
Optimization of Potassium Promoted Molybdenum Carbide Catalyst for the Low Temperature Reverse Water Gas Shift Reaction


Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
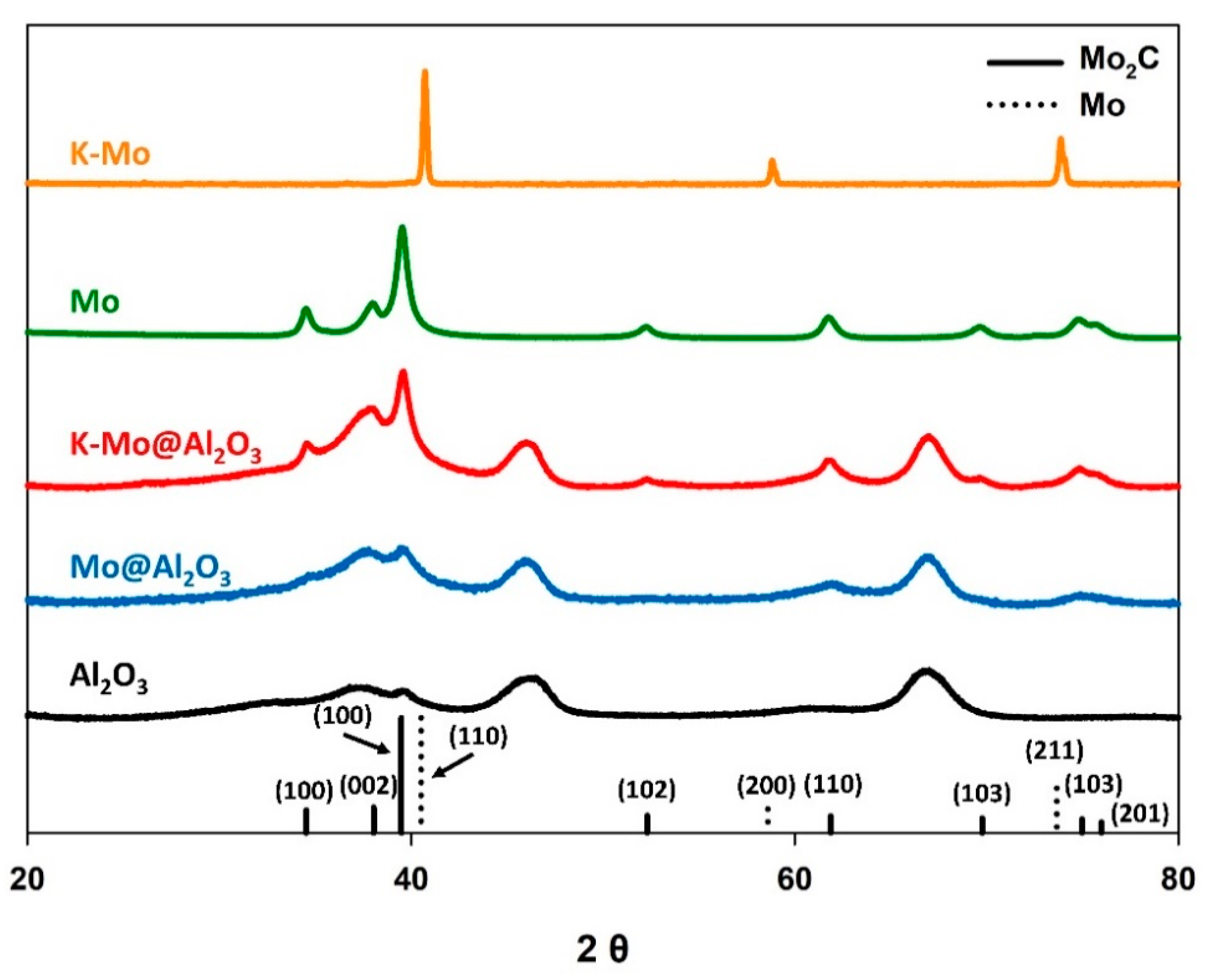
3.1. Influence of γ-Al2O3 Support
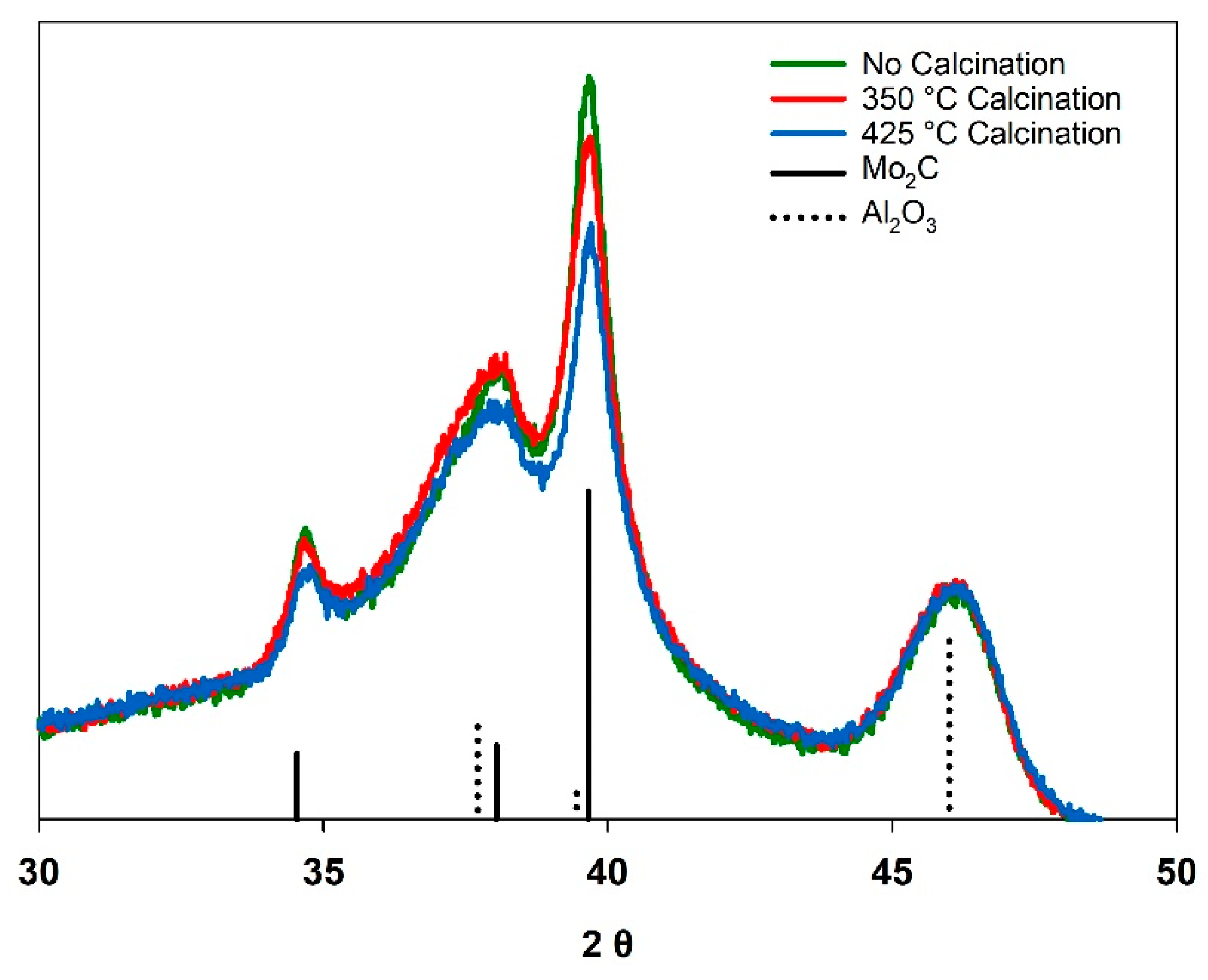
3.2. Calcination
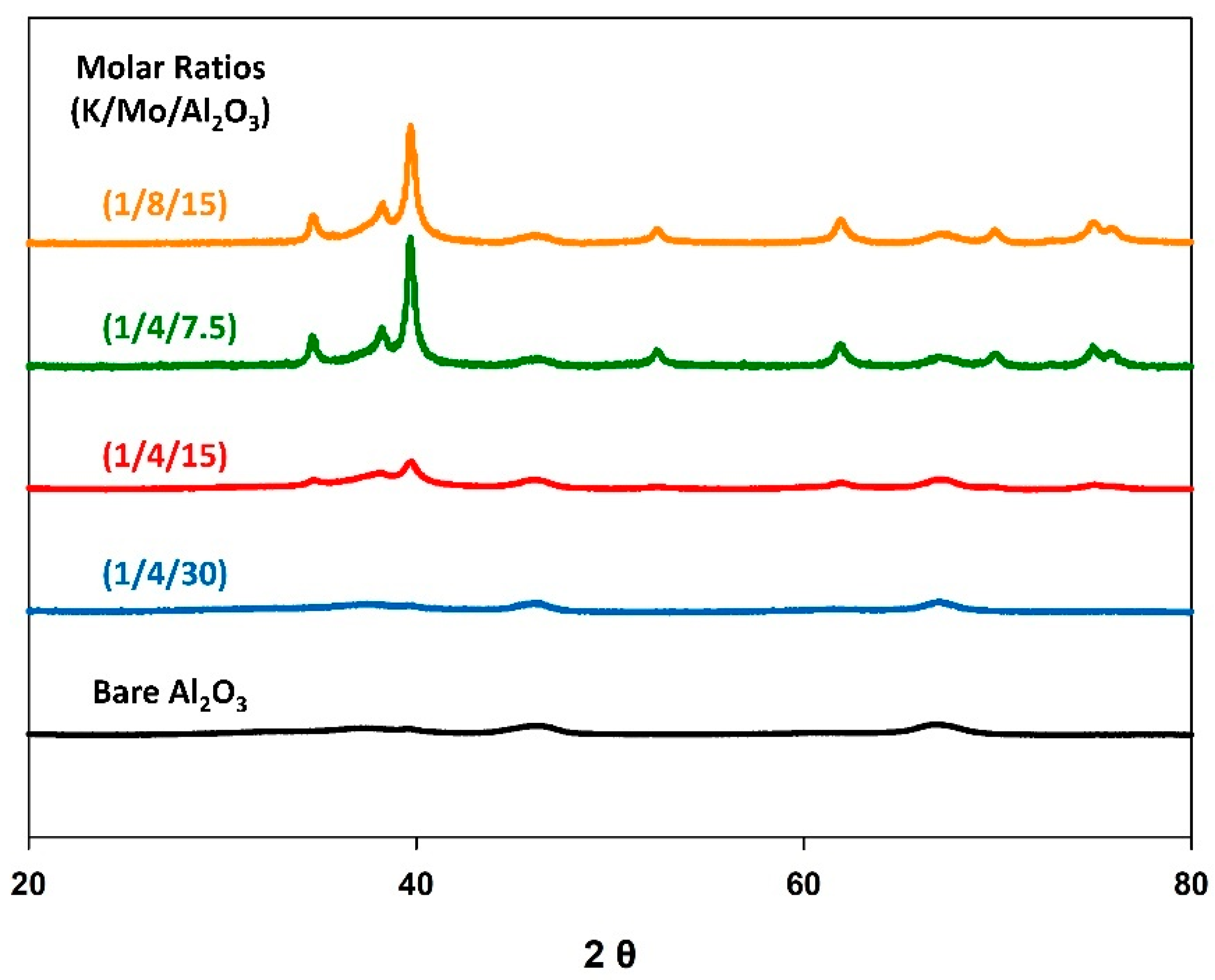
3.3. Catalyst Loading
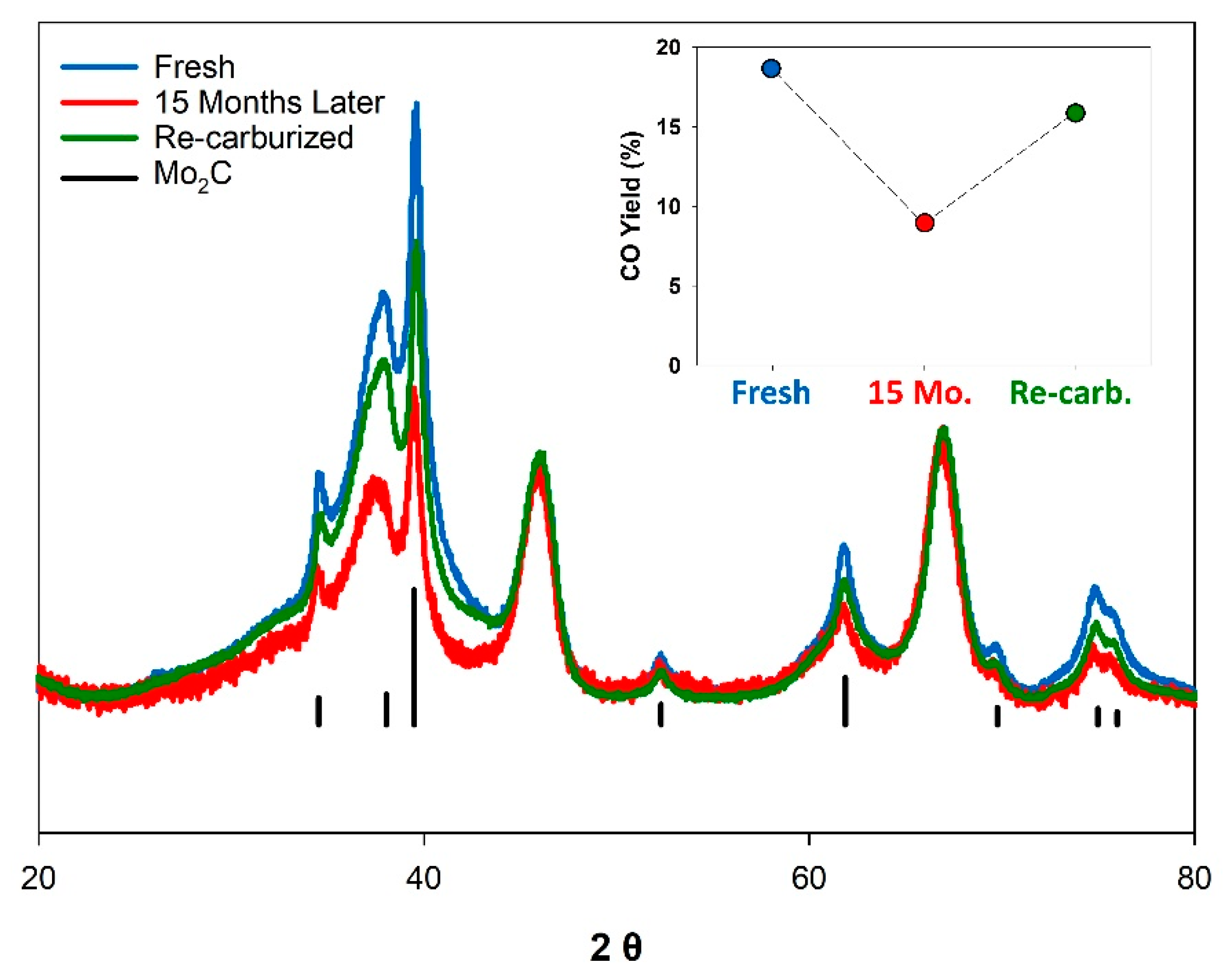
3.4. Shelf Life
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Department of Defense. Operational Energy Strategy. 2016. Available online: http://www.acq.osd.mil/eie/Downloads/OE/2016%20DoD%20Operational%20Energy%20Strategy%20WEBc.pdf (accessed on 28 January 2022).
- National, G.; Pillars, H. Summary of the 2018 National Defense Strategy of the United States of America. Available online: https://dod.defense.gov/Portals/1/Documents/pubs/2018-National-Defense-Strategy-Summary.pdf (accessed on 28 January 2022).
- S.1790—116th Congress (2019–2020): National Defense Authorization Act for Fiscal Year 2020. 20 December 2019. Available online: http://www.congress.gov/ (accessed on 28 January 2022).
- Dorner, R.; Hardy, D.; Williams, F. Influence of Gas Feed Composition and Pressure on the Catalytic Conversion of CO2 to Hydrocarbons Using a Traditional Cobalt-Based Fischer- Tropsch Catalyst. Energy Fuels 2009, 23, 4190–4195. [Google Scholar] [CrossRef]
- Willauer, H.D.; Hardy, D.R.; Lewis, M.K.; Ndubizu, E.C.; Williams, F.W. Effects of pressure on the recovery of CO2 by phase transition from a seawater system by means of multilayer gas permeable membranes. J. Phys. Chem. A 2010, 114, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Willauer, H.D.; Dimascio, F.; Hardy, D.R.; Williams, F.W. Feasibility of CO2 extraction from seawater and simultaneous hydrogen gas generation using a novel and robust electrolytic cation exchange module based on continuous electrodeionization technology. Ind. Eng. Chem. Res. 2014, 53, 12192–12200. [Google Scholar] [CrossRef]
- Willauer, H.D.; DiMascio, F.; Hardy, D.R.; Lewis, M.K.; Williams, F.W. Development of an Electrochemical Acidification Cell for the Recovery of CO2 and H2 from Seawater. Ind. Eng. Chem. Res. 2011, 50, 9876–9882. [Google Scholar] [CrossRef]
