
On the Kinetic Mechanisms of the Reduction and Oxidation Reactions of Iron Oxide/Iron Pellets for a Hydrogen Storage Process



Abstract
1. Introduction
2. Kinetic Models
2.1. Geometry Model
- In Reaction order models, the reaction rate is proportional to the concentration, where the remaining amount or fraction of the reactant is raised to a particular power (integral or fractional), which represents the reaction order . The general formulation for a reaction-based model is:
- Geometric contraction models assume that the adsorption and desorption of the reaction gas occurs uniformly on the surface of the particle. The reaction rate is controlled by the resulting expansion of the contact area towards the centre of the reactant. Assuming that all particles have the same shape and size, this model is easy to parameterise and shows good agreement for metal oxide/metal redox systems. The two relevant kinetic parameters, the generalised rate constant and the morphology (cubic, cylindrical, spherical), can be determined by fitting a single isothermal curve. In general, this can be described with the following algebraic equation:
- Diffusion models describe complex concentration equilibrium processes through porous solid matrices or simply pore diffusion, which can play an important role in gas–solid reactions. If the reacting solid B has a porosity (), the description of diffusion through the micro- and macro-pore volume is necessary for the reacting gas A to access the surface of the solid B; the removal of the gaseous product D also occurs in the same way. However, pore diffusion can also be an important component in the reaction of non-porous solids. If the solid product layer C formed is itself porous, the supply of the gaseous reactant A and the removal of the gaseous product D can take place by diffusion through the porous product layer. Depending on the model used, e.g., Gingstling–Brounshtein, Jander or Chou, the mathematical description differs and will be considered in more detail later.
