

X-ray and Synchrotron FTIR Studies of Partially Decomposed Magnesium Borohydride


 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
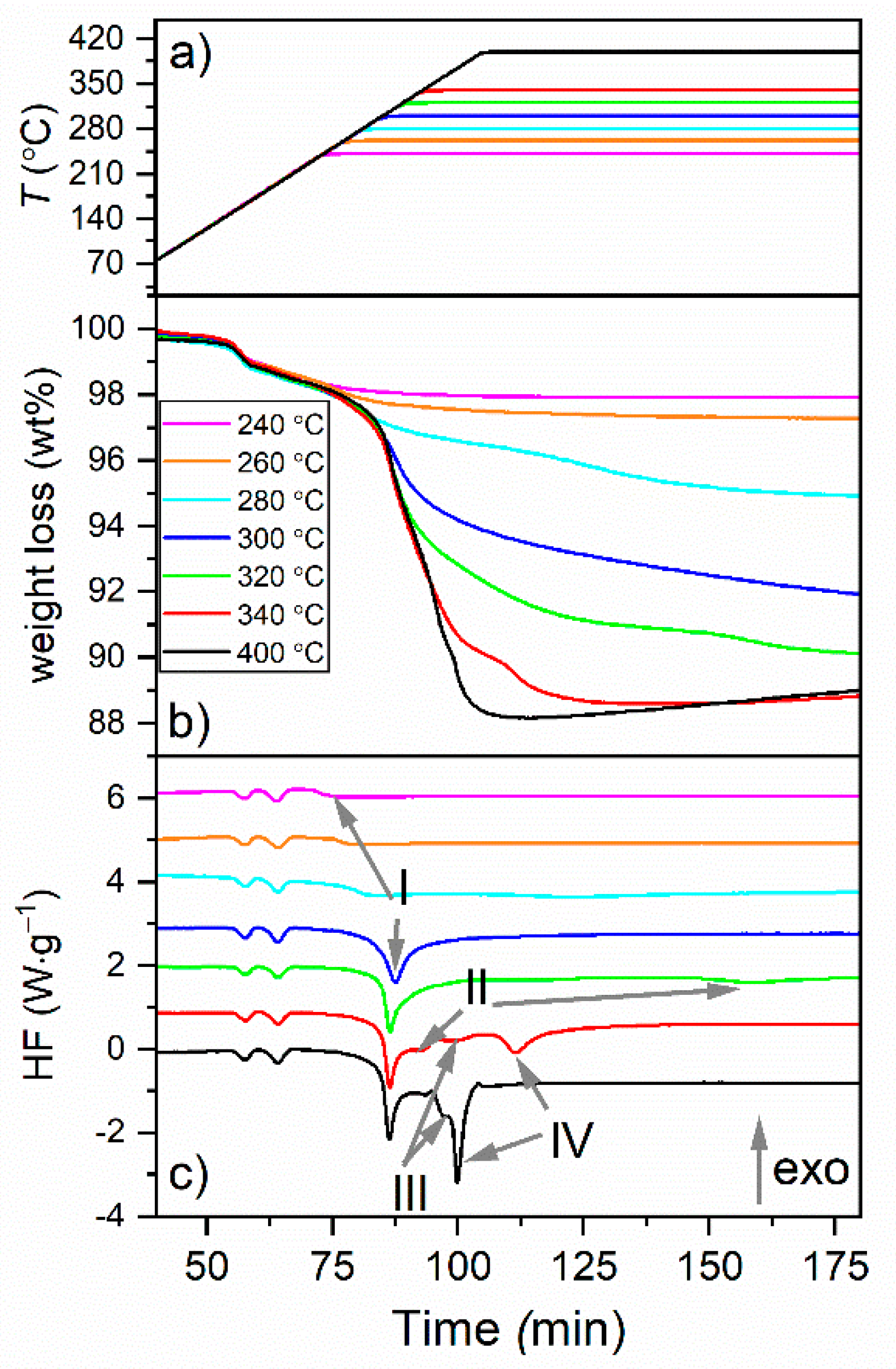
3.1. TGA-DSC Analysis of Decomposition of γ-Mg(BH4)2
- Some weight loss, ~0.5 wt%, occurred between RT and the phase-transition temperature region (Tons = 152 °C).
- ~0.8 wt% occurred during the two phase transitions at 163 and 196 °C.
- The number of the reaction steps is the same for the 240 °C and 260 °C isotherms. The DSC profiles of these reactions show the onset of the first decomposition reaction peak (I). This peak is more pronounced in the 280 °C isotherm and is complete in the 300 °C isotherm. Thus, similar reaction products can be expected after the decomposition at 240 and 260 °C, and slightly different at 280 °C. These reaction products decompose as shown by the strong exothermic peak at 305 °C. Moreover, since a larger weight loss was observed at 260 °C than that at 240 °C, we could expect the presence of Mg(BH4)2 in the latter sample due to the incomplete decomposition reaction at this step. The final weight loss for reaction step I was, thus, observed as 8.8% after decomposition at 300 °C for 5 h when the step was complete.