- Willauer, H.D.; Dimascio, F.; Hardy, D.R.; Lewis, M.K.; Williams, F.W. Development of an electrochemical acidification cell for the recovery of CO2 and H2 from seawater II. Evaluation of the cell by natural seawater. Ind. Eng. Chem. Res. 2012, 51, 11254–11260. [Google Scholar] [CrossRef]
- Willauer, H.D.; DiMascio, F.; Hardy, D.R.; Williams, F.W. Development of an Electrolytic Cation Exchange Module for the Simultaneous Extraction of Carbon Dioxide and Hydrogen Gas from Natural Seawater. Energy Fuels 2017, 31, 1723–1730. [Google Scholar] [CrossRef]
- Willauer, H.D.; Hardy, D.R.; Lewis, M.K.; Ndubizu, E.C.; Williams, F.W. Extraction of CO2 from seawater and aqueous bicarbonate systems by ion-exchange resin processes. Energy Fuels 2010, 24, 6682–6688. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yan, B.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62. [Google Scholar] [CrossRef]
- Daza, Y.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts and mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Xie, C.; Chen, C.; Yu, Y.; Su, J.; Li, Y.; Somorjai, G.A.; Yang, P. Tandem Catalysis for CO2 Hydrogenation to C2–C4 Hydrocarbons. Nano Lett. 2017, 17, 3798–3802. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703. [Google Scholar] [CrossRef]
- Kim, S.S.; Lee, H.H.; Hong, S.C. A study on the effect of support’s reducibility on the reverse water-gas shift reaction over Pt catalysts. Appl. Catal. A Gen. 2012, 423–424, 100–107. [Google Scholar] [CrossRef]
- Lu, B.; Kawamoto, K. Preparation of mesoporous CeO2 and monodispersed NiO particles in CeO2, and enhanced selectivity of NiO/CeO2 for reverse water gas shift reaction. Mater. Res. Bull. 2014, 53, 70–78. [Google Scholar] [CrossRef]
- Chen, C.-S.; Cheng, W.-H.; Lin, S. Mechanism of CO Formation in Reverse Water–Gas Shift Reaction over Cu/Al2O3 Catalyst. Catal. Lett. 2000, 68, 45–48. [Google Scholar] [CrossRef]
- Joo, O.S.; Jung, K.D.; Moon, I.; Rozovskii, A.Y.; Lin, G.I.; Han, S.H.; Uhm, S.J. Carbon dioxide hydrogenation to form methanol via a reverse-water-gas-shift reaction (the CAMERE process). Ind. Eng. Chem. Res. 1999, 38, 1808–1812. [Google Scholar] [CrossRef]