- Nucleation and nuclei growth models describe the formation and growth of nuclei, which are finite quantities of the product inside the reactant lattice. This includes crystallisation, crystallographic transition, decomposition, adsorption, hydration and desolvation. In general, the rate-controlling step can be the solid diffusion or movement of the interface.
- Isothermal condition dominates ;
- Reactions proceed independently and without interaction between the gaseous species;
- No crack formation occurs during the reaction;
- Pressure is uniform inside and around the pellets;
- Pellet has a uniform and constant porosity ;
- Chemical reaction-controlled process inside each grain is reversible and proceeds topochemically.
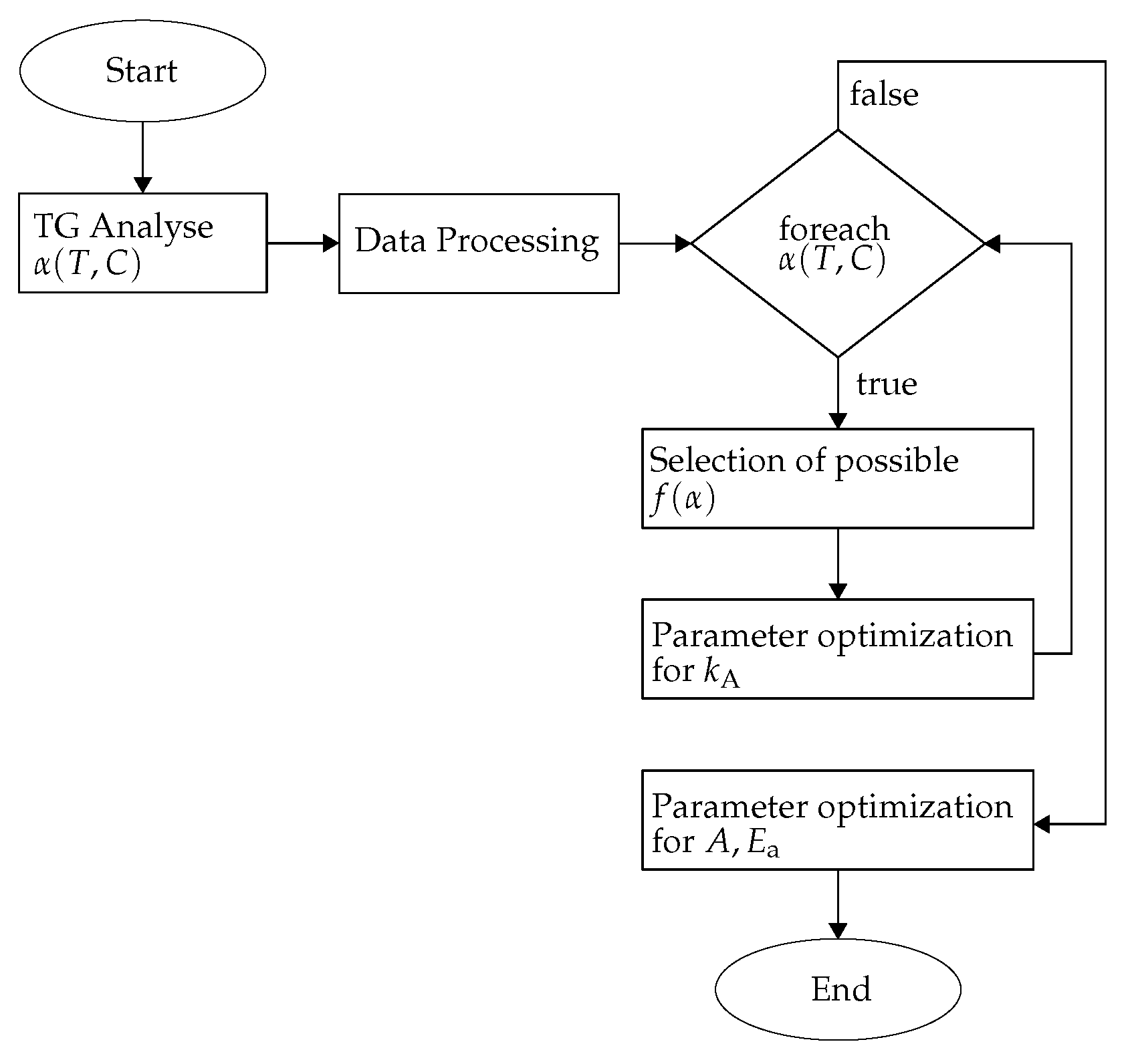
2.2. Estimation of the Kinetic Parameters
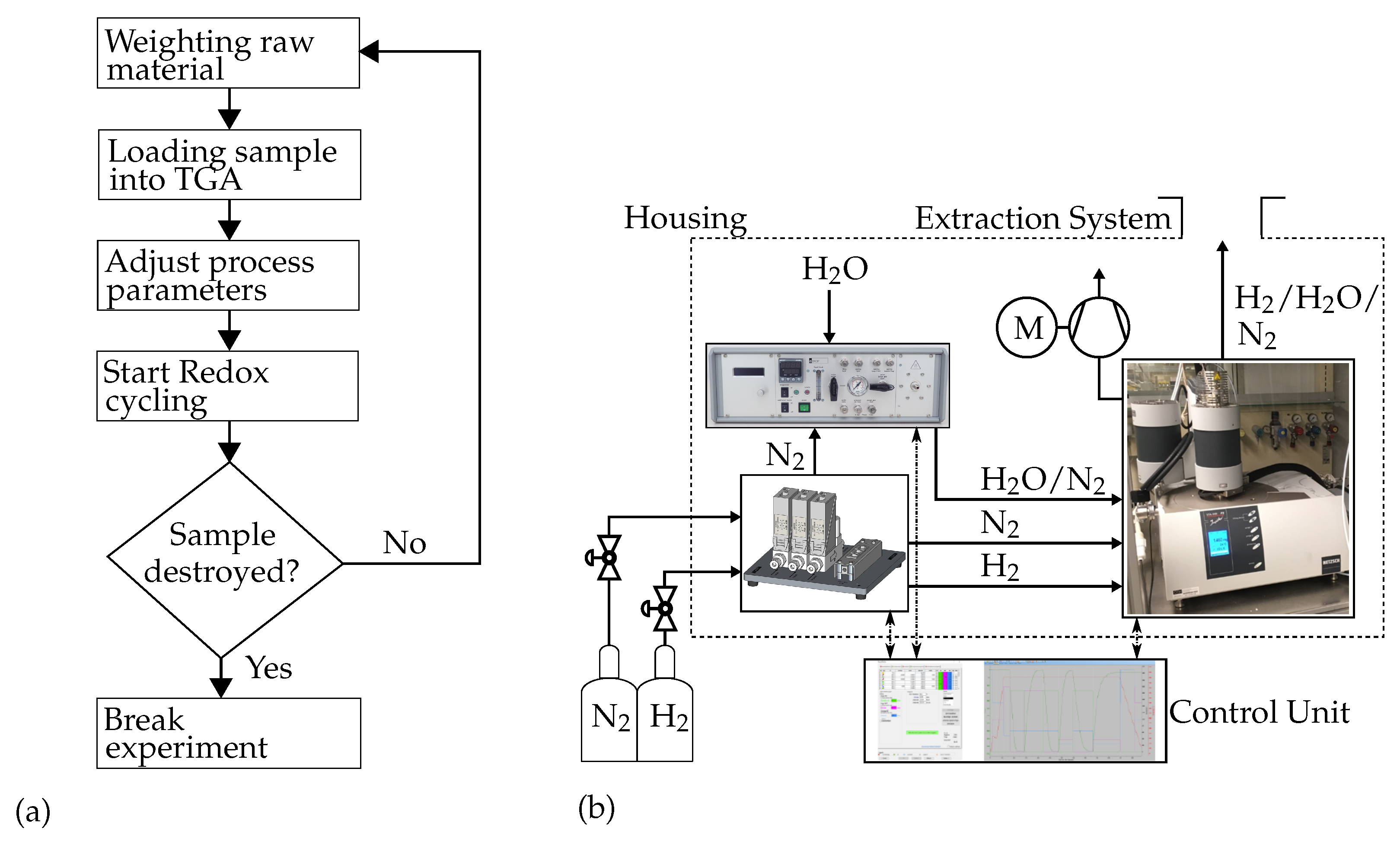