- The DSC profile of the isotherm obtained at 320 °C shows the onset of yet another peak (II). The weight loss at this temperature was larger (9.9 vs. 8.8% than that at 300 °C) Thus, the reaction products are expected to be somewhat different after the 320 °C isotherm.
- Between 320 and 340 °C, the reaction step II was completed, and two new reactions corresponding to the thermal events III and IV occurred where the latter one was incomplete. The reaction step IV was completed during the decomposition at 400 °C. Thus, yet another and different reaction products can be expected after the decomposition after 340 and 400 °C.
- The most significant weight losses occurred during the decomposition at 300 and 340 °C. These should correspond to the formation and decomposition of MgH2.
- The decomposition at 400 °C was not complete as it resulted in 12 wt% weight loss compared to the 14.9 wt% of the theoretical content of H2. A significant release of borane gases is, thus, not expected because it would correspond to larger weight losses with respect to the theoretical value. As all MgH2 was decomposed at this stage, the remaining hydrogen might be trapped in higher borane intermediates.
- Some isotherms showed weight gain at elevated temperatures. This could likely be related to oxygen contamination and formation of MgO. For these isotherms, the weight loss values (summarized in Table 5) refer to the maximum observed values.
- The TGA-DSC results are discussed in Section 4 after the analysis of the decomposition phases obtained at each of the steps.
3.2. PXRD Analysis of the Reaction Products
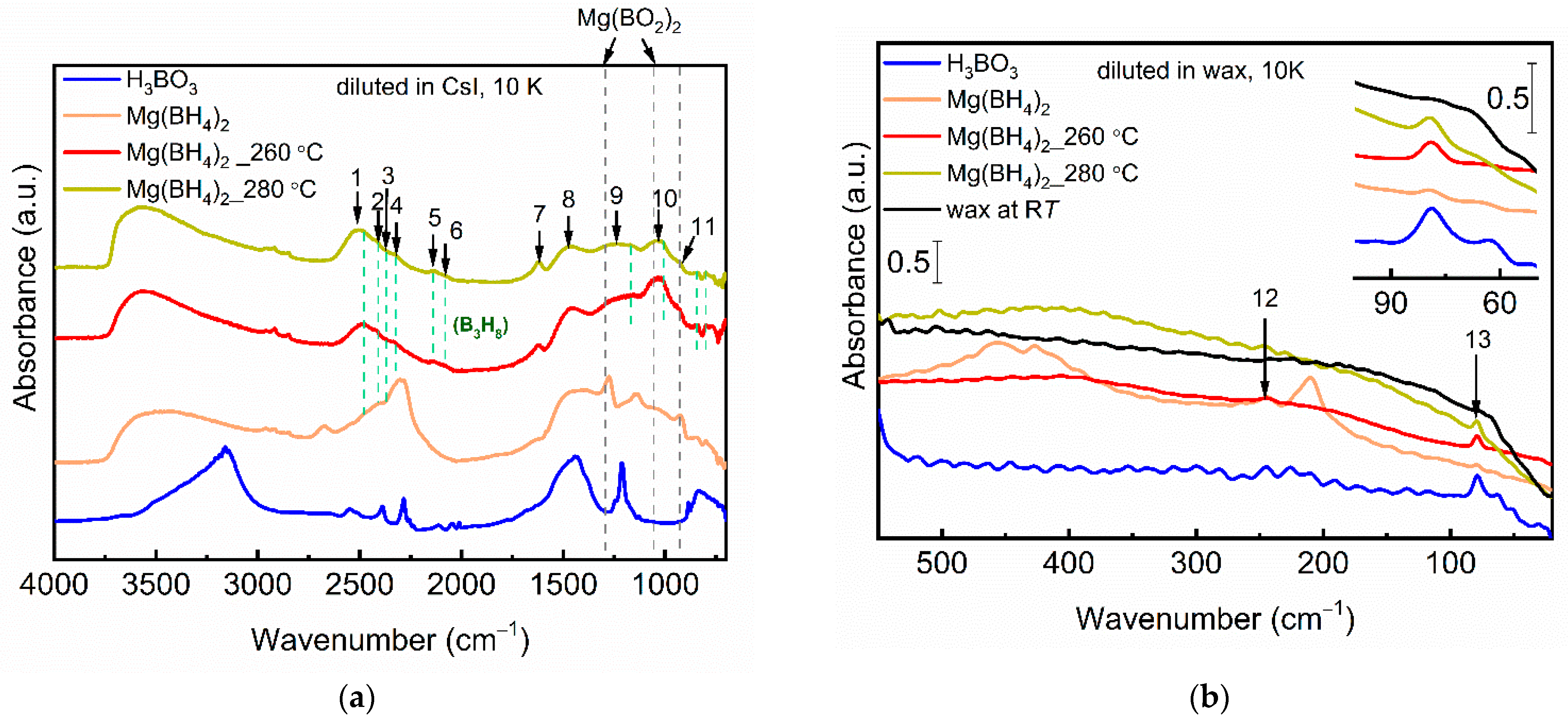
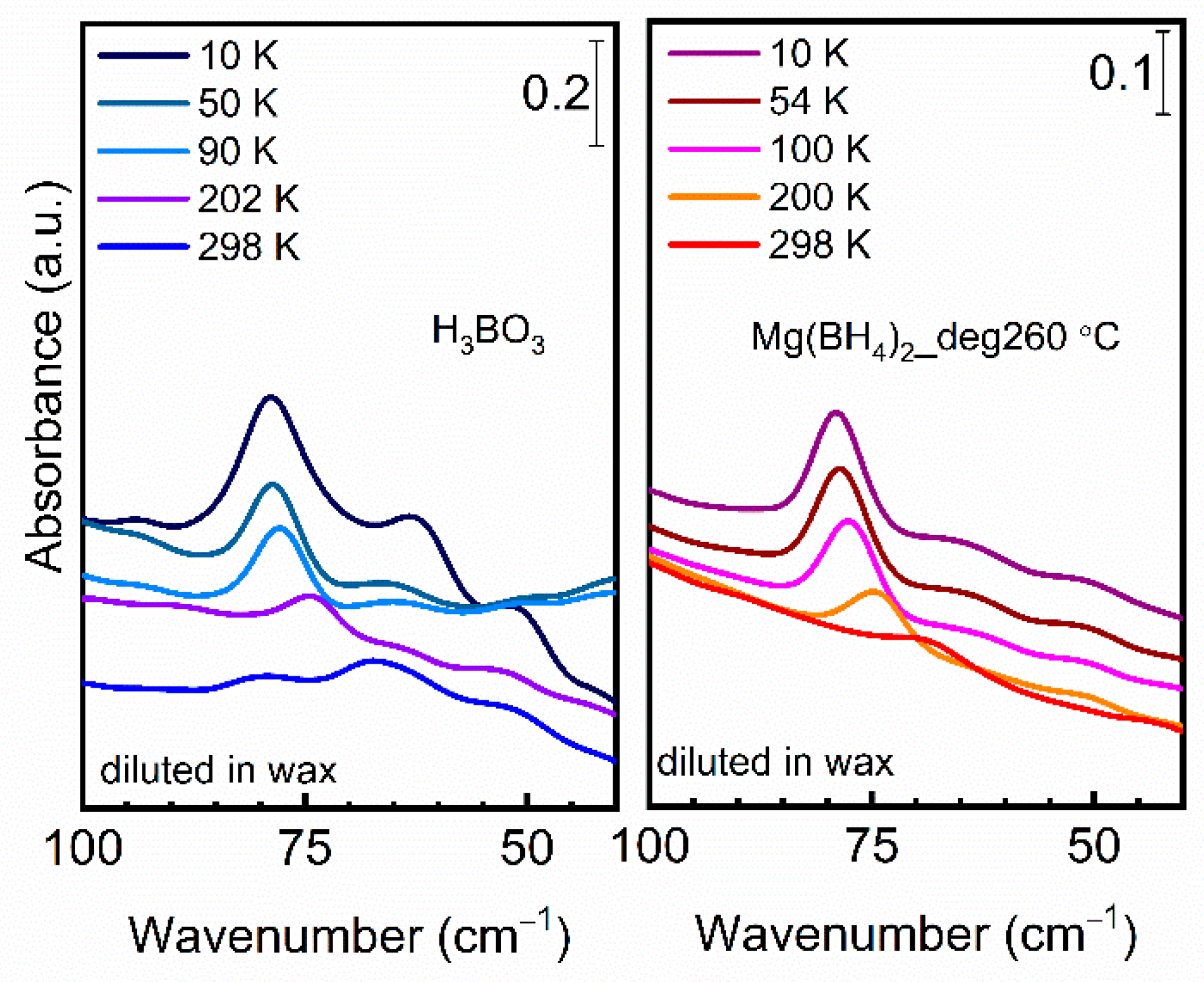
3.3. Synchrotron FTIR Analysis of the Decomposition Reaction Products
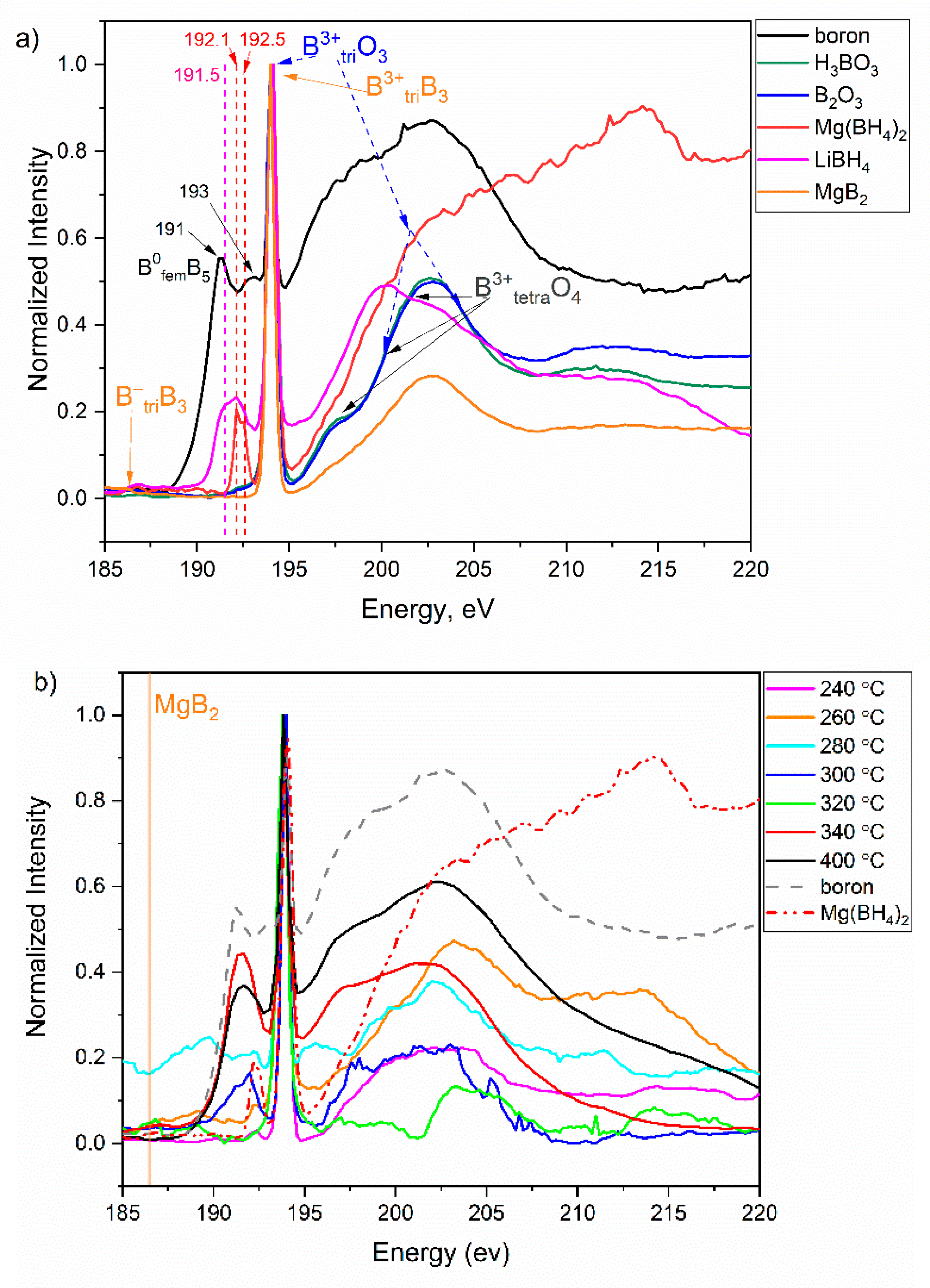
3.4. Soft X-ray Absorption Spectroscopy at the B K-Edge of the Decomposition Products
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rifkin, J. The Hydrogen Economy: The Creation of the Worldwide Energy Web and the Redistribution of Power on Earth; Wiley: New York, NY, USA, 2002; p. 304. [Google Scholar]
- Lovisolo, L.; Serdoner, A.; Whiriskey, K.; Larsson, R. Electrolysis Hydrogen Production in Europe; Bellona Europa, 2021. Available online: https://network.bellona.org/content/uploads/sites/3/2021/04/Electrolysis-Hydrogen-Production-In-Europe-5.pdf (accessed on 10 August 2022).
- Andersson, J.; Grönkvist, S. Large-scale storage of hydrogen. Int. J. Hydrogen Energy 2019, 44, 11901–11919. [Google Scholar] [CrossRef]
- Hydrogen and Fuel Cell Technologies Office. Hydrogen Storage- Basics. Available online: https://www.energy.gov/eere/fuelcells/hydrogen-storage (accessed on 10 August 2022).