- Chen, C.S.; Cheng, W.H.; Lin, S.S. Study of iron-promoted Cu/SiO2 catalyst on high temperature reverse water gas shift reaction. Appl. Catal. A Gen. 2004, 257, 97–106. [Google Scholar] [CrossRef]
- Chen, C.S.; Cheng, W.H.; Lin, S.S. Enhanced activity and stability of a Cu/SiO2 catalyst for the reverse water gas shift reaction by an iron promoter. Chem. Commun. 2001, 1, 1770–1771. [Google Scholar] [CrossRef]
- Su, X.; Yang, X.; Zhao, B.; Huang, Y. Designing of highly selective and high-temperature endurable RWGS heterogeneous catalysts: Recent advances and the future directions. J. Energy Chem. 2017, 26, 854–867. [Google Scholar] [CrossRef]
- Willauer, H.D.; Bradley, M.J.; Baldwin, W.; Hartvigsen, J.J.; Frost, L.; Morse, J.R.; Dimascio, F.; Hardy, D.R.; Hasler, D.J. Evaluation of CO2 Hydrogenation in a Modular Fixed-Bed Reactor Prototype. Catalysts 2020, 10, 970. [Google Scholar] [CrossRef]
- Juneau, M.; Vonglis, M.; Hartvigsen, J.; Frost, L.; Bayerl, D.; Dixit, M.; Mpourmpakis, G.; Morse, J.R.; Baldwin, J.W.; Willauer, H.D.; et al. Assessing the viability of K-Mo2C for reverse water-gas shift scale-up: Molecular to laboratory to pilot scale. Energy Environ. Sci. 2020, 13, 2524–2539. [Google Scholar] [CrossRef]
- Kattel, S.; Yu, W.; Yang, X.; Yan, B.; Huang, Y.; Wan, W.; Liu, P.; Chen, J.G. CO2 Hydrogenation over Oxide-Supported PtCo Catalysts: The Role of the Oxide Support in Determining the Product Selectivity. Angew. Chem. Int. Ed. 2016, 55, 7968–7973. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Su, X.; Duan, H.; Liang, B.; Huang, Y.; Zhang, T. Catalytic performance of the Pt/TiO2 catalysts in reverse water gas shift reaction: Controlled product selectivity and a mechanism study. Catal. Today 2017, 281, 312–318. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Chen, J.G. Trends in the catalytic reduction of CO2 by hydrogen over supported monometallic and bimetallic catalysts. J. Catal. 2013, 301, 30–37. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. Heterogeneous catalysis on atomically dispersed supported metals: CO2 reduction on multifunctional Pd catalysts. ACS Catal. 2013, 3, 2094–2100. [Google Scholar] [CrossRef]