3. Materials and Methods
4. Results and Discussion
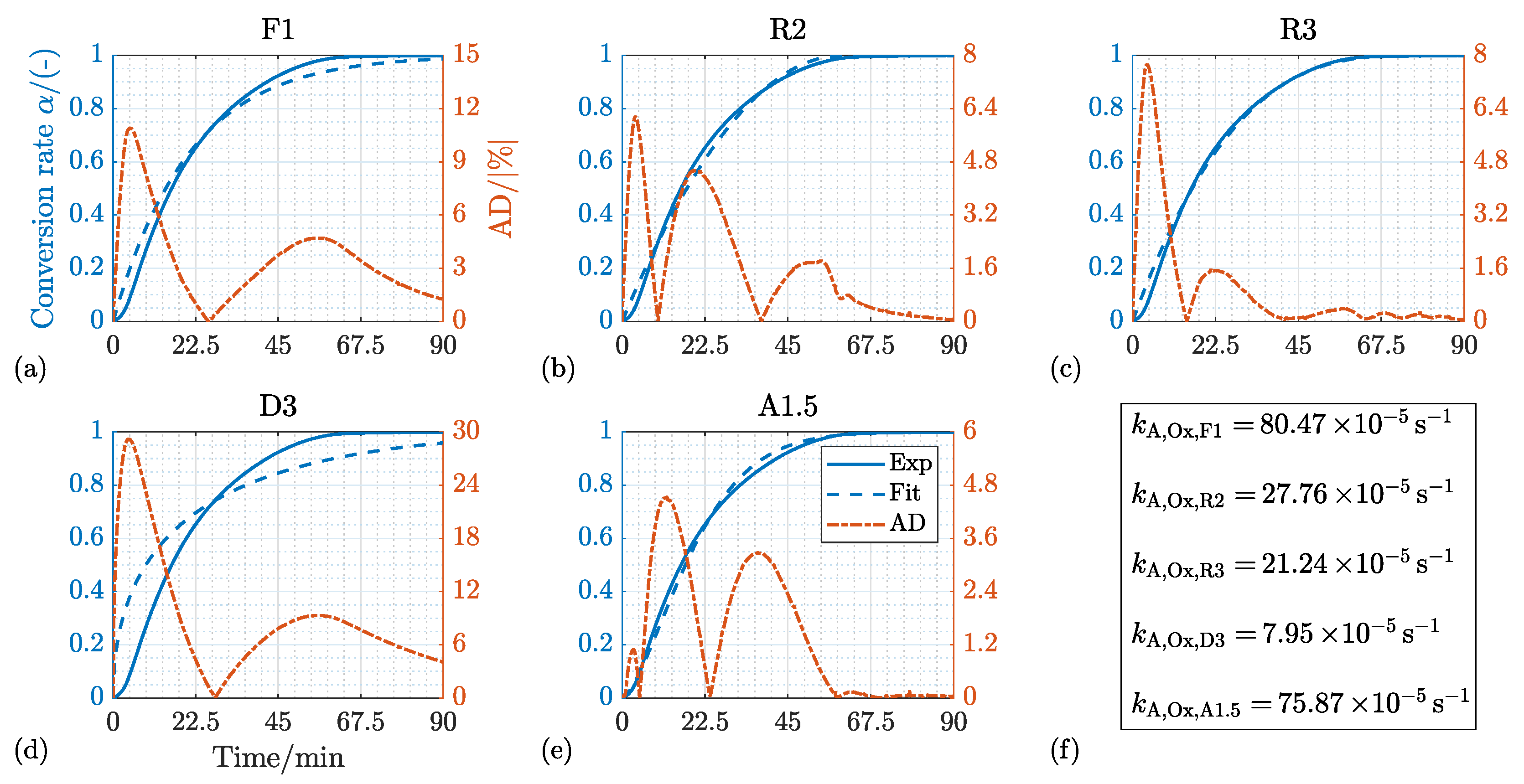
4.1. Reduction
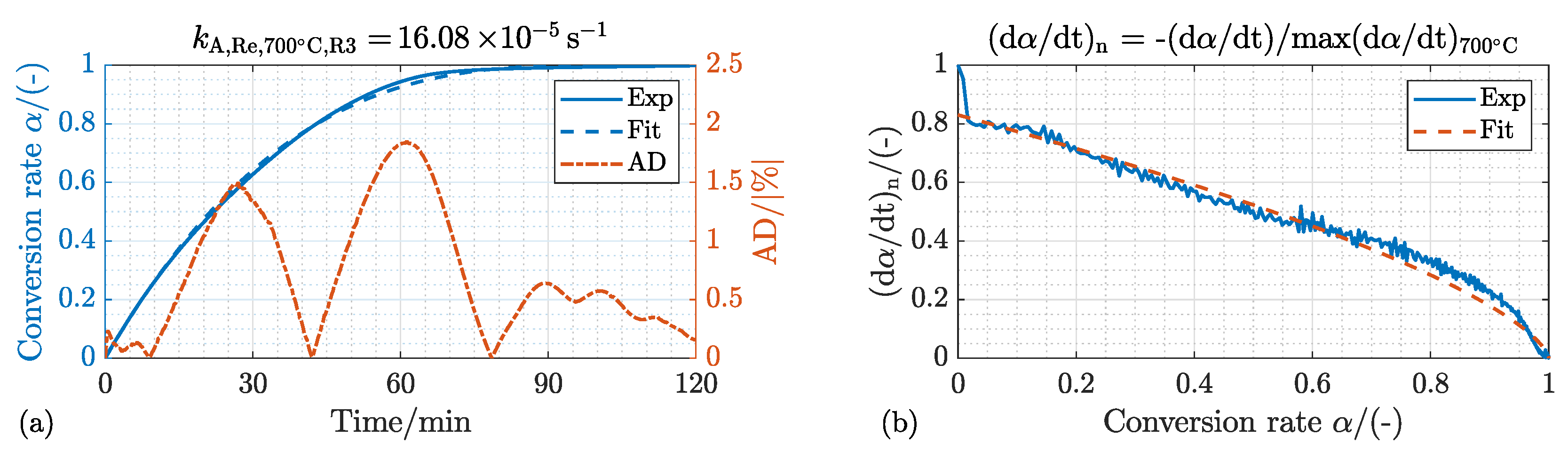
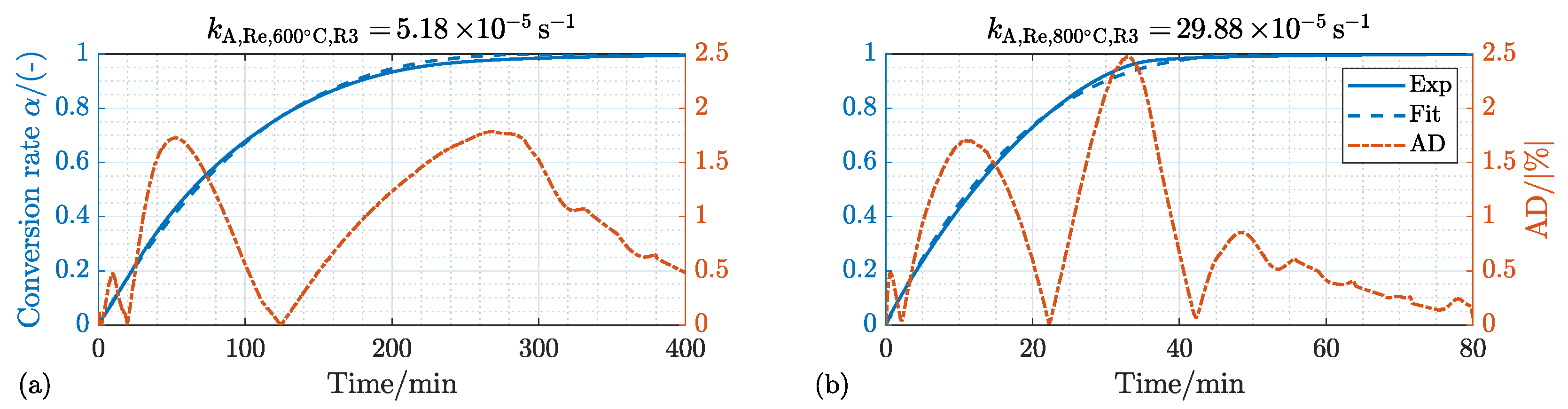
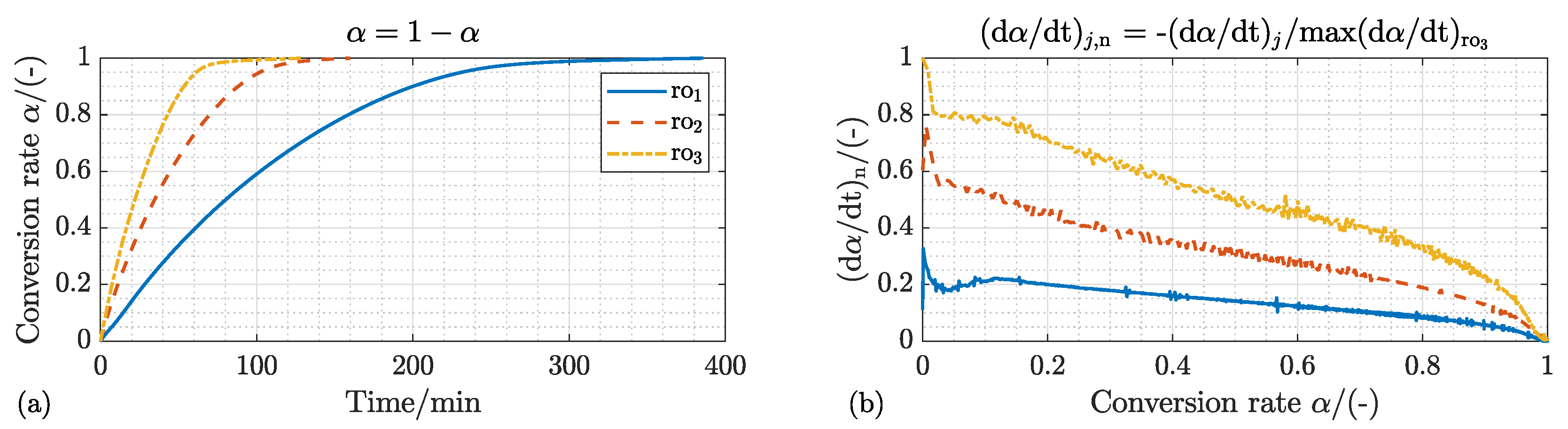
4.1.1. Kinetic Mechanism and Effect of the Process Temperature
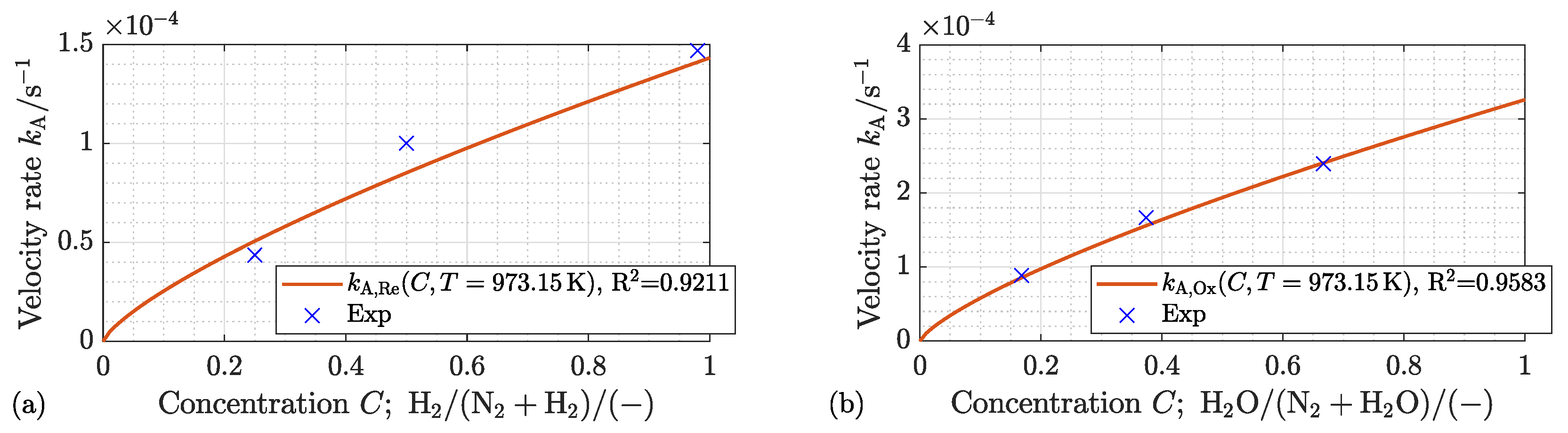
4.1.2. Effect of the Process Gas Composition
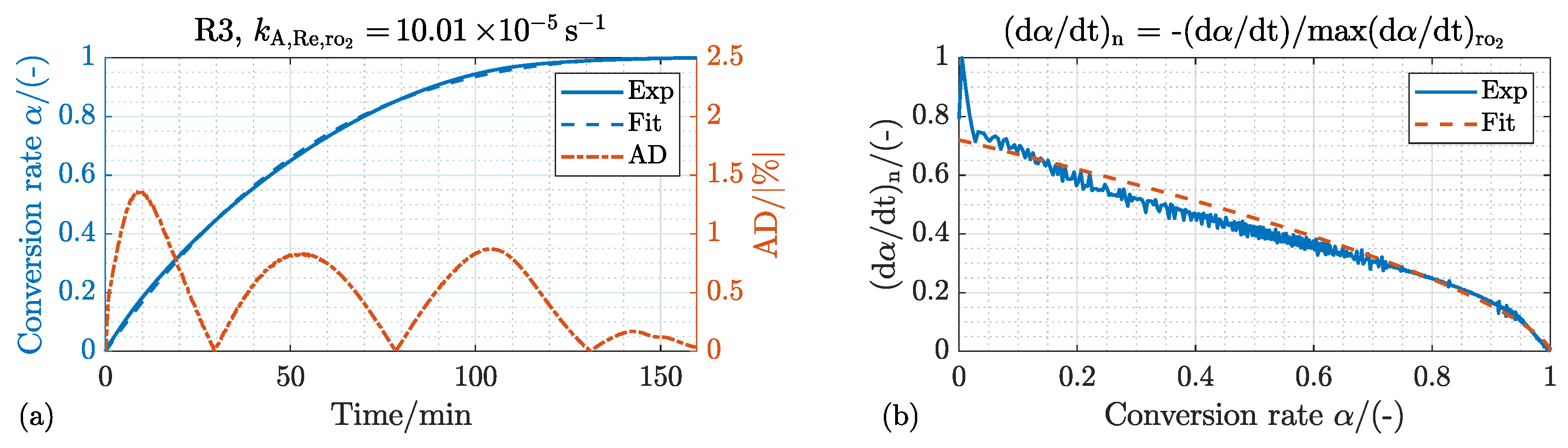
4.2. Oxidation
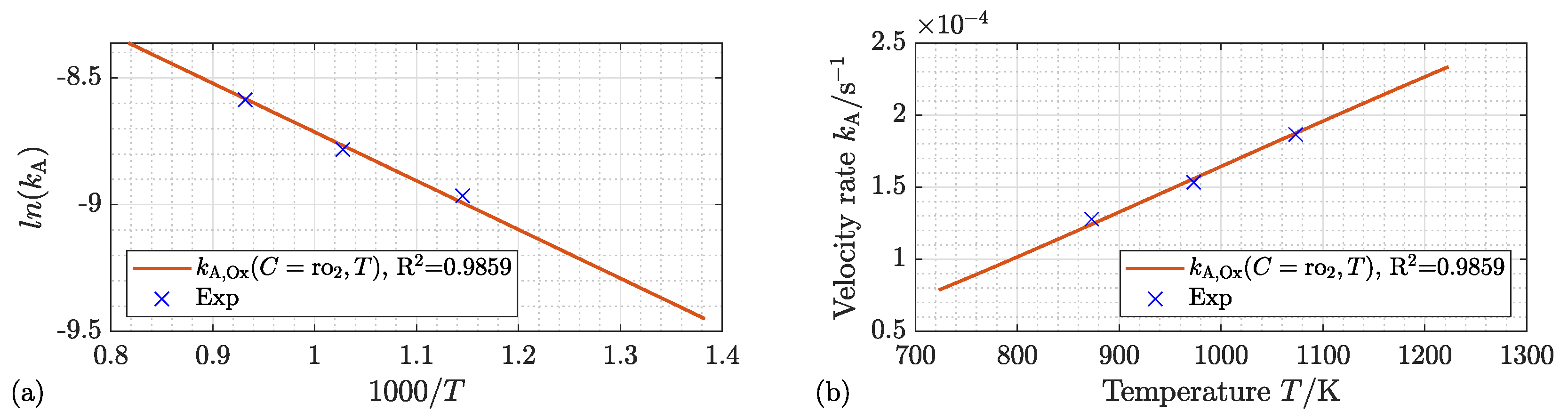
4.2.1. Kinetic Mechanism and Effect of the Process Temperature
4.2.2. Effect of the Process Gas Composition
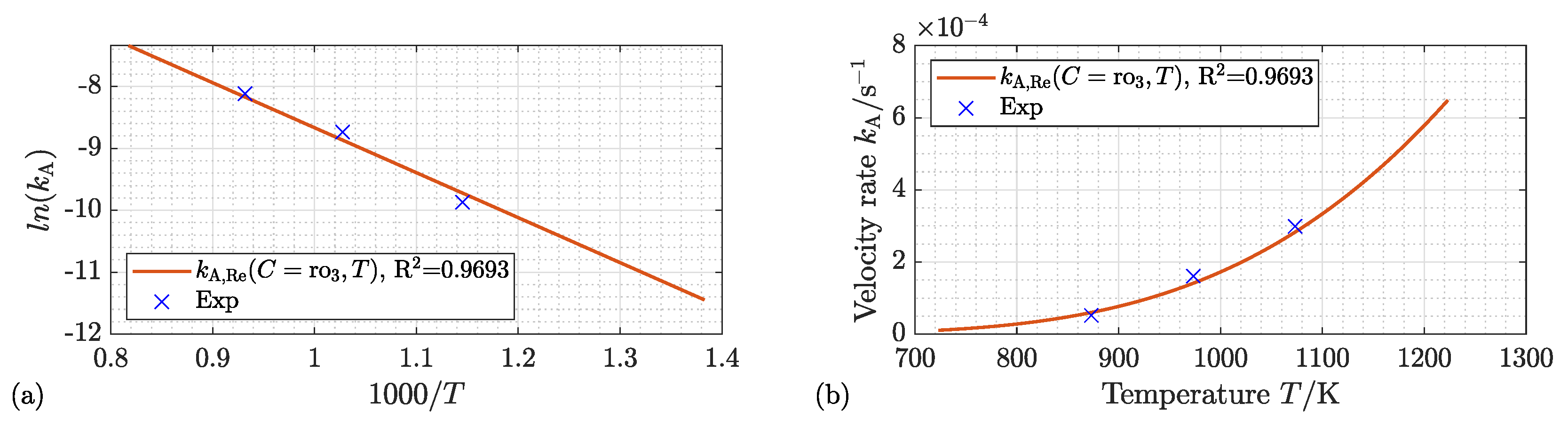
4.3. Activation Energy and Kinetic Velocity Rate
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- US National Aeronautics and Space - Climate Change Evidence: How Do We Know? Global Climate Change Vital Signs of the Planet. Available online: https://climate.nasa.gov/evidence/ (accessed on 2 October 2022).