- Mohtadi, R.; Orimo, S.-I. The renaissance of hydrides as energy materials. Nat. Rev. Mater. 2016, 2, 16091. [Google Scholar] [CrossRef] [Green Version]
- Rivard, E.; Trudeau, M.; Zaghib, K. Hydrogen Storage for Mobility: A Review. Materials 2019, 12, 1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yartys, V.A.; Lototsky, M.V. An Overview of Hydrogen Storage Methods. In Hydrogen Materials Science and Chemistry of Carbon Nanomaterials; Veziroglu, T.N., Yu. Zaginaichenko, S., Schur, D.V., Baranowski, B., Shpak, A.P., Skorokhod, V.V., Eds.; Springer: Dordrecht, The Netherlands, 2005; pp. 75–104. [Google Scholar]
- Dematteis, E.M.; Amdisen, M.B.; Autrey, T.; Barale, J.; Bowden, M.E.; Buckley, C.E.; Cho, Y.W.; Deledda, S.; Dornheim, M.; de Jongh, P.; et al. Hydrogen storage in complex hydrides: Past activities and new trends. Prog. Energy 2022, 4, 032009. [Google Scholar] [CrossRef]
- Chong, M.; Karkamkar, A.; Autrey, T.; Orimo, S.; Jalisatgi, S.; Jensen, C.M. Reversible dehydrogenation of magnesium borohydride to magnesium triborane in the solid state under moderate conditions. Chem. Commun. 2011, 47, 1330–1332. [Google Scholar] [CrossRef] [PubMed]
- Zavorotynska, O.; El-Kharbachi, A.; Deledda, S.; Hauback, B.C. Recent progress in magnesium borohydride Mg(BH4)2: Fundamentals and applications for energy storage. Int. J. Hydrogen Energy 2016, 41, 14387–14403. [Google Scholar] [CrossRef] [Green Version]
- Zavorotynska, O.; Deledda, S.; Li, G.Q.; Matsuo, M.; Orimo, S.; Hauback, B.C. Isotopic Exchange in Porous and Dense Magnesium Borohydride. Angew. Chem. Int. Ed. 2015, 54, 10592–10595. [Google Scholar] [CrossRef]
- Heere, M.; Zavorotynska, O.; Deledda, S.; Sorby, M.H.; Book, D.; Steriotis, T.; Hauback, B.C. Effect of additives, ball milling and isotopic exchange in porous magnesium borohydride. RSC Adv. 2018, 8, 27645–27653. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Chan, H.; Reed, D.; Book, D. Investigation of dehydrogenation processes in disordered γ-Mg(BH4)2. J. Alloys Compd. 2013, 580, S296–S300. [Google Scholar] [CrossRef]
- Hanada, N.; Chłopek, K.; Frommen, C.; Lohstroh, W.; Fichtner, M. Thermal decomposition of Mg(BH4)2 under He flow and H2 pressure. J. Mater. Chem. 2008, 18, 2611–2614. [Google Scholar] [CrossRef]
- Li, H.W.; Kikuchi, K.; Nakamori, Y.; Miwa, K.; Towata, S.; Orimo, S. Effects of ball milling and additives on dehydriding behaviors of well-crystallized Mg(BH4)2. Scr. Mater. 2007, 57, 679–682. [Google Scholar] [CrossRef]
- Paskevicius, M.; Pitt, M.P.; Webb, C.J.; Sheppard, D.A.; Filsø, U.; Gray, E.M.; Buckley, C.E. In-Situ X-ray Diffraction Study of γ-Mg(BH4)2 Decomposition. J. Phys. Chem. C 2012, 116, 15231–15240. [Google Scholar] [CrossRef]
- Soloveichik, G.L.; Gao, Y.; Rijssenbeek, J.; Andrus, M.; Kniajanski, S.; Bowman, R.C.; Hwang, S.-J.; Zhao, J.-C. Magnesium borohydride as a hydrogen storage material: Properties and dehydrogenation pathway of unsolvated Mg(BH4)2. Int. J. Hydrogen Energy 2009, 34, 916–928. [Google Scholar] [CrossRef]
- Vitillo, J.G.; Bordiga, S.; Baricco, M. Spectroscopic and Structural Characterization of Thermal Decomposition of γ-Mg(BH4)2: Dynamic Vacuum versus H2 Atmosphere. J. Phys. Chem. C 2015, 119, 25340–25351. [Google Scholar] [CrossRef]
- Zavorotynska, O.; Saldan, I.; Hino, S.; Humphries, T.D.; Deledda, S.; Hauback, B.C. Hydrogen cycling in γ-Mg(BH4)2 with cobalt-based additives. J. Mater. Chem. A 2015, 3, 6592–6602. [Google Scholar] [CrossRef] [Green Version]
