
Identifying the Active Species in Li-Na Dual-Ion “Saltwater Battery” Based on Spinel Lithium Manganese Oxide, Sodium Titanium Phosphate and Aqueous Electrolyte

 , , and
, , and 
Abstract
1. Introduction
2. Experimental
2.1. Electrode Preparation
2.2. Electrolyte Preparation
2.3. Cyclic Voltammetry
2.4. Full Cell Measurement
2.5. Electrolyte Ion Analysis
2.6. X-ray Diffraction (XRD)
3. Results and Discussion
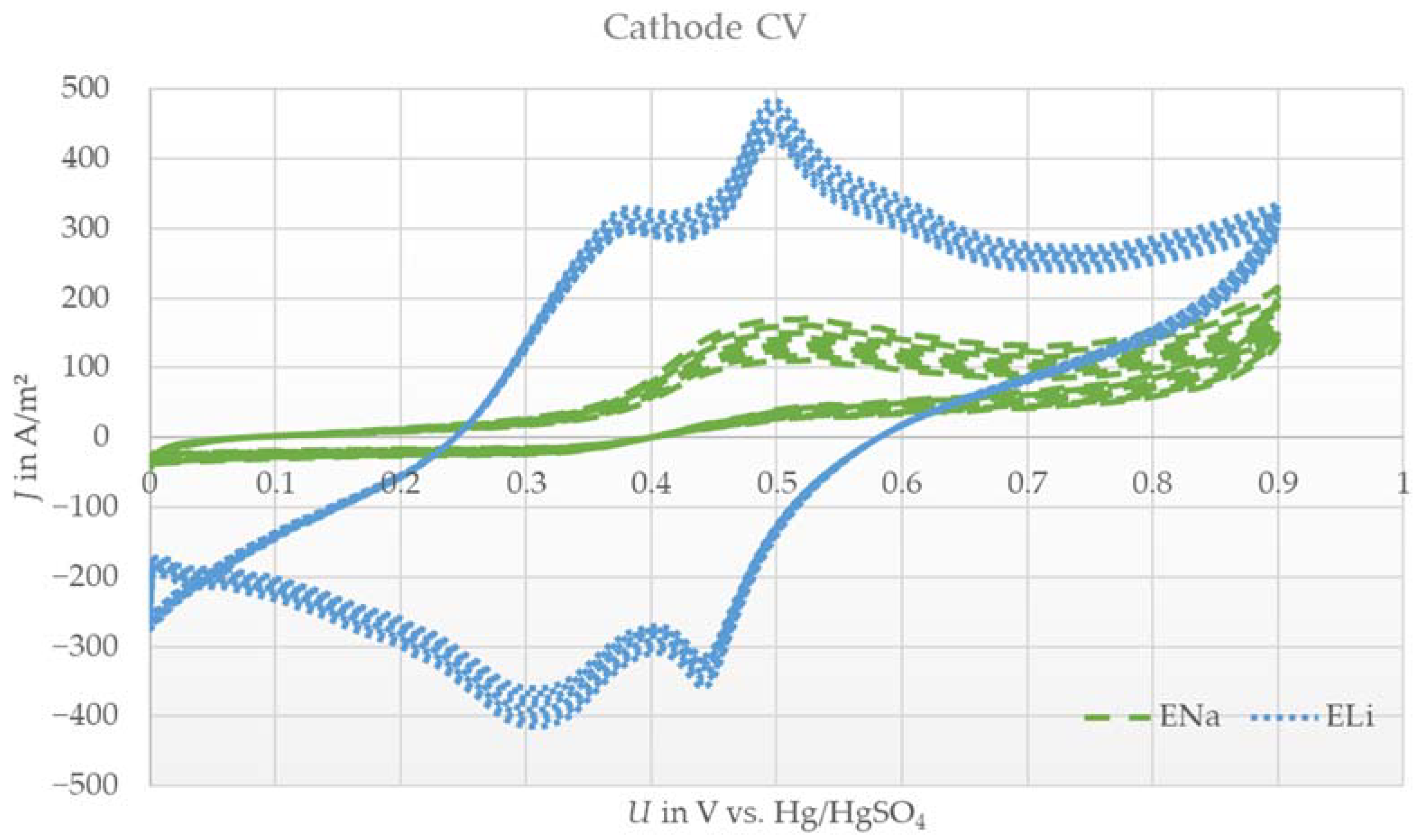
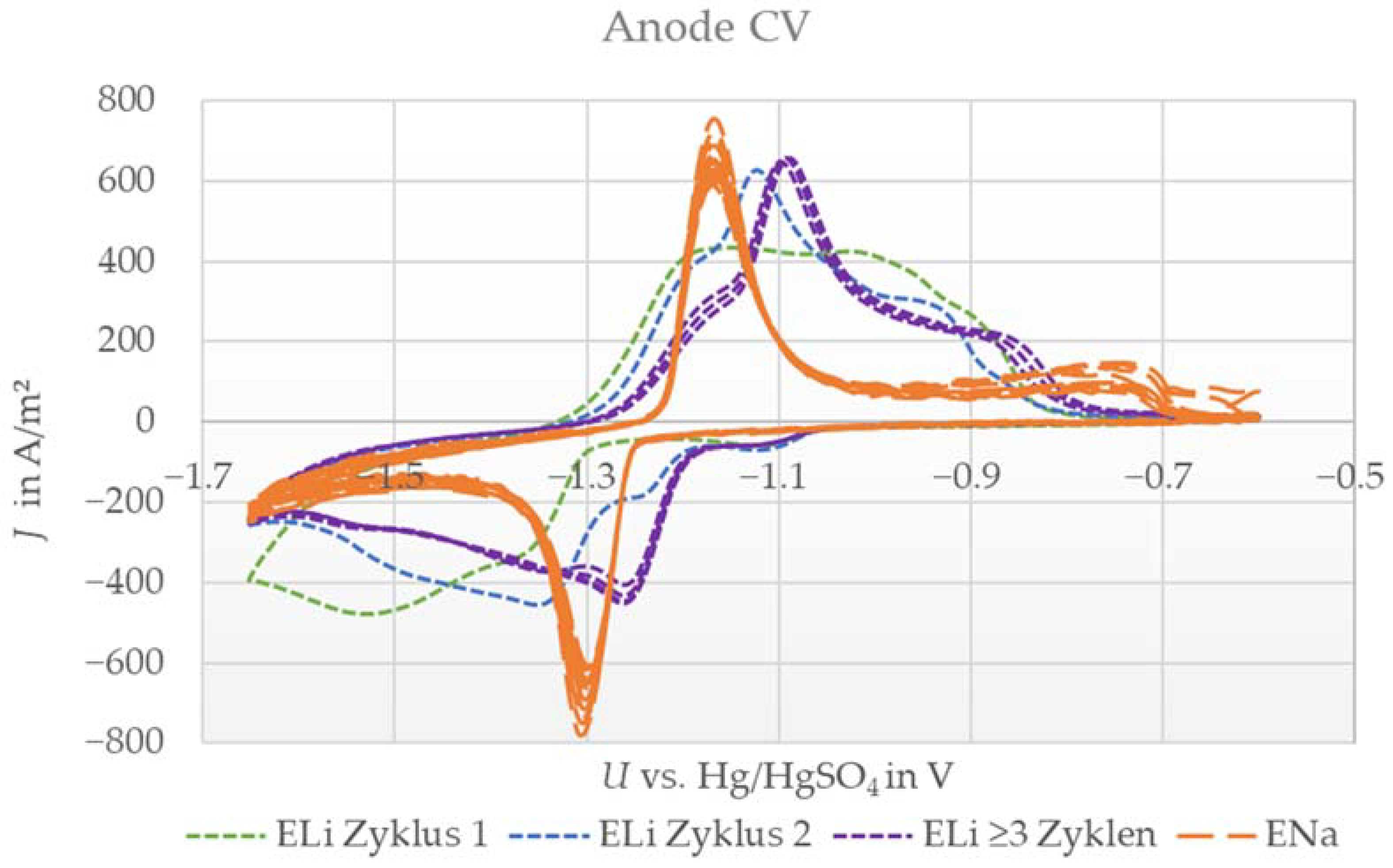