- International Renewable Energy Agency. Global Renewables Outlook: Energy Transformation 2050; International Renewable Energy Agency: Abu Dhabi, United Arab Emirates, 2020. [Google Scholar]
- Thess, A.; Trieb, F.; Wörner, A.; Zunft, S. Herausforderung Wärmespeicher. Phys. J. 2015, 33–39. [Google Scholar]
- Sterner, M.; Stadler, I. Handbook of Energy Storage; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar] [CrossRef]
- Klell, M.; Eichlseder, H.; Trattner, A. Wasserstoff in der Fahrzeugtechnik: Erzeugung Speicherung Anwendung, 4. aufl. 2018 ed.; ATZ/MTZ-Fachbuch; Springer: Wiesbaden, Germany, 2018. [Google Scholar]
- Lorente, E.; Peña, J.A.; Herguido, J. Cycle Behaviour of Iron Ores in the Steam-Iron Process. Int. J. Hydrogen Energy 2011, 36, 7043–7050. [Google Scholar] [CrossRef]
- Berger, C.M.; Mahmoud, A.; Hermann, R.P.; Braun, W.; Yazhenskikh, E.; Sohn, Y.J.; Menzler, N.H.; Guillon, O.; Bram, M. Calcium-Iron Oxide as Energy Storage Medium in Rechargeable Oxide Batteries. J. Am. Ceram. Soc. 2016, 99, 4083–4092. [Google Scholar] [CrossRef]
- Müller, K. Technologies for the Storage of Hydrogen Part 1: Hydrogen Storage in the Narrower Sense. Chem. Ing. Tech. 2019, 91, 383–392. [Google Scholar] [CrossRef]
- Voitic, G.; Hacker, V. Recent Advancements in Chemical Looping Water Splitting for the Production of Hydrogen. RSC Adv. 2016, 6, 98267–98296. [Google Scholar] [CrossRef]
- Hossain, M.M.; de Lasa, H.I. Chemical-Looping Combustion (CLC) for Inherent CO2 Separations—A Review. Chem. Eng. Sci. 2008, 63, 4433–4451. [Google Scholar] [CrossRef]
- Thaler, M.; Hacker, V. Storage and Separation of Hydrogen with the Metal Steam Process. Int. J. Hydrogen Energy 2012, 37, 2800–2806. [Google Scholar] [CrossRef]
- Lane, H. Apparatus for Producing Hydrogen Gas. U.S. Patent 1,028,366, 4 June 1912. [Google Scholar]
- Messerschmitt, A. Process of Producing Hydrogen. U.S. Patent 971,206, 27 September 1910. [Google Scholar]
- Heidari, A.; Niknahad, N.; Iljana, M.; Fabritius, T. A Review on the Kinetics of Iron Ore Reduction by Hydrogen. Materials 2021, 14, 7540. [Google Scholar] [CrossRef]
- Spreitzer, D.; Schenk, J. Reduction of Iron Oxides with Hydrogen—A Review. Steel Res. Int. 2019, 90, 1900108. [Google Scholar] [CrossRef]
- Feilmayr, C.; Thurnhofer, A.; Winter, F.; Mali, H.; Schenk, J. Reduction Behavior of Hematite to Magnetite under Fluidized Bed Conditions. ISIJ Int. 2004, 44, 1125–1133. [Google Scholar] [CrossRef]
- Levenspiel, O. Chemical Reaction Engineering, 3rd ed.; Wiley: New York, NY, USA, 1999. [Google Scholar]
- Gamisch, B.; Gaderer, M.; Dawoud, B. On the Development of Thermochemical Hydrogen Storage: An Experimental Study of the Kinetics of the Redox Reactions under Different Operating Conditions. Appl. Sci. 2021, 11, 1623. [Google Scholar] [CrossRef]
- Jin, X.; Zhao, X.; White, R.E.; Huang, K. Heat Balance in a Planar Solid Oxide Iron-Air Redox Battery: A Computational Analysis. J. Electrochem. Soc. 2015, 162, F821–F833. [Google Scholar] [CrossRef]
- Braun, W.; Erfurt, V.; Thaler, F.; Menzler, N.H.; Spatschek, R.; Singheiser, L. Kinetic Study of Iron Based Storage Materials for the Use in Rechargeable Oxide Batteries (ROB). ECS Trans. 2017, 75, 59–73. [Google Scholar] [CrossRef]
- Berger, C.M.; Tokariev, O.; Orzessek, P.; Hospach, A.; Fang, Q.; Bram, M.; Quadakkers, W.J.; Menzler, N.H.; Buchkremer, H.P. Development of Storage Materials for High-Temperature Rechargeable Oxide Batteries. J. Energy Storage 2015, 1, 54–64. [Google Scholar] [CrossRef]
- Bock, S.; Zacharias, R.; Hacker, V. High Purity Hydrogen Production with a 10 kWth RESC Prototype System. Energy Convers. Manag. 2018, 172, 418–427. [Google Scholar] [CrossRef]
- Zacharias, R.; Visentin, S.; Bock, S.; Hacker, V. High-Pressure Hydrogen Production with Inherent Sequestration of a Pure Carbon Dioxide Stream via Fixed Bed Chemical Looping. Int. J. Hydrogen Energy 2019, 44, 7943–7957. [Google Scholar] [CrossRef]
- Otsuka, K.; Yamada, C.; Kaburagi, T.; Takenaka, S. Hydrogen Storage and Production by redox of Iron Oxide for Polymer Electrolyte Fuel Cell Vehicles. Int. J. Hydrogen Energy 2003, 28, 335–342. [Google Scholar] [CrossRef]
- Takenaka, S.; Nomura, K.; Hanaizumi, N.; Otsuka, K. Storage and Formation of pure Hydrogen mediated by the redox of modified Iron Oxides. Appl. Catal. A Gen. 2005, 282, 333–341. [Google Scholar] [CrossRef]
- Rusman, N.; Dahari, M. A Review on the Current Progress of Metal Hydrides Material for Solid-State Hydrogen Storage Applications. Int. J. Hydrogen Energy 2016, 41, 12108–12126. [Google Scholar] [CrossRef]
- Müller, K. Technologies for the Storage of Hydrogen Part 2: Irreversible Conversion and Comparison. Chem. Ing. Tech. 2019, 91, 393–402. [Google Scholar] [CrossRef]
- Williams, P.T. Pyrolysis of Waste Tyres: A Review. Waste Manag. 2013, 33, 1714–1728. [Google Scholar] [CrossRef] [PubMed]
- Posdziech, O.; Schwarze, K.; Brabandt, J. Efficient Hydrogen Production for Industry and Electricity Storage via High-Temperature Electrolysis. Int. J. Hydrogen Energy 2019, 44, 19089–19101. [Google Scholar] [CrossRef]
- Betsch, K.; Knauf, G.; Kobow, C.; Martin, H.; Petzold, D.; Ufer, J.; Wolf, B. Verfahren zur Erzeugung von Wasserstoff oder Wasserstoffreichen Gasen. DD 000,000,094,984, 1 March 1972. [Google Scholar]