- Patterson, P.M.; Das, T.K.; Davis, B.H. Carbon monoxide hydrogenation over molybdenum and tungsten carbides. Appl. Catal. A Gen. 2003, 251, 449–455. [Google Scholar] [CrossRef]
- Oshikawa, K.; Nagai, M.; Omi, S. Characterization of molybdenum carbides for methane reforming by TPR, XRD, and XPS. J. Phys. Chem. B 2001, 105, 9124–9131. [Google Scholar] [CrossRef]
- Claridge, J.B.; York, A.P.E.; Brungs, A.J.; Marquez-alvarez, C.; Sloan, J.; Tsang, S.C.; Green, M.L.H. New Catalysts for the Conversion of Methane to Synthesis Gas: Molybdenum and Tungsten Carbide. J. Catal. 1998, 100, 85–100. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yang, X.; Boscoboinik, J.A.; Chen, J.G. Molybdenum Carbide as Alternative Catalysts to Precious Metals for Highly Selective Reduction of CO2 to CO. Angew. Chemie 2014, 126, 6823–6827. [Google Scholar] [CrossRef]
- Ma, Y.; Guo, Z.; Jiang, Q.; Wu, K.H.; Gong, H.; Liu, Y. Molybdenum carbide clusters for thermal conversion of CO2 to CO via reverse water-gas shift reaction. J. Energy Chem. 2020, 50, 37–43. [Google Scholar] [CrossRef]
- Juneau, M.; Pope, C.; Liu, R.; Porosoff, M.D. Support acidity as a descriptor for reverse water-gas shift over Mo2C-based catalysts. Appl. Catal. A Gen. 2021, 620, 118034. [Google Scholar] [CrossRef]
- Roy, S.; Cherevotan, A.; Peter, S.C. Thermochemical CO2 Hydrogenation to Single Carbon Products: Scientific and Technological Challenges. ACS Energy Lett. 2018, 3, 1938–1966. [Google Scholar] [CrossRef]
- Dietz, L.; Piccinin, S.; Maestri, M. Mechanistic insights into CO2 activation via reverse water—Gas shift on metal surfaces. J. Phys. Chem. C 2015, 119, 4959–4966. [Google Scholar] [CrossRef]
- Morse, J.R.; Juneau, M.; Baldwin, J.W.; Porosoff, M.D.; Willauer, H.D. Alkali promoted tungsten carbide as a selective catalyst for the reverse water gas shift reaction. J. CO2 Util. 2020, 35, 38–46. [Google Scholar] [CrossRef]
- Correlation, T.; Oosthuizen, G.J. The Correlation Between Catalyst Surface Basicity and Hydrocarbon Selectivity in the Fischer Tropsch Synthesis. J. Catal. 1968, 11, 18–24. [Google Scholar]