3.1. Cyclic Voltammetry
3.2. Full Cell Measurement
3.3. Ion Analysis of Electrolyte
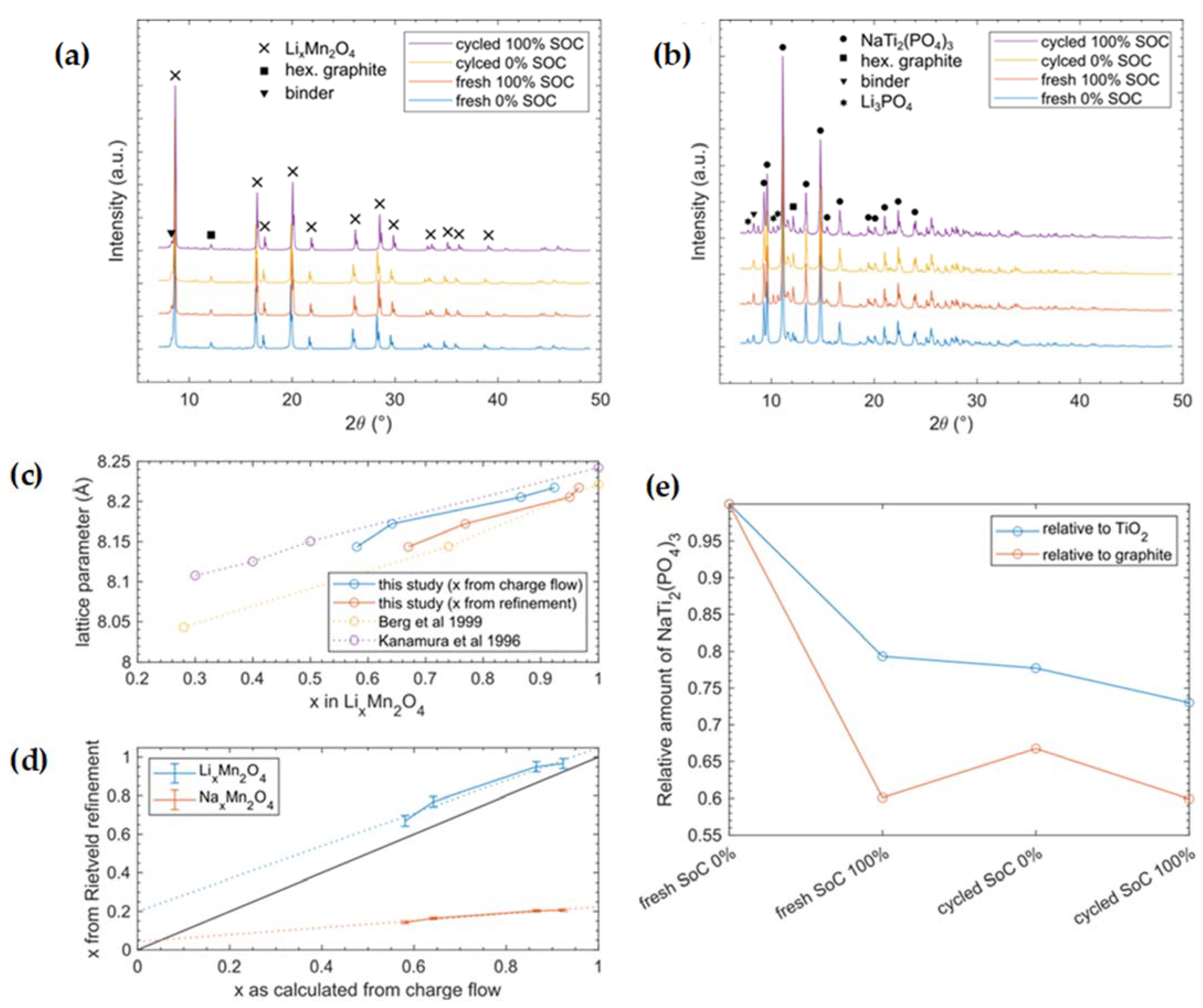
3.4. X-ray Diffraction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
- LiMn2O4(LMO): COD #1514006
- Graphite: ICSD #76767
- LiNaSO4: COD #2106021
- Li2SO4(H2O): COD #1008190
- Na3Li(SO4)2(H20)6: COD #2243890
- NaTi2(PO4)3 (NaTiPO): ICSD #19995
- Li3PO4: ICSD #10257
- TiO2 (Anatase): COD #9008214
- Na2SO4: COD #9004092
- Six coefficients of the Chebyshev-1 background polynomial model;
- Two separate background peaks at 2θ = 8.29° and 8.17° that originated from the binder;
- Lattice parameters and phase fractions of all phases;
- One isotropic microstrain parameter for each phase except for NaTiPO;
- Three anisotropic microstrain parameters for NaTiPO using the generalized model;
- One isotropic atomic displacement factor U for all atoms of the NaTiPO and Li3PO4 phases, respectively;
- All possible atomic positions of the NaTiPO and Li3PO4 phases.
