ion Transport in PEM Fuel Cell Catalyst Layers. Molecules 2020, 25, 1523. [Google Scholar] [CrossRef]
- Park, J.-H.; Shin, M.-S.; Park, J.-S. Effect of dispersing solvents for ionomers on the performance and durability of catalyst layers in proton exchange membrane fuel cells. Electrochim. Acta 2021, 391, 138971. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, D.; Tang, H.; Li, B.; Yang, D.; Ming, P.; Zhang, C. Solvent effects on the rheology of fuel cell catalyst ink and the adsorption of ionomers on the particles. Phys. Fluids 2022, 34, 103113. [Google Scholar] [CrossRef]
- Biddinger, E.J.; von Deak, D.; Marsh, H.S.; Ozkan, U.S. RRDE Catalyst Ink Aging Effects on Selectivity to Water Formation in ORR. Electrochem. Solid-State Lett. 2010, 13, B98–B100. [Google Scholar] [CrossRef]
- Shirlaine Koh, P.S. Dealloyed Pt Nanoparticle Fuel Cell Electrocatalysts: Stability and Aging Study of Cata-lyst Powders, Thin Films, and Inks. J. Electrochem. Soc. 2010, 157, B585–B591. [Google Scholar]
- Uemura, S.; Kameya, Y.; Iriguchi, N.; Yoshida, T.; Hirai, S. Time Lapse Visualization of Catalyst Ink Degradation by X-ray CT. ECS Trans. 2017, 80, 403–407. [Google Scholar] [CrossRef]
- Uemura, S.; Yoshida, T.; Koga, M.; Matsumoto, H.; Yang, X.; Shinohara, K.; Sasabe, T.; Hirai, S. Ink Degradation and Its Effects on the Crack Formation of Fuel Cell Catalyst Layers. J. Electrochem. Soc. 2019, 166, F89–F92. [Google Scholar] [CrossRef]
- Uemura, S.; Sasabe, T.; Sakai, K.; Shinohara, K.; Hirai, S. Relation between Degradation Reaction and Mixing at Fuel Cell Catalyst Ink Fabrication Process. ECS Trans. 2019, 92, 183–187. [Google Scholar] [CrossRef]
- Uemura, S.; Sakai, K.; Sasabe, T.; Matsumoto, H.; Sugimori, H.; Shinohara, K.; Hirai, S. Effect of Reaction Products on the PEFC Catalyst Ink Property and Catalyst Layer Quality. ECS Trans. 2020, 98, 61–65. [Google Scholar] [CrossRef]
- Uemura, S.; Sasabe, T.; Sakai, K.; Matsumoto, H.; Sugimori, H.; Shinohara, K.; Hirai, S. Reaction Products Affecting the PEFC Catalyst Ink Property. J. Electrochem. Soc. 2021, 168, 104504. [Google Scholar] [CrossRef]
- Liu, P.; Yang, D.; Li, B.; Zhang, C.; Ming, P. Influence of Degassing Treatment on the Ink Properties and Performance of Proton Exchange Membrane Fuel Cells. Membranes 2022, 12, 541. [Google Scholar] [CrossRef]
- Ren, H.; Meng, X.; Lin, Y.; Shao, Z. Structural stability of catalyst ink and its effects on the catalyst layer microstructure and fuel cell performance. J. Power Sources 2022, 517, 230698. [Google Scholar] [CrossRef]
- Grandi, M.; Mayer, K.; Gatalo, M.; Kapun, G.; Ruiz-Zepeda, F.; Marius, B.; Gaberšček, M.; Hacker, V. The Influence Catalyst Layer Thickness on Resistance Contributions of PEMFC Determined by Electrochemical Impedance Spectroscopy. Energies 2021, 14, 7299. [Google Scholar] [CrossRef]
- Ferstl, E.; Gabriel, M.; Gomernik, F.; Müller, S.M.; Selinger, J.; Thaler, F.; Bauer, W.; Uhlig, F.; Spirk, S.; Chemelli, A. Investigation of the Adsorption Behavior of Jet-Cooked Cationic Starches on Pulp Fibers. Polymers 2020, 12, 2249. [Google Scholar] [CrossRef] [PubMed]