- Saeidi, S.; Amin, N.A.S.; Rahimpour, M.R. Hydrogenation of CO2 to value-added products—A review and potential future developments. J. CO2 Util. 2014, 5, 66–81. [Google Scholar] [CrossRef]
- Gnanamani, M.K.; Hamdeh, H.H.; Shafer, W.D.; Hopps, S.D.; Davis, B.H. Hydrogenation of carbon dioxide over iron carbide prepared from alkali metal promoted iron oxalate. Appl. Catal. A Gen. 2018, 564, 243–249. [Google Scholar] [CrossRef]
- Woo, H.C.; Park, K.Y.; Kim, Y.G.; Namau]Jong ShikChung, I.S.; Lee, J.S. Mixed alcohol synthesis from carbon monoxide and dihydrogen over potassium-promoted molybdenum carbide catalysts. Appl. Catal. 1991, 75, 267–280. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Baldwin, J.W.; Peng, X.; Mpourmpakis, G.; Willauer, H.D. Potassium-Promoted Molybdenum Carbide as a Highly Active and Selective Catalyst for CO2 Conversion to CO. ChemSusChem 2017, 10, 2408–2415. [Google Scholar] [CrossRef]
- Porosoff, M.; Willauer, H.D. Alkali metal doped molybdenum carbide supported on gamma-alumina for selective CO2 hydrogenation into CO 2019. U.S. Patent 11,266,980, 11 July 2019. [Google Scholar]
- Ro, I.; Resasco, J.; Christopher, P. Approaches for Understanding and Controlling Interfacial Effects in Oxide-Supported Metal Catalysts. ACS Catal. 2018, 8, 7368–7387. [Google Scholar] [CrossRef]
- Munnik, P.; De Jongh, P.E.; De Jong, K.P. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef]
- Christensen, J.M.; Duchstein, L.D.L.; Wagner, J.B.; Jensen, P.A.; Temel, B.; Jensen, A.D. Catalytic conversion of syngas into higher alcohols over carbide catalysts. Ind. Eng. Chem. Res. 2012, 51, 4161–4172. [Google Scholar] [CrossRef]
- Kojima, R.; Aika, K.I. Molybdenum nitride and carbide catalysts for ammonia synthesis. Appl. Catal. A Gen. 2001, 219, 141–147. [Google Scholar] [CrossRef]
- Said, A.A. Mutual influences between ammonium heptamolybdate and γ-alumina during their thermal treatments. Thermochim. Acta 1994, 236, 93–104. [Google Scholar] [CrossRef]