- Six coefficients of the Chebyshev-1 background polynomial model;
- One separate background peak at 2θ = 8.29° that originated from the binder;
- Lattice parameters and phase fractions of all phases;
- One isotropic microstrain parameter for each phase except for LMO and Na3Li(SO4)2(H2O)6;
- Two anisotropic microstrain parameters for LMO using the generalized model;
- No broadening model was used for Na3Li(SO4)2(H2O)6 because its phase fraction was too low;
- One isotropic atomic displacement factor U for all atoms of the LMO phase;
- The x-position of the oxygen atom in the LMO phase;
- The fraction of Li in the LMO phase was set to a value extracted from the summed transferred charge measured by the potentiostat. We assume that the same amount of Li+ was removed from LMO by assuming a transfer ratio of 1 Li+/e−.
References
- Zugschwert, C.; Dundálek, J.; Leyer, S.; Hadji-Minaglou, J.-R.; Kosek, J.; Pettinger, K.-H. The Effect of Input Parameter Variation on the Accuracy of a Vanadium Redox Flow Battery Simulation Model. Batteries 2021, 7, 7. [Google Scholar] [CrossRef]
- Oh, H.G.; Park, S.-K. Co-MOF Derived MoSe2@CoSe2/N-Doped Carbon Nanorods as High-Performance Anode Materials for Potassium Ion Batteries. Int. J. Energy Res. 2022, 46, 10677–10688. [Google Scholar] [CrossRef]
- Deng, Q.; Wang, M.; Liu, X.; Fan, H.; Zhang, Y.; Yang, H.Y. Ultrathin Cobalt Nickel Selenides (Co0.5Ni0.5Se2) Nanosheet Arrays Anchoring on Ti3C2 MXene for High-Performance Na+/K+ Batteries. J. Colloid Interface Sci. 2022, 626, 700–709. [Google Scholar] [CrossRef]
- Li, X.; Liang, H.; Qin, B.; Wang, M.; Zhang, Y.; Fan, H. Rational Design of Heterostructured Bimetallic Sulfides (CoS2/NC@VS4) with VS4 Nanodots Decorated on CoS2 Dodecahedron for High-Performance Sodium and Potassium Ion Batteries. J. Colloid Interface Sci. 2022, 625, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Bin, D.; Wang, F.; Tamirat, A.G.; Suo, L.; Wang, Y.; Wang, C.; Xia, Y. Progress in Aqueous Rechargeable Sodium-Ion Batteries. Adv. Energy Mater. 2018, 8, 1703008. [Google Scholar] [CrossRef]
- Li, W.-H.; Wu, X.-L. Advanced Cathode Materials in Dual-Ion Batteries: Progress and Prospect. Electrochem. Sci. Adv. 2022, 2, e2100127. [Google Scholar] [CrossRef]
- Wang, X.-T.; Yang, Y.; Guo, J.-Z.; Gu, Z.-Y.; Ang, E.H.; Sun, Z.-H.; Li, W.-H.; Liang, H.-J.; Wu, X.-L. An Advanced Cathode Composite for Co-Utilization of Cations and Anions in Lithium Batteries. J. Mater. Sci. Technol. 2022, 102, 72–79. [Google Scholar] [CrossRef]
- Wang, H.; Wang, R.; Song, Z.; Zhang, H.; Zhang, H.; Wang, Y.; Li, X. A Novel Aqueous Li+ (or Na+)/Br− Hybrid-Ion Battery with Super High Areal Capacity and Energy Density. J. Mater. Chem. A 2019, 7, 13050–13059. [Google Scholar] [CrossRef]