- Roschger, M.; Wolf, S.; Hasso, R.; Genorio, B.; Gorgieva, S.; Hacker, V. Influence of the Electrode Deposition Method of Graphene-Based Catalyst Inks for ADEFC on Performance. ACS Appl. Mater. Interfaces 2023, 15, 40687–40699. [Google Scholar] [CrossRef] [PubMed]
- Hicks, L.M. (Ed.) Antimicrobial Peptides; Academic Press: Cambridge, MA, USA; San Diego, CA, USA; Oxford, UK; London, UK, 2022. [Google Scholar]
- Cooper, K. Laboratory #4—Fuel Crossover by Linear Sweep Voltammetry & Electrochemical Surface Area by Cyclic Voltammetry. Fuel Cell Magazine 2009. Available online: https://www.scribner.com/files/tech-papers/Scribner-on-Crossover-Fuel-Cell-Magazine-2008.pdf (accessed on 4 September 2023).
- Wolf, S.; Roschger, M.; Genorio, B.; Garstenauer, D.; Radić, J.; Hacker, V. Ce-modified Co–Mn oxide spinel on reduced graphene oxide and carbon black as ethanol tolerant oxygen reduction electrocatalyst in alkaline media. RSC Adv. 2022, 12, 35966–35976. [Google Scholar] [CrossRef]
- Wei, Y.-C.; Liu, C.-W.; Wang, K.-W. Improvement of oxygen reduction reaction and methanol tolerance characteristics for PdCo electrocatalysts by Au alloying and CO treatment. Chem. Commun. 2011, 47, 11927–11929. [Google Scholar] [CrossRef]
- Santana, J.; Espinoza-Andaluz, M.; Li, T.; Andersson, M. A Detailed Analysis of Internal Resistance of a PEFC Comparing High and Low Humidification of the Reactant Gases. Front. Energy Res. 2020, 8, 1–12. [Google Scholar] [CrossRef]
- Ferreira, R.B.; Falcão, D.; Oliveira, V.; Pinto, A. Experimental study on the membrane electrode assembly of a proton exchange membrane fuel cell: Effects of microporous layer, membrane thickness and gas diffusion layer hydrophobic treatment. Electrochim. Acta 2017, 224, 337–345. [Google Scholar] [CrossRef]
- Xu, R.; Wu, C.; Xu, H. Particle size and zeta potential of carbon black in liquid media. Carbon 2007, 45, 2806–2809. [Google Scholar] [CrossRef]
- Kim, O.; Oh, S.; Ahn, C.; Kim, S.; Kim, J.K.; Yang, S.; Choi, M.; Cho, Y.; Sung, Y. Enhanced Performance of Ionomer Binder with Shorter Side-Chains, Higher Dispersibility, and Lower Equivalent Weight. Fuel Cells 2018, 18, 711–722. [Google Scholar] [CrossRef]
- Esplandiu, M.J.; Reguera, D.; Romero-Guzmán, D.; Gallardo-Moreno, A.M.; Fraxedas, J. From radial to unidirectional water pumping in zeta-potential modulated Nafion nanostructures. Nat. Commun. 2022, 13, 2812. [Google Scholar] [CrossRef] [PubMed]
- Berens, S.J.; Yahya, A.; Fang, J.; Angelopoulos, A.P.; Nickels, J.D.; Vasenkov, S. Transition between Different Diffusion Regimes and Its Relationship with Structural Properties in Nafion by High Field Diffusion NMR in Combination with Small-Angle X-ray and Neutron Scattering. J. Phys. Chem. B 2020, 124, 8943–8950. [Google Scholar] [CrossRef] [PubMed]