- Zavorotynska, O.; Deledda, S.; Vitillo, J.G.; Saldan, I.; Guzik, M.N.; Baricco, M.; Walmsley, J.C.; Muller, J.; Hauback, B.C. Combined X-ray and Raman Studies on the Effect of Cobalt Additives on the Decomposition of Magnesium Borohydride. Energies 2015, 8, 9173–9190. [Google Scholar] [CrossRef] [Green Version]
- Newhouse, R.J.; Stavila, V.; Hwang, S.-J.; Klebanoff, L.E.; Zhang, J.Z. Reversibility and Improved Hydrogen Release of Magnesium Borohydride. J. Phys. Chem. C 2010, 114, 5224–5232. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Remhof, A.; Rentsch, D.; Züttel, A. The role of MgB12H12 in the hydrogen desorption process of Mg(BH4)2. Chem. Commun. 2015, 51, 700–702. [Google Scholar] [CrossRef] [Green Version]
- Zavorotynska, O.; Sorby, M.H.; Vitillo, J.G.; Deledda, S.; Frommen, C.; Hauback, B.C. Experimental and computational characterization of phase transitions in CsB3H8. Phys. Chem. Chem. Phys. 2021, 23, 17836–17847. [Google Scholar] [CrossRef]
- Sethio, D.; Daku, L.M.L.; Hagemann, H.; Kraka, E. Quantitative Assessment of B-B-B, B-H(b) -B, and B-H(t) Bonds: From BH(3) to B(12) H(12) (2). Chemphyschem 2019, 20, 1967–1977. [Google Scholar] [CrossRef]
- Severa, G.; Rönnebro, E.; Jensen, C.M. Direct hydrogenation of magnesium boride to magnesium borohydride: Demonstration of >11 weight percent reversible hydrogen storage. Chem. Commun. 2010, 46, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Zavorotynska, O.; Deledda, S.; Hauback, B.C. Kinetics studies of the reversible partial decomposition reaction in Mg(BH4)2. Int. J. Hydrogen Energy 2016, 41, 9885–9892. [Google Scholar] [CrossRef]
- Saldan, I.; Hino, S.; Humphries, T.D.; Zavorotynska, O.; Chong, M.; Jensen, C.M.; Deledda, S.; Hauback, B.C. Structural Changes Observed during the Reversible Hydrogenation of Mg(BH4)(2) with Ni-Based Additives. J. Phys. Chem. C 2014, 118, 23376–23384. [Google Scholar] [CrossRef] [Green Version]
- Ray, K.G.; Klebanoff, L.E.; Lee, J.R.I.; Stavila, V.; Heo, T.W.; Shea, P.; Baker, A.A.; Kang, S.; Bagge-Hansen, M.; Liu, Y.-S.; et al. Elucidating the mechanism of MgB2 initial hydrogenation via a combined experimental–theoretical study. Phys. Chem. Chem. Phys. 2017, 19, 22646–22658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.; Heo, T.; Oktawiec, J.; Shi, R.; Kang, S.; White, J.; Schneemann, A.; Zaia, E.; Wan, L.; Ray, K.; et al. A Mechanistic Analysis of Phase Evolution and Hydrogen Storage Behavior in Nanocrystalline Mg(BH4)2 within Reduced Graphene Oxide. ACS Nano 2020, 14, 1745–1756. [Google Scholar] [CrossRef] [Green Version]
- Sahle, C.J.; Kujawski, S.; Remhof, A.; Yan, Y.; Stadie, N.P.; Al-Zein, A.; Tolan, M.; Huotari, S.; Krisch, M.; Sternemann, C. In situ characterization of the decomposition behavior of Mg(BH4)2 by X-ray Raman scattering spectroscopy. Phys. Chem. Chem. Phys. 2016, 18, 5397–5403. [Google Scholar] [CrossRef] [Green Version]
- Solovev, M.V.; Chashchikhin, O.V.; Dorovatovskii, P.V.; Khrustalev, V.N.; Zyubin, A.S.; Zyubina, T.S.; Kravchenko, O.V.; Zaytsev, A.A.; Dobrovolsky, Y.A. Hydrolysis of Mg(BH4)2 and its coordination compounds as a way to obtain hydrogen. J. Power Sources 2018, 377, 93–102. [Google Scholar] [CrossRef]
- Jun, L.; Shuping, X.; Shiyang, G. FT-IR and Raman spectroscopic study of hydrated borates. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1995, 51, 519–532. [Google Scholar] [CrossRef]
- Nakamoto, K. Applications in Inorganic Chemistry. In Infrared and Raman Spectra of Inorganic and Coordination Compounds, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2008; Volume A, pp. 149–354. [Google Scholar]
- Mirabella, F.M. Principles, Theory and Practice of Internal Reflection Spectroscopy. In Handbook of Vibrational Spectroscopy; John Wiley & Sons, Ltd.: New York, NY, USA, 2006. [Google Scholar]