- Zhang, Z.; Hu, X.; Zhou, Y.; Wang, S.; Yao, L.; Pan, H.; Su, C.-Y.; Chen, F.; Hou, X. Aqueous Rechargeable Dual-Ion Battery Based on Fluoride Ion and Sodium Ion Electrochemistry. J. Mater. Chem. A 2018, 6, 8244–8250. [Google Scholar] [CrossRef]
- Whitacre, J.F.; Shanbhag, S.; Mohamed, A.; Polonsky, A.; Carlisle, K.; Gulakowski, J.; Wu, W.; Smith, C.; Cooney, L.; Blackwood, D.; et al. A Polyionic, Large-Format Energy Storage Device Using an Aqueous Electrolyte and Thick-Format Composite NaTi 2 (PO 4 ) 3 /Activated Carbon Negative Electrodes. Energy Technol. 2015, 3, 20–31. [Google Scholar] [CrossRef]
- Kalapsazova, M.; Rasheev, H.; Zhecheva, E.; Tadjer, A.; Stoyanova, R. Insights into the Function of Electrode and Electrolyte Materials in a Hybrid Lithium–Sodium Ion Cell. J. Phys. Chem. C 2019, 123, 11508–11521. [Google Scholar] [CrossRef]
- Kim, H.; Hong, J.; Park, K.-Y.; Kim, H.; Kim, S.-W.; Kang, K. Aqueous Rechargeable Li and Na Ion Batteries. Chem. Rev. 2014, 114, 11788–11827. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.I.; Whitacre, J.F. Capacity Fade of NaTi2(PO4)3 in Aqueous Electrolyte Solutions: Relating PH Increases to Long Term Stability. Electrochim. Acta 2017, 235, 730–739. [Google Scholar] [CrossRef]
- Whitacre, J.F.; Wiley, T.; Shanbhag, S.; Wenzhuo, Y.; Mohamed, A.; Chun, S.E.; Weber, E.; Blackwood, D.; Lynch-Bell, E.; Gulakowski, J.; et al. An Aqueous Electrolyte, Sodium Ion Functional, Large Format Energy Storage Device for Stationary Applications. J. Power Sources 2012, 213, 255–264. [Google Scholar] [CrossRef]
- Wu, W.; Yan, J.; Wise, A.; Rutt, A.; Whitacre, J.F. Using Intimate Carbon to Enhance the Performance of NaTi2(PO4)3 Anode Materials: Carbon Nanotubes vs Graphite. J. Electrochem. Soc. 2014, 161, 561–567. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Cheng, Y.; Feng, K.; Li, X.; Zhang, H. Rational Design and Synthesis of LiTi 2 (PO 4 ) 3−x F x Anode Materials for High-Performance Aqueous Lithium Ion Batteries. J. Mater. Chem. A 2017, 5, 593–599. [Google Scholar] [CrossRef]
- Wu, W.; Mohamed, A.; Whitacre, J.F. Microwave Synthesized NaTi2(PO4)3 as an Aqueous Sodium-Ion Negative Electrode. J. Electrochem. Soc. 2013, 160, 497–504. [Google Scholar] [CrossRef]
- Hou, Z.; Li, X.; Liang, J.; Zhu, Y.; Qian, Y. An Aqueous Rechargeable Sodium Ion Battery Based on a NaMnO2–NaTi2(PO4)3 Hybrid System for Stationary Energy Storage. J. Mater. Chem. A 2015, 3, 1400–1404. [Google Scholar] [CrossRef]