- Vo, D.V.N.; Adesina, A.A. Kinetics of the carbothermal synthesis of Mo carbide catalyst supported on various semiconductor oxides. Fuel Process. Technol. 2011, 92, 1249–1260. [Google Scholar] [CrossRef]
- Hiraishi, J.; Nishijima, A. Support Effect on the Catalytic Activity and Properties Molybdenum Catalysts. J. Catal. 1988, 110, 275–284. [Google Scholar]
- Thomazeau, C.; Martin, V.; Afanasiev, P. Effect of support on the thermal decomposition of (NH4)6Mo7O24·4H2O in the inert gas atmosphere. Appl. Catal. A Gen. 2000, 199, 61–72. [Google Scholar] [CrossRef]
- Liu, X.; Kunkel, C.; Ramírez De La Piscina, P.; Homs, N.; Viñes, F.; Illas, F. Effective and Highly Selective CO Generation from CO2 Using a Polycrystalline α-Mo2C Catalyst. ACS Catal. 2017, 7, 4323–4335. [Google Scholar] [CrossRef]
- Marquart, W.; Raseale, S.; Prieto, G.; Zimina, A.; Sarma, B.B.; Grunwaldt, J.D.; Claeys, M.; Fischer, N. CO2 Reduction over Mo2C-Based Catalysts. ACS Catal. 2021, 11, 1624–1639. [Google Scholar] [CrossRef]
- Lee, J.S.; Oyama, S.T.; Boudart, M. Molybdenum Carbide Catalysts 1. Synthesis of Unsupported Powders. J. Catal. 1987, 106, 125–133. [Google Scholar] [CrossRef]
- Ribeiro, F.H.; Betta, R.A.D.; Guskey, G.J.; Boudart, M. Preparation and Surface Composition of Tungsten Carbide Powders with High Specific Surface Area. Chem. Mater. 1991, 3, 805–812. [Google Scholar] [CrossRef]
- Li, S.; Kim, W.B.; Lee, J.S. Effect of the Reactive Gas on the Solid-State Transformation of Molybdenum Trioxide to Carbides and Nitrides. Chem. Mater. 1998, 10, 1853–1862. [Google Scholar] [CrossRef]
- Volpe, L.; Boudart, M. Compounds of molybdenum and tungsten with high specific surface area. II. Carbides. J. Solid State Chem. 1985, 59, 348–356. [Google Scholar] [CrossRef]
- Thiollier, A.; Afanasiev, P.; Cattenot, M.; Vrinat, M. Preparation and properties of chromium-containing hydrotreating catalysts (Ni-Mo)/ZrO2-Cr2O3. Catal. Lett. 1998, 55, 39–45. [Google Scholar] [CrossRef]
- Fountzoula, C.; Spanos, N.; Matralis, H.K.; Kordulis, C. Molybdenum-titanium oxide catalysts: The influence of the preparation conditions on their activity for the selective catalytic reduction of NO by NH3. Appl. Catal. B Environ. 2002, 35, 295–304. [Google Scholar] [CrossRef]