- Chong, M.; Autrey, T.; Jensen, C.M. Lewis Base Complexes of Magnesium Borohydride: Enhanced Kinetics and Product Selectivity upon Hydrogen Release. Inorganics 2017, 5, 89. [Google Scholar] [CrossRef] [Green Version]
- Gigante, A.; Leick, N.; Lipton, A.S.; Tran, B.; Strange, N.A.; Bowden, M.; Martinez, M.B.; Moury, R.; Gennett, T.; Hagemann, H.; et al. Thermal Conversion of Unsolvated Mg(B3H8)2 to BH4– in the Presence of MgH2. ACS Appl. Energy Mater. 2021, 4, 3737–3747. [Google Scholar] [CrossRef]
- Palumbo, O.; Nguyen, P.; Jensen, C.M.; Paolone, A. Determination of the molecular structure of amorphous Mg(B3H8)2(THF)2 through infrared spectroscopic and computational studies. Int. J. Hydrogen Energy 2016, 41, 5986–5993. [Google Scholar] [CrossRef] [Green Version]
- Matsui, Y.; Taylor, R.C. Vibrational spectra and structure of the heptahydrodiborate ion, [B2H7]− in glycol ethers. Spectrochim. Acta Part A Mol. Spectrosc. 1989, 45, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Lutz, C.A.; Ritter, D.M. Observations on alkylboranes. Can. J. Chem. 1963, 41, 1344–1358. [Google Scholar] [CrossRef]
- Hrostowski, H.J.; Pimentel, G.C. The Infrared Spectra of Stable Pentaborane and Deuterated Pentaborane. J. Am. Chem. Soc. 1954, 76, 998–1003. [Google Scholar] [CrossRef]
- Johnson, H.D.; Geanangel, R.A.; Shore, S.G. Alkali metal salts derived from pentaborane(9) and hexaborane(10): MB5H8 and MB6H9. Inorg. Chem. 1970, 9, 908–912. [Google Scholar] [CrossRef]
- Brint, P.; Sangchakr, B.; Fowler, P.W.; Weldon, V.J. Bonding in clusters Part 10. The nature of the bh groups in closo-borane anions—A study by vibrational spectroscopy, electronic spectroscopy for chemical-analysis, and abinitio computation. J. Chem. Soc. Dalton Trans. 1989, 11, 2253–2260. [Google Scholar] [CrossRef]
- Leites, L.A. Vibrational spectroscopy of carboranes and parent boranes and its capabilities in carborane chemistry. Chem. Rev. 1992, 92, 279–323. [Google Scholar] [CrossRef]
- Erbil, A.; Cargill, G.S.; Frahm, R.; Boehme, R.F. Total-electron-yield current measurements for near-surface extended X-ray-absorption fine-structure. Phys. Rev. B 1988, 37, 2450–2464. [Google Scholar] [CrossRef] [Green Version]
- Esposto, F.J.; Aebi, P.; Tyliszczak, T.; Hitchcock, A.P.; Kasrai, M.; Bozek, J.D.; Jackman, T.E.; Rolfe, S.R. Boron K-shell spectroscopy of boron-doped silicon. J. Vac. Sci. Technol. A 1991, 9, 1663–1669. [Google Scholar] [CrossRef]
- Blau, W.; Dudde, R.; Petersen, H. Photoyield measurements of the boron k-edge fine structure of amorphous Fe80B20 alloys. Solid State Commun. 1989, 69, 147–150. [Google Scholar] [CrossRef]
- Henderson, G.S.; de Groot, F.M.F.; Moulton, B.J.A. X-ray Absorption Near-Edge Structure (XANES) Spectroscopy. Rev. Mineral. Geochem. 2014, 78, 75–138. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, E.; Iwata, S.; Suzuki, Y.; Mikuni, A.; Sasaki, T. The boron K photoabsorption spectra of BF3, BCl3, and BBr3. J. Phys. B At. Mol. Phys. 1982, 15, 1841–1854. [Google Scholar] [CrossRef]
- Fleet, M.E.; Muthupari, S. Boron K-edge XANES of borate and borosilicate minerals. Am. Mineral. 2000, 85, 1009–1021. [Google Scholar] [CrossRef]
- Guerfi, T. Out-of-plane ionicity versus in-plane covalency interplay and electron–phonon coupling in MgB2 superconductor. Chin. J. Phys. 2020, 64, 287–294. [Google Scholar] [CrossRef]
- Li, D.; Bancroft, G.M.; Fleet, M.E. B K-edge XANES of crystalline and amorphous inorganic materials. J. Electron Spectrosc. Relat. Phenom. 1996, 79, 71–73. [Google Scholar] [CrossRef]