- Sun, D.; Jin, G.; Tang, Y.; Zhang, R.; Xue, X.; Huang, X.; Chu, H.; Wang, H. NaTi2 (PO4)3 Nanoparticles Embedded in Carbon Matrix as Long-Lived Anode for Aqueous Lithium Ion Battery. J. Electrochem. Soc. 2016, 163, A1388–A1393. [Google Scholar] [CrossRef]
- Park, S.I.; Gocheva, I.; Okada, S.; Yamaki, J.-I. Electrochemical Properties of NaTi2(PO4)3 Anode for Rechargeable Aqueous Sodium-Ion Batteries. J. Electrochem. Soc. 2011, 158, 1067–1070. [Google Scholar] [CrossRef]
- Whitacre, J.F.; Tevar, A.; Sharma, S. Na4Mn9O18 as a Positive Electrode Material for an Aqueous Electrolyte Sodium-Ion Energy Storage Device. Electrochem. Commun. 2010, 12, 463–466. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, L.; Wang, Y.; Xia, Y. Cycling Stability of Spinel LiMn2O4 with Different Particle Sizes in Aqueous Electrolyte. Electrochim. Acta 2015, 173, 178–183. [Google Scholar] [CrossRef]
- Zhu, Z.; Peelaers, H.; Van de Walle, C.G. Hydrogen-Induced Degradation of NaMnO2. Chem. Mater. 2019, 31, 5224–5228. [Google Scholar] [CrossRef]
- Li, Z.; Young, D.; Xiang, K.; Carter, W.C.; Chiang, Y.-M. Towards High Power High Energy Aqueous Sodium-Ion Batteries: The NaTi2 (PO4)3 /Na0.44MnO2 System. Adv. Energy Mater. 2013, 3, 290–294. [Google Scholar] [CrossRef]
- He, X.; Wang, J.; Qiu, B.; Paillard, E.; Ma, C.; Cao, X.; Liu, H.; Stan, M.C.; Liu, H.; Gallash, T.; et al. Durable High-Rate Capability Na0.44MnO2 Cathode Material for Sodium-Ion Batteries. Nano Energy 2016, 27, 602–610. [Google Scholar] [CrossRef]
- Zhan, X.; Shirpour, M. Evolution of Solid/Aqueous Interface in Aqueous Sodium-Ion Batteries. Chem. Commun. 2017, 53, 204–207. [Google Scholar] [CrossRef]
- Sauvage, F.; Laffont, L.; Tarascon, J.-M.; Baudrin, E. Study of the Insertion/Deinsertion Mechanism of Sodium into Na0.44MnO2. Inorg. Chem. 2007, 46, 3289–3294. [Google Scholar] [CrossRef]
- Luo, J.-Y.; Xia, Y.-Y. Aqueous Lithium-Ion Battery LiTi2(PO4)3/LiMn2O4 with High Power and Energy Densities as Well as Superior Cycling Stability**. Adv. Funct. Mater. 2007, 17, 3877–3884. [Google Scholar] [CrossRef]
- Tang, W.; Hou, Y.; Wang, F.; Liu, L.; Wu, Y.; Zhu, K. LiMn 2 O 4 Nanotube as Cathode Material of Second-Level Charge Capability for Aqueous Rechargeable Batteries. Nano Lett. 2013, 13, 2036–2040. [Google Scholar] [CrossRef]
- Chen, L.; Liu, J.; Guo, Z.; Wang, Y.; Wang, C.; Xia, Y. Electrochemical Profile of LiTi2(PO4)3 and NaTi2(PO4)3 in Lithium, Sodium or Mixed Ion Aqueous Solutions. J. Electrochem. Soc. 2016, 163, A904. [Google Scholar] [CrossRef]
- DIN EN ISO/IEC 17025:2018-03; Allgemeine Anforderungen an Die Kompetenz von Prüf- Und Kalibrierlaboratorien. Deutsche Beuth Verlag GmbH: Berlin, Germany, 2018.