- Massoth, F.E. Characterization of Molybdena Catalysts. Adv. Catal. 1979, 27, 265–310. [Google Scholar]
- Okamoto, Y.; Imanaka, T. Interaction chemistry between molybdena and alumina: Infrared studies of surface hydroxyl groups and adsorbed carbon dioxide on aluminas modified with molybdate, sulfate, or fluorine anions. J. Phys. Chem. 1988, 92, 7102–7112. [Google Scholar] [CrossRef]
- Okamoto, Y.; Ishihara, S.Y.; Kawano, M.; Satoh, M.; Kubota, T. Preparation of Co-Mo/Al2O3 model sulfide catalysts for hydrodesulfurization and their application to the study of the effects of catalyst preparation. J. Catal. 2003, 217, 12–22. [Google Scholar] [CrossRef]
- Okamoto, Y.; Maezawa, A.; Imanaka, T. Active sites of molybdenum sulfide catalysts supported on Al2O3 and TiO2 for hydrodesulfurization and hydrogenation. J. Catal. 1989, 120, 29–45. [Google Scholar] [CrossRef]
- Mehdad, A.; Jentoft, R.E.; Jentoft, F.C. Passivation agents and conditions for Mo2C and W2C: Effect on catalytic activity for toluene hydrogenation. J. Catal. 2017, 347, 89–101. [Google Scholar] [CrossRef]
- Leary, K.J.; Michaels, J.N.; Stacy, A.M. Carbon and Oxygen Atom Mobility during Activation of Mo2C Catalysts. J. Catal. 1986, 101, 301–313. [Google Scholar] [CrossRef]
- Wu, W.; Wu, Z.; Liang, C.; Chen, X.; Ying, P.; Li, C. In situ FT-IR spectroscopic studies of CO adsorption on fresh Mo2C/Al2O3 catalyst. J. Phys. Chem. B 2003, 107, 7088–7094. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wu, Z.; Liang, C.; Ying, P.; Feng, Z.; Li, C.; Academy, C. An IR study on the surface passivation of Mo2C/Al2O3 catalyst with O2, H2O and CO2. Phys. Chem. Chem. Phys. 2004, 6, 5603–5608. [Google Scholar] [CrossRef]
- Shou, H.; Ferrari, D.; Barton, D.G.; Jones, C.W.; Davis, R.J. Influence of passivation on the reactivity of unpromoted and Rb-promoted Mo2C nanoparticles for CO hydrogenation. ACS Catal. 2012, 2, 1408–1416. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Starting Material (Molar Ratios) | Mo Phase Following Carburization | BET Surface Area (m2 g−1) | CO2 Conv. (%) | CO Selectivity (%) | CO Yield (%) |
|---|---|---|---|---|---|