- Ertl, G. Handbook of Heterogeneous Catalysis, 2nd, completely rev. and enl. ed.; WILEY-VCH: Weinheim, Germany; Chichester, UK, 2008. [Google Scholar]
- Engell, H.J.; von Bogdandy, L. (Eds.) Die Reduktion der Eisenerze: Wissenschaftliche Grundlagen und Technische Durchführung; Springer: Berlin/Heidelberg, Germany, 1967. [Google Scholar] [CrossRef]
- Ebrahimi, A.A.; Ebrahim, H.A.; Hatam, M.; Jamshidi, E. Finite Element Solution for Gas–Solid Reactions: Application to the Moving Boundary Problems. Chem. Eng. J. 2008, 144, 110–118. [Google Scholar] [CrossRef]
- Szekely, J.; Evans, J.W.; Sohn, H.Y. Gas-Solid Reactions; Academic Press: New York, NY, USA, 1976. [Google Scholar]
- Kim, S.H.; Zhang, X.; Ma, Y.; Souza Filho, I.R.; Schweinar, K.; Angenendt, K.; Vogel, D.; Stephenson, L.T.; El-Zoka, A.A.; Mianroodi, J.R.; et al. Influence of Microstructure and Atomic-Scale Chemistry on the Direct Reduction of Iron Ore with Hydrogen at 700 °C. Acta Mater. 2021, 212, 116933. [Google Scholar] [CrossRef]
- Emig, G.; Klemm, E. Chemische Reaktionstechnik; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar] [CrossRef]
- Chorkendorff, I.; Niemantsverdriet, J.W. Concepts of Modern Catalysis and Kinetics; Wiley: Hoboken, NJ, USA, 2003. [Google Scholar] [CrossRef]
- Gómez-Barea, A.; Ollero, P. An approximate Method for solving Gas–Solid Non-Catalytic Reactions. Chem. Eng. Sci. 2006, 61, 3725–3735. [Google Scholar] [CrossRef]
- Melchiori, T.; Canu, P. Improving the Quantitative Description of Reacting Porous Solids: Critical Analysis of the Shrinking Core Model by Comparison to the Generalized Grain Model. Ind. Eng. Chem. Res. 2014, 53, 8980–8995. [Google Scholar] [CrossRef]
- Khawam, A.; Flanagan, D.R. Solid-State Kinetic Models: Basics and Mathematical Fundamentals. J. Phys. Chem. B 2006, 110, 17315–17328. [Google Scholar] [CrossRef]
- Khawam, A.; Flanagan, D.R. Basics and Applications of Solid-State Kinetics: A Pharmaceutical Perspective. J. Pharm. Sci. 2006, 95, 472–498. [Google Scholar] [CrossRef]
- Li, Q.; Lin, X.; Luo, Q.; Chen, Y.; Wang, J.; Jiang, B.; Pan, F. Kinetics of the Hydrogen Absorption and Desorption Processes of Hydrogen Storage Alloys: A Review. Int. J. Miner. Metall. Mater. 2022, 29, 32–48. [Google Scholar] [CrossRef]
- Pang, Y.; Li, Q. A Review on Kinetic Models and Corresponding Analysis Methods for Hydrogen Storage Materials. Int. J. Hydrogen Energy 2016, 41, 18072–18087. [Google Scholar] [CrossRef]
- Faraji, F.; Alizadeh, A.; Rashchi, F.; Mostoufi, N. Kinetics of Leaching: A Review. Rev. Chem. Eng. 2022, 38, 113–148. [Google Scholar] [CrossRef]
- El-Geassy, A.A. Gaseous Reduction of Fe2O3 Compacts at 600 to 1050 °C. J. Mater. Sci. 1986, 21, 3889–3900. [Google Scholar] [CrossRef]
- Parisi, D.R.; Laborde, M.A. Modeling of Counter Current Moving Bed Gas-Solid Reactor Used in Direct Reduction of Iron Ore. Chem. Eng. J. 2004, 104, 35–43. [Google Scholar] [CrossRef]
- Pineau, A.; Kanari, N.; Gaballah, I. Kinetics of Reduction of Iron Oxides by H2: Part I: Low temperature reduction of hematite. Thermochim. Acta 2006, 447, 89–100. [Google Scholar] [CrossRef]
- Turkdogan, E.T.; Vinters, J.V. Gaseous Reduction of Iron Oxides: Part I. Reduction of Hematite in Hydrogen. Metall. Mater. Trans. B 1971, 2, 3175–3188. [Google Scholar] [CrossRef]
- Usui, T.; Ohmi, M.; Yamamura, E. Analysis of Rate of Hydrogen Reduction of Porous Wustite Pellets Basing on Zone-Reaction Models. ISIJ Int. 1990, 30, 347–355. [Google Scholar] [CrossRef]
- King, P.L.; Wheeler, V.M.; Renggli, C.J.; Palm, A.B.; Wilson, S.A.; Harrison, A.L.; Morgan, B.; Nekvasil, H.; Troitzsch, U.; Mernagh, T.; et al. Gas–Solid Reactions: Theory, Experiments and Case Studies Relevant to Earth and Planetary Processes. Rev. Mineral. Geochem. 2018, 84, 1–56. [Google Scholar] [CrossRef]
- Szekely, J.; Evans, J.W. A Structural Model for Gas-Solid Reactions with a Moving Boundary-II. Chem. Eng. Sci. 1971, 26, 1901–1913. [Google Scholar] [CrossRef]
- Schüth, F. Handbook of Porous Solids; WILEY-VCH: Weinheim, Germany, 2002. [Google Scholar] [CrossRef]
- Valipour, M.S.; Motamed Hashemi, M.Y.; Saboohi, Y. Mathematical Modeling of the Reaction in an Iron Ore Pellet Using a Mixture of Hydrogen, Water Vapor, Carbon Monoxide and Carbon Dioxide: An Isothermal Study. Adv. Powder Technol. 2006, 17, 277–295. [Google Scholar] [CrossRef]
- Hossain, M.M.; de Lasa, H.I. Reduction and Oxidation Kinetics of Co–Ni/Al2O3 Oxygen Carrier Involved in a Chemical-Looping Combustion Cycles. Chem. Eng. Sci. 2010, 65, 98–106. [Google Scholar] [CrossRef]
- Piotrowski, K.; Mondal, K.; Wiltowski, T.; Dydo, P.; Rizeg, G. Topochemical Approach of Kinetics of the Reduction of Hematite to Wüstite. Chem. Eng. J. 2007, 131, 73–82. [Google Scholar] [CrossRef]
- Nouri, S.; Ale Ebrahim, H.; Jamshidi, E. Simulation of Direct Reduction Reactor by the Grain Model. Chem. Eng. J. 2011, 166, 704–709. [Google Scholar] [CrossRef]
- Jeong, M.H.; Lee, D.H.; Bae, J.W. Reduction and Oxidation Kinetics of Different Phases of Iron Oxides. Int. J. Hydrogen Energy 2015, 40, 2613–2620. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. ICTAC Kinetics Committee Recommendations for Performing Kinetic Computations on Thermal Analysis Data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Kang, K.S.; Kim, C.H.; Bae, K.K.; Cho, W.C.; Jeong, S.U.; Lee, Y.J.; Park, C.S. Reduction and Oxidation Properties of Fe2O3/ZrO2 Oxygen Carrier for Hydrogen Production. Chem. Eng. Res. Des. 2014, 92, 2584–2597. [Google Scholar] [CrossRef]