- Wieckowski, A.; Zelenay, P.; Szklarczyk, M.; Sobkowski, J. The Pt-acetone-water system investigated by radiotracer and electrochemical methods. J. Electroanal. Chem. Interfacial Electrochem. 1982, 135, 285–299. [Google Scholar] [CrossRef]
- Akterian, S. Evaluating the Vapour Evaporation from the Surface of Pure Organic Solvents and Their Mixtures. Food Sci. Appl. Biotechnol. 2020, 3, 77–84. [Google Scholar] [CrossRef]
- Shinozaki, K.; Zack, J.W.; Pylypenko, S.; Pivovar, B.S.; Kocha, S.S. Oxygen reduction reaction measurements on platinum electrocatalysts utilizing rotating disk electrode technique: II. Influence of ink formulation, catalyst layer uniformity and thickness. J. Electrochem. Soc. 2015, 162, F1384–F1396. [Google Scholar] [CrossRef]
- Shinozaki, K.; Zack, J.W.; Richards, R.M.; Pivovar, B.S.; Kocha, S.S. Oxygen Reduction Reaction Measurements on Platinum Electrocatalysts Utilizing Rotating Disk Electrode Technique: I. Impact of impurities, measurement protocols and applied corrections. J. Electrochem. Soc. 2015, 162, F1144–F1158. [Google Scholar] [CrossRef]
- Fisher Scientific, Summary of Key Physical Data for Solvents. Available online: https://www.fishersci.co.uk/gb/en/scientific-products/technical-tools/summary-key-physical-data-solvents.html (accessed on 1 September 2023).
- Gmehling, J.; Kleiber, M.; Kolbe, B.; Rarey, J. Chemical Thermodynamics for Process Simulation, 4th ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015. [Google Scholar]
- Miller, C.G.; Maass, O. Determination of dielectric constant in binary organic sys-tems. Can. J. Chem. 1960, 38, 1606–1616. [Google Scholar] [CrossRef]
- Gong, Q.; Li, C.; Liu, Y.; Ilavsky, J.; Guo, F.; Cheng, X.; Xie, J. Effects of Ink Formulation on Construction of Catalyst Layers for High-Performance Polymer Electrolyte Membrane Fuel Cells. ACS Appl. Mater. Interfaces 2021, 13, 37004–37013. [Google Scholar] [CrossRef]
- Lei, C.; Yang, F.; Macauley, N.; Spinetta, M.; Purdy, G.M.; Jankovic, J.; Cullen, D.A.; More, K.L.; Kim, Y.S.; Xu, H. Impact of Catalyst Ink Dispersing Solvent on PEM Fuel Cell Performance and Durability. J. Electrochem. Soc. 2021, 168, 044517. [Google Scholar] [CrossRef]
- Horsley, L.H. (Ed.) Azeotropic Data; American Chemical Society: Washington, DC, USA, 1973; Volume 116, ISBN 8412-0166-8. [Google Scholar]
- Liu, H.; Ney, L.; Zamel, N.; Li, X. Effect of Catalyst Ink and Formation Process on the Multiscale Structure of Catalyst Layers in PEM Fuel Cells. Appl. Sci. 2022, 12, 3776. [Google Scholar] [CrossRef]
- Moore, M.; Shukla, S.; Voss, S.; Karan, K.; Weber, A.; Zenyuk, I.; Secanell, M. A Numerical Study on the Impact of Cathode Catalyst Layer Loading on the Open Circuit Voltage in a Proton Exchange Membrane Fuel Cell. J. Electrochem. Soc. 2021, 168, 044519. [Google Scholar] [CrossRef]
- Bondue, C.J.; Liang, Z.; Koper, M.T.M. Dissociative Adsorption of Acetone on Platinum Single-Crystal Electrodes. J. Phys. Chemistry C Nanomater. Interfaces 2021, 125, 6643–6649. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).