| 300 °C, 3:1 H2:CO2, GHSV: 3600 L kg−1 h−1 | |||||
| K-Mo (1/4) | Metallic Mo | 1.0 | 0.0 | N.A. | N.A. |
| K-Mo@γ-Al2O3 (1/4/15) | Mo2C | 142 | 20.0 | 93.0 | 18.6 |
| Mo | Mo2C | 13.7 | 32.6 | 8.2 | 2.7 |
| Mo@γ-Al2O3 (4/15) | Mo2C | 154 | 18.7 | 73.1 | 13.7 |
| Bare γ-Al2O3 | Mo2C | 160 | N.A. | N.A. | N.A. |
| K-Mo2C@γ-Al2O3 | CO2 Conv. (%) | CO Selectivity (%) | CO Yield (%) | CO2 Conv. (%) | CO Selectivity (%) | CO Yield (%) |
|---|---|---|---|---|---|---|
| GHSV: 3600 L kg−1 h−1 | GHSV: 18,100 L kg−1 h−1 | |||||
| No calcination | 20.6 | 90.6 | 18.7 | 14.1 | 97.2 | 13.7 |
| 350 °C Calcination | 20.0 | 93.0 | 18.6 | 12.5 | 98.0 | 12.3 |
| 425 °C Calcination | 18.5 | 94.0 | 17.4 | 10.0 | 97.6 | 9.8 |
| Catalyst (K/Mo/Al2O3) Molar Ratio | Scherrer Crystallite Size (nm) | CO2 Conv. (%) | CO Selectivity (%) | CO Yield (%) |
|---|---|---|---|---|
| 300 °C, 3:1 H2: CO2, GHSV: 18,100 L kg−1 h−1 | ||||
| (1/4/30) | N.A. | 6.9 | 95.5 | 6.6 |
| (1/4/15) | 9 | 14.1 | 97.2 | 13.7 |
| (1/4/7.5) | 17 | 11.5 | 98.8 | 11.4 |
| (1/8/15) | 16 | 13.1 | 97.0 | 12.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morse, J.R.; Holder, C.F.; Baldwin, J.W.; Willauer, H.D. Optimization of Potassium Promoted Molybdenum Carbide Catalyst for the Low Temperature Reverse Water Gas Shift Reaction. Energies 2022, 15, 7109. https://doi.org/10.3390/en15197109
Morse JR, Holder CF, Baldwin JW, Willauer HD. Optimization of Potassium Promoted Molybdenum Carbide Catalyst for the Low Temperature Reverse Water Gas Shift Reaction. Energies. 2022; 15(19):7109. https://doi.org/10.3390/en15197109
Chicago/Turabian StyleMorse, James R., Cameron F. Holder, Jeffrey W. Baldwin, and Heather D. Willauer. 2022. "Optimization of Potassium Promoted Molybdenum Carbide Catalyst for the Low Temperature Reverse Water Gas Shift Reaction" Energies 15, no. 19: 7109. https://doi.org/10.3390/en15197109
APA StyleMorse, J. R., Holder, C. F., Baldwin, J. W., & Willauer, H. D. (2022). Optimization of Potassium Promoted Molybdenum Carbide Catalyst for the Low Temperature Reverse Water Gas Shift Reaction. Energies, 15(19), 7109. https://doi.org/10.3390/en15197109