- Binnewies, M.; Finze, M.; Jäckel, M.; Schmidt, P.; Willner, H.; Rayner-Canham, G. Allgemeine und Anorganische Chemie; Springer: Berlin/Heidelberg, Germany, 2016; ISBN 978-3-662-45066-6. [Google Scholar]
- Luo, F.; Wei, C.; Zhang, C.; Gao, H.; Niu, J.; Ma, W.; Peng, Z.; Bai, Y.; Zhang, Z. Operando X-Ray Diffraction Analysis of the Degradation Mechanisms of a Spinel LiMn2O4 Cathode in Different Voltage Windows. J. Energy Chem. 2020, 44, 138–146. [Google Scholar] [CrossRef]
- Marchini, F.; Rubi, D.; del Pozo, M.; Williams, F.J.; Calvo, E.J. Surface Chemistry and Lithium-Ion Exchange in LiMn 2 O 4 for the Electrochemical Selective Extraction of LiCl from Natural Salt Lake Brines. J. Phys. Chem. C 2016, 120, 15875–15883. [Google Scholar] [CrossRef]
- Berg, H. Neutron Diffraction Study of Electrochemically Delithiated LiMn2O4 Spinel. Solid State Ion. 1999, 126, 227–234. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Yano, M.; Kuze, S.; Komaba, S. Electrochemical Behavior and Structural Change of Spinel-Type Li[LixMn2−x]O4 (X = 0 and 0.2) in Sodium Cells. Electrochim. Acta 2012, 82, 296–301. [Google Scholar] [CrossRef]
- Yang, J.; Wang, H.; Hu, P.; Qi, J.; Guo, L.; Wang, L. A High-Rate and Ultralong-Life Sodium-Ion Battery Based on NaTi2(PO4)3 Nanocubes with Synergistic Coating of Carbon and Rutile TiO2. Small 2015, 11, 3744–3749. [Google Scholar] [CrossRef] [PubMed]
- Kabbour, H.; Coillot, D.; Colmont, M.; Masquelier, C.; Mentré, O. α-Na3M2 (PO4)3(M = Ti, Fe): Absolute Cationic Ordering in NASICON-Type Phases. J. Am. Chem. Soc. 2011, 133, 11900–11903. [Google Scholar] [CrossRef]
- Kanamura, K.; Naito, H.; Yao, T.; Takehara, Z. Structural Change of the LiMn2O4 Spinel Structure Induced by Extraction of Lithium. J. Mater. Chem. 1996, 6, 33. [Google Scholar] [CrossRef]
- Jazouli, A.E.; Nadiri, A.; Dance, J.M.; Delmas, C.; Flem, L. Relationships between Structure and Magnetic Properties of Titanium (III) NASICON-Type Phosphates. J. Phys. Chem. Solids 1988, 7, 779–783. [Google Scholar] [CrossRef]
- Luo, J.-Y.; Cui, W.-J.; He, P.; Xia, Y.-Y. Raising the Cycling Stability of Aqueous Lithium-Ion Batteries by Eliminating Oxygen in the Electrolyte. Nat. Chem. 2010, 2, 760–765. [Google Scholar] [CrossRef]
- Chen, L.; Cao, L.; Ji, X.; Hou, S.; Li, Q.; Chen, J.; Yang, C.; Eidson, N.; Wang, C. Enabling Safe Aqueous Lithium Ion Open Batteries by Suppressing Oxygen Reduction Reaction. Nat. Commun. 2020, 11, 2638. [Google Scholar] [CrossRef]
- Song, Y.-J. Recovery of Lithium as Li3PO4 from Waste Water in a LIB Recycling Process. Korean J. Met. Mater. 2018, 56, 755–762. [Google Scholar] [CrossRef]
- Toby, B.H.; Von Dreele, R.B. It GSAS-II: The Genesis of a Modern Open-Source All Purpose Crystallography Software Package. J. Appl. Crystallogr. 2013, 46, 544–549. [Google Scholar] [CrossRef]
- Rietveld, H.M. A Profile Refinement Method for Nuclear and Magnetic Structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Function | Weight Percentage in Anode/% | Weight Percentage in Cathode/% |
|---|---|---|---|
| NaTiPO | Anode intercalation material | 70 | 0 |
| LMO | Cathode intercalation material | 0 | 80 |