- Guo, D.; Hu, M.; Pu, C.; Xiao, B.; Hu, Z.; Liu, S.; Wang, X.; Zhu, X. Kinetics and Mechanisms of Direct Reduction of Iron Ore-Biomass Composite Pellets with Hydrogen Gas. Int. J. Hydrogen Energy 2015, 40, 4733–4740. [Google Scholar] [CrossRef]
- Alonso, E.; Pérez-Rábago, C. Isothermal and Non-Isothermal Kinetics of Metal Oxide Redox Reactions Performed in a Solar Furnace. In AIP Conference Proceedings; AIP Publishing LLC.: Melville, NY, USA, 2018; Volume 2033, p. 100002. [Google Scholar] [CrossRef]
- Luo, L.; Zhang, Z.; Li, C.; Nishu; He, F.; Zhang, X.; Cai, J. Insight into Master Plots Method for Kinetic Analysis of Lignocellulosic Biomass Pyrolysis. Energy 2021, 233, 121194. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Dollimore, D. Linear and Nonlinear Procedures in Isoconversional Computations of the Activation Energy of Nonisothermal Reactions in Solids. J. Chem. Inf. Comput. Sci. 1996, 36, 42–45. [Google Scholar] [CrossRef]
- Šesták, J.; Berggren, G. Study of the Kinetics of the Mechanism of Solid-State Reactions at Increasing Temperatures. Thermochim. Acta 1971, 3, 1–12. [Google Scholar] [CrossRef]
- Pérez-Maqueda, L.A.; Criado, J.M.; Sanchez-Jiménez, P.E. Combined Kinetic Analysis of Solid-State Reactions: A Powerful Tool for the Simultaneous Determination of Kinetic Parameters and the Kinetic Model Without Previous Assumptions on the Reaction Mechanism. J. Phys. Chemistry. A 2006, 110, 12456–12462. [Google Scholar] [CrossRef]
- DIN EN 14647:2005; Calcium Aluminate Cement—Composition, Specifications and Conformity Criteria. Beuth Verlag GmbH: Berlin, Germany, 2007.
- Huber, L.; Gamisch, B.; Dawoud, B. Experimental Investigation of the Cycle Stability of Different Iron Oxide Composites for a Redox Hydrogen Storage Process. In Proceedings of the International Renewable Energy Storage Conference 2021 (IRES 2021), Paris, France, 18–22 November 2022. [Google Scholar] [CrossRef]
- Mehmood, T.; Liland, K.H.; Snipen, L.; Sæbø, S. A Review of Variable Selection Methods in Partial Least Squares Regression. Chemom. Intell. Lab. Syst. 2012, 118, 62–69. [Google Scholar] [CrossRef]
- Heumann, T. Diffusion in Metallen: Grundlagen Theorie Vorgänge in Reinmetallen und Legierungen; Springer: Berlin/Heidelberg, Germany, 1992; Volume 10. [Google Scholar]
- Piotrowski, K.; Mondal, K.; Lorethova, H.; Stonawski, L.; Szymanski, T.; Wiltowski, T. Effect of Gas Composition on the Kinetics of Iron Oxide Reduction in a Hydrogen Production Process. Int. J. Hydrogen Energy 2005, 30, 1543–1554. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, J.; Li, Q.; He, Y.; Tang, B.; Li, M.; Zhang, Z.; Zou, Z. Thermodynamic Analyses of Iron Oxides Redox Reactions. In Proceedings of the 8th Pacific Rim International Congress on Advanced Materials and Processing, Waikoloa, HI, USA, 4–9 August 2013; Marquis, F., Ed.; Springer International Publishing: Cham, Switzerland, 2016; Volume 44, pp. 777–789. [Google Scholar] [CrossRef]
- Takenaka, Y.; Kimura, Y.; Narita, K.; Kaneko, D. Mathematical Model of Direct Reduction Shaft Furnace and its Application to Actual Operations of a Model Plant. Comput. Chem. Eng. 1986, 10, 67–75. [Google Scholar] [CrossRef]
- Pineau, A.; Kanari, N.; Gaballah, I. Kinetics of Reduction of Iron Oxides by H2: Part II. Low temperature reduction of magnetite. Thermochim. Acta 2007, 456, 75–88. [Google Scholar] [CrossRef]
- El-Geassy, A.A.; Shehata, K.A.; Ezz, S.Y. Mechanism of Iron Oxide Reduction with Hydrogen/Carbon Monoxide Mixtures. Trans. Iron Steel Inst. Jpn. 1977, 17, 629–635. [Google Scholar] [CrossRef]
- Lin, H.Y.; Chen, Y.W.; Li, C. The Mechanism of Reduction of Iron Oxide by Hydrogen. Thermochim. Acta 2003, 400, 61–67. [Google Scholar] [CrossRef]
- Manchili, S.K.; Wendel, J.; Hryha, E.; Nyborg, L. Analysis of Iron Oxide Reduction Kinetics in the Nanometric Scale Using Hydrogen. Nanomaterials 2020, 10, 1276. [Google Scholar] [CrossRef]
- Munteanu, G.; Ilieva, L.; Andreeva, D. TPR Data regarding the Effect of Sulfur on the Reducibility of Alpha-Fe2O3. Thermochim. Acta 1999, 329, 157–162. [Google Scholar] [CrossRef]
- Munteanu, G.; Ilieva, L.; Andreeva, D. Kinetic Parameters obtained from TPR Data for Alpha-Fe2O3 and Systems. Thermochim. Acta 1997, 291, 171–177. [Google Scholar] [CrossRef]
- Kuila, S.K.; Chatterjee, R.; Ghosh, D. Kinetics of Hydrogen Reduction of Magnetite Ore Fines. Int. J. Hydrogen Energy 2016, 41, 9256–9266. [Google Scholar] [CrossRef]
- Coetsee, T.; Pistorius, P.; de Villiers, E. Rate-Determining Steps for Reduction in Magnetite-Coal Pellets. Miner. Eng. 2002, 15, 919–929. [Google Scholar] [CrossRef]
- Wen, F.; Wang, H.; Tang, Z. Kinetic Study of the Redox Process of Iron Oxide for Hydrogen Production at Oxidation Step. Thermochim. Acta 2011, 520, 55–60. [Google Scholar] [CrossRef]
- Go, K.; Son, S.; Kim, S. Reaction Kinetics of Reduction and Oxidation of Metal Oxides for Hydrogen Production. Int. J. Hydrogen Energy 2008, 33, 5986–5995. [Google Scholar] [CrossRef]
- Tsay, Q.T.; Ray, W.H.; Szekely, J. The Modeling of Hematite Reduction with Hydrogen Plus Carbon Monoxide Mixtures: Part I. The Behavior of Single Pellets. AIChE J. 1976, 22, 1064–1072. [Google Scholar] [CrossRef]