- Peatman, W.; Schade, U. A brilliant infrared light source at BESSY. Rev. Sci. Instrum. 2001, 72, 1620–1624. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | ν1 | ν2 | ν3 | ν4 |
|---|---|---|---|---|
| [BO3]3− | 1060 (R) 1 | 668, 648 (IR) | 1490–1428 (R, IR) | 545 (R, IR) |
| [BO4]5− | 880 (R) | 371 (R) | 886 (R, IR) | 627 (R, IR) |
| H3BO3 | Mg(BO2)2 · 3H2O | H3BO3 |
|---|---|---|
| Ref. [32] | Ref. [32] | This work |
| 3571, vs 1 | ||
| 3491, vs | ||
| 3202, br | 3179, vs | 3160 vs, br |
| 2515, m | 2531, m | 2550 m, br |
| 2362, m | 2388 m | |
| 2262, s | 2283 m | |
| 2030, w | 2046–2010 w | |
| 1423 (ν3) 2, s, br | 1448 vs, br | |
| 1297, vs | 1245 w | |
| 1221, m | ||
| 1193 (ν3?), vs | 1163, vs | 1212 vs |
| 1042, vs | ||
| 953, vs | ||
| 883, m | 873 (ν3), vs | 886 m |
| 811, m | 802, w | 815 m, br |
| 644 (ν2), s | ||
| 546 (ν4), vs | 586 (ν4), vs | 550, s |
| n/a | 79, m |
| Observed Peak Refs. [10,11] | Mode | Assignment 2 | This Work |
|---|---|---|---|
| n/a | ν1 | νs (B-H) | − |
| 1370, m, br 1 | ν2 | δs (H-B-H) | − |
| 2270 ± 2, vs | ν3 | νa (B-H) | 2300 s, br |
| 1260, s | ν4 | δa (H-B-H) | 1276 s, br |
| 1120 ± 10, m | ν4 | δa (H-B-H) | 1144 s, br |
| 2660, m | ν2 + ν4 | overtone | 2672 m |
| 434, m | Mg-B stretching | 455 m, br | |
| 409, m | Mg-B stretching | 425 m, br | |
| 257, 231, 208, m | B-Mg-B bending | 240 w, 210 m |
| Peak No. | Energy, cm−1 | Tentative Assignment |
|---|---|---|
| 1 | 2510 s, br | ν(B-H) |
| 2 | 2410, s | ν(B-H) |
| 3 | 2375, m | ν(B-H) |
| 4 | 2320, m | ν(B-H), /ν(Mg-H-B) |
| 5 | 2140, w | ν(B-H-B) |
| 6 | 2075, w | ν(B-H-B) |
| 7 | 1620, m | δ(H-O-H)/δ(B-H) |
| 8 | 1460, s, br | νa(Btri-O)/δ(B-H) |
| 9 | ca. 1230, br | νs(Btri-O)/ν(Btetra-O)/δ(B-H) |
| 10 | ~1030, s, br | ν(Btetra-O)/δ(B-H) |
| 11 | 930, sh | ν(B-B)/δ(B-H) |
| 13 | 79, m | T (Btri-O) |
| Tdec (°C) | Maximum Weight Loss, wt% (TGA) | Released No. of H2 Molecules | Nominal Composition after H2 Desorption | Reaction Step (TGA-DSC) | Crystalline Reaction Products (PXRD) | FTIR | NEXAFS |
|---|---|---|---|---|---|---|---|
| 240 | 2.1 | 1.1 | MgB2H7 | I | ht-Mg(BH4)2 | n/a | Btri-O, (BH4)− |
| 260 | 4.5 | 2.4 | MgB2H5.6 | I | No | - several BxHy phases- B-H-B species - B3-B5 clusters | Btri-O, (BH4)− |
| 280 | 5.2 | 2.8 | MgB2H5.2 | I | No | Btri-O | |
| 300 | 8.8 | 4.7 | MgB2H3.3 | I | MgH2 | n/a | Btri-O, boron species between (BH4)− and B0 |
| 320 | 9.9 | 5.3 | MgB2H2.8 | II | Mg, MgO | n/a | Btri-O |
| 340 | 11.6 | 6.2 | MgB2H1.8 | III, IV | Mg, MgO | n/a | Btri-O, B0 |
| 400 | 12 | 6.5 | MgB2H1.5 | IV | Mg, MgO | n/a | Btri-O, B0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahal, R.; Vitillo, J.G.; Åsland, A.C.; Frommen, C.; Deledda, S.; Zavorotynska, O. X-ray and Synchrotron FTIR Studies of Partially Decomposed Magnesium Borohydride. Energies 2022, 15, 7998. https://doi.org/10.3390/en15217998
Dahal R, Vitillo JG, Åsland AC, Frommen C, Deledda S, Zavorotynska O. X-ray and Synchrotron FTIR Studies of Partially Decomposed Magnesium Borohydride. Energies. 2022; 15(21):7998. https://doi.org/10.3390/en15217998
Chicago/Turabian StyleDahal, Rashmi, Jenny G. Vitillo, Anna C. Åsland, Christoph Frommen, Stefano Deledda, and Olena Zavorotynska. 2022. "X-ray and Synchrotron FTIR Studies of Partially Decomposed Magnesium Borohydride" Energies 15, no. 21: 7998. https://doi.org/10.3390/en15217998
APA StyleDahal, R., Vitillo, J. G., Åsland, A. C., Frommen, C., Deledda, S., & Zavorotynska, O. (2022). X-ray and Synchrotron FTIR Studies of Partially Decomposed Magnesium Borohydride. Energies, 15(21), 7998. https://doi.org/10.3390/en15217998