| Carbon black | Electrical conduction | 7 | 2 |
| Graphite | Electrical conduction | 3 | 10 |
| Polytetrafluoroethylene | Binder material | 5 | 8 |
| Activated Carbon | Anode additive for cycle stability | 15 | 0 |
| Type | Electrode | Product Name | Supplier |
|---|---|---|---|
| LMO | Cathode | HLM-Y01 | Eachem, Hunan, China |
| NaTiPO | Anode | Customized synthesis according to [10] | |
| Activated carbon | Anode | PAK C-1000C | CarboTech, Essen, Germany |
| Carbon black | Anode and cathode | Super P | Imerys, Willebroek, Belgium |
| Graphite | Anode and cathode | KS6 | Imerys, Bodio, Switzerland |
| Polytetrafluoroethylene | Anode | Algoflon L203 | Solvay, Bollate, Italy |
| Polytetrafluoroethylene | Cathode | Dyneon TF 2021Z | 3M, Burgkirchen, Germany |
| Fresh Electrolyte before Filling | Sample 0% SoC | Sample 100% SoC | |
|---|---|---|---|
| Lithium content in g/L | 11.1 | 13.1 | 14.7 |
| Sodium content in g/L | 58.4 | 69.2 | 70.4 |
| Sample | LixMn2O4 in wt.% | Graphite in wt.% | LiNaSO4 in wt.% | Li2SO4(H2O) in wt.% | Na3Li(SO4)2(H2O)6 in wt.% |
|---|---|---|---|---|---|
| Fresh SoC 0% | 86.9(2) | 7.8(2) | 1.95(9) | 4.5(8) | 2.9(2) |
| Fresh SoC 100% | 87.4(2) | 8.6(2) | 1.3(1) | 2.7(2) | 0.000 |
| Cycled SoC 0% | 86.8(2) | 9.5(2) | 3.2(1) | 0.000 | 0.5(1) |
| Cycled SoC 100% | 87.4(2) | 8.4(2) | 1.4(2) | 2.8(2) | 0.000 |
| Sample | NaTi2(PO4)3 in wt.% | Graphite in wt.% | Na3Li(SO4)2(H2O)6 in wt.% | Li3PO4 in wt.% | TiO2 in wt.% | Na2SO4 in wt.% |
|---|---|---|---|---|---|---|
| Fresh SoC 0% | 86.4(2) | 6.5(2) | 4.4(2) | 0.000 | 2.7(1) | 0.000 |
| Fresh SoC 100% | 76.5(3) | 9.5(2) | 0.000 | 11.0(3) | 3.0(1) | 0.000 |
| Cycled SoC 0% | 86.8(2) | 9.7(2) | 0.000 | 0.000 | 3.5(1) | 0.000 |
| Cycled SoC 100% | 73.5(2) | 9.2(2) | 0.000 | 6.8(2) | 3.1(1) | 7.4(1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schubert, J.; Grossmann, L.; Seidlmayer, S.; Pettinger, K.-H.; Gilles, R.; Danzer, M.A. Identifying the Active Species in Li-Na Dual-Ion “Saltwater Battery” Based on Spinel Lithium Manganese Oxide, Sodium Titanium Phosphate and Aqueous Electrolyte. Energies 2023, 16, 4485. https://doi.org/10.3390/en16114485
Schubert J, Grossmann L, Seidlmayer S, Pettinger K-H, Gilles R, Danzer MA. Identifying the Active Species in Li-Na Dual-Ion “Saltwater Battery” Based on Spinel Lithium Manganese Oxide, Sodium Titanium Phosphate and Aqueous Electrolyte. Energies. 2023; 16(11):4485. https://doi.org/10.3390/en16114485
Chicago/Turabian StyleSchubert, Jonathan, Lukas Grossmann, Stefan Seidlmayer, Karl-Heinz Pettinger, Ralph Gilles, and Michael A. Danzer. 2023. "Identifying the Active Species in Li-Na Dual-Ion “Saltwater Battery” Based on Spinel Lithium Manganese Oxide, Sodium Titanium Phosphate and Aqueous Electrolyte" Energies 16, no. 11: 4485. https://doi.org/10.3390/en16114485
APA StyleSchubert, J., Grossmann, L., Seidlmayer, S., Pettinger, K.-H., Gilles, R., & Danzer, M. A. (2023). Identifying the Active Species in Li-Na Dual-Ion “Saltwater Battery” Based on Spinel Lithium Manganese Oxide, Sodium Titanium Phosphate and Aqueous Electrolyte. Energies, 16(11), 4485. https://doi.org/10.3390/en16114485