- Beheshti, R.; Moosberg-Bustnes, J.; Kennedy, M.W.; Aune, R.E. Reduction of Commercial Hematite Pellet in Isothermal Fixed Bed—Experiments and Numerical Modelling. Ironmak. Steelmak. 2016, 43, 31–38. [Google Scholar] [CrossRef]
- Chen, Z.; Zeilstra, C.; van der Stel, J.; Sietsma, J.; Yang, Y. Reduction Mechanism of Fine Hematite Ore Particles in Suspension. Metall. Mater. Trans. B 2021, 52, 2239–2252. [Google Scholar] [CrossRef]
- Yu, X.; Ji, Z. Grain Boundary in Oxide Scale During High-Temperature Metal Processing. In Study of Grain Boundary Character; Tanski, T., Borek, W., Eds.; InTech: London, UK, 2017. [Google Scholar] [CrossRef]
- Laidler, K.J. The Development of the Arrhenius Equation. J. Chem. Educ. 1984, 61, 494. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Mechanism | Symbol | Kinetic Mechanism Function | |
|---|---|---|---|
| Differential Form | Integral Form | ||
| Nucleation models (Avarami–Erofe’ev) | |||
| Nucleation and growth, n = order of reaction | An | ||
| Phase boundary-controlled reaction | |||
| Geometrical contracting infinite slab (Film Diffusion control) | R1 | 1 | |
| Geometrical contracting cylinder (Chemical reaction control) | R2 | ||
| Geometrical contracting sphere (Chemical reaction control) | R3 | ||
| Diffusion models | |||
| 3D Diffusion Jander eqn. | D3 | ||
| Ginstling–Brounshtein (Ash Layer, Diffusion control) | D4 | ||
| Reaction order-based models | |||
| First-order | F1 | ||
| Second-order | F2 | ||
| o | m | p | Is Suitable for: |
|---|---|---|---|
| − | − | − | Phase boundary controlled reaction |
| ✓ | − | − | Phase boundary-controlled reaction (R2, R3), Reaction order models (F1) |
| − | ✓ | − | Mampel Equation (P2, P3), linear nucleation and diffusion models |
| − | − | ✓ | Gingstling–Brounshtein (D4), 2D diffusion models |
| ✓ | ✓ | − | Nucleation, latter stages of linear growth of nuclei |
| ✓ | − | ✓ | Growth of nulcei (A1.5–A4) |
| − | ✓ | ✓ | Unjustified as yet |
| ✓ | ✓ | ✓ | Any complicated case, unjustified as yet |
| Device | Physical Unit | Measurement Accuracy |
|---|---|---|
| STA 449-F3, Netzsch | Mass | 35,000 mg ± 0.0001 mg |
| Temperature | ||
| EL-FLOW, Bronkhorst | Gas flow (H2, N2) | ± 0.5% |
| Steam generator DV2MK, ADROP | Mass flow (H2O) | ± 1% |
| Label | Reduction | Oxidation | ||
|---|---|---|---|---|
| H2/N2 | H2O/N2 | |||
| / | mol%/mol% | / | mol%/mol% | |
| ro | 63/187 | 25/75 | 1.8/162 | 16/84 |
| ro | 125/125 | 50/50 | 3.6/125 | 33/67 |
| ro | 245/5 | 98/2 | 5.8/60 | 67/33 |
| Reaction | Temperature | Conc. | Geometry | Model | Experiment | Ref. | |
|---|---|---|---|---|---|---|---|
| Pellet, | R3 | Isothermal | [53,72] | ||||
| Pellet, | R3 | Isothermal | [53,72] | ||||
(Fe2O3-20% ZrO2) | 1023–1173 K | –25% | Powder, | R3 | Isothermal | [59] | |
| 973–1173 K | 10% + 90% | Powder, | A2 | Isothermal | [70] | ||
| 610–877 K | 100% | Powder, | A2/A3 R1 | 39– | Isothermal | [47] | |
| 493–956 K | 10% + 90% | Powder, | A2/A3 Rx | 36– | Isothermal | [47] | |
| 473–723 K | 100% | Powder, | R2/R3 | Isothermal | [73] | ||
| 523–663 K | 100% | Powder, | R2/R3 | Isothermal | [73] | ||
| >663 K | 100% | Powder, | A2 | Isothermal | [73] | ||
| 973–1373 K | 100% | Cyl., | - | Isothermal | [74] | ||
| 1123–1323 K | 100% | Pellet, – | R3 | Isothermal | [60] | ||
| 603–1003 K | 5% / | Powder, – | A2 | Nonisothermal | [75] | ||
| 703–953 K | 83% /Ar | Powder, | - | 45– | Nonisothermal | [76] | |
| 600–1000 K | 10% /Ar | Powder, | - | 93.2– | Nonisothermal | [77] | |
| 450–1000 K | 10% /Ar | Powder, | - | Nonisothermal | [78] | ||
| 450–1000 K | 10% H2/Ar | Powder, | - | Nonisothermal | [78] | ||
| 973–1273 K | 100% H2 | Powder, | - | Isothermal | [79] | ||
| 973–1273 K | 100% H2 | Powder, | - | Isothermal | [79] | ||
| 1533–1573 K | Pellet, | F1 | Isothermal | [80] | |||
| 1533–1573 K | Pellet, | F1 | Isothermal | [80] | |||
(Fe2O3-20% ZrO2) | 1023–1173 K | H2O/Ar = 5–20% | Powder, | R3 | Isothermal | [59] | |
| (Fe2O3) | 633–713 K | H2O/Ar = 1:4 | Powder, | D3 | 55.5– | Isothermal | [81] |
(Fe2O3-8% Mo) | 633–713 K | H2O/Ar = 1:4 | Powder, | D3 | 36.2– | Isothermal | [81] |
| 1023–1173 K | , | Powder, | D3 | Isothermal | [57] | ||
| (Fe2O3) | 773–1173 K | Powder, –8 mg | D3 | Isothermal | [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamisch, B.; Huber, L.; Gaderer, M.; Dawoud, B. On the Kinetic Mechanisms of the Reduction and Oxidation Reactions of Iron Oxide/Iron Pellets for a Hydrogen Storage Process. Energies 2022, 15, 8322. https://doi.org/10.3390/en15218322
Gamisch B, Huber L, Gaderer M, Dawoud B. On the Kinetic Mechanisms of the Reduction and Oxidation Reactions of Iron Oxide/Iron Pellets for a Hydrogen Storage Process. Energies. 2022; 15(21):8322. https://doi.org/10.3390/en15218322
Chicago/Turabian StyleGamisch, Bernd, Lea Huber, Matthias Gaderer, and Belal Dawoud. 2022. "On the Kinetic Mechanisms of the Reduction and Oxidation Reactions of Iron Oxide/Iron Pellets for a Hydrogen Storage Process" Energies 15, no. 21: 8322. https://doi.org/10.3390/en15218322
APA StyleGamisch, B., Huber, L., Gaderer, M., & Dawoud, B. (2022). On the Kinetic Mechanisms of the Reduction and Oxidation Reactions of Iron Oxide/Iron Pellets for a Hydrogen Storage Process. Energies, 15(21), 8322. https://doi.org/10.3390/en15218322