Abstract
To date, the world has accumulated a large amount of long-lived radioactive materials that need to be disposed of or reprocessed. Such materials include nuclear legacy objects containing 226Ra, which is an important material for obtaining a wide range of isotopes for nuclear medicine via irradiation in reactors, cyclotrons, and electron accelerators. For the selective recovery of 226Ra from waste materials, crown-ether (CE) 18-crown-6 (18C6) or its derivatives can be used, which, however, have not been widely studied for these purposes. In our work, the key property of 18C6 and its derivatives, the phase distribution, was studied using tritium labeling. The possibility of introducing a tritium label into CEs molecules using thermal activation of tritium has been demonstrated; a high specific activity of the obtained compounds was achieved (from 18 to 108 TBq/mol). Methods for chromatographic purification of the studied CEs were developed. The distribution of 18C6 and its derivatives between various organic solvents and water was studied in detail for the first time. Subsequently, the obtained data will allow us to choose conditions for the selective recovery of 226Ra from aged sources.
1. Introduction
The amounts of fissile materials accumulated over decades necessitate the search for ways to dispose of or reprocess them [1]. One of the most important isotopes contained in recycled materials and suitable for reuse is 226Ra. Its irradiation in a reactor, cyclotron, or electron accelerator can result in the formation of sought-after and hardly accessible 225Ac and other isotopes for nuclear medicine [2,3,4]. According to the IAEA reports, the world has accumulated several kg of 226Ra in the form of waste, particularly needles and tubes for brachytherapy filled with radium salts [5]. Thus, the search for ways to recover 226Ra from such waste to be used further as a material for irradiation is an urgent task today.
The difficulty of 226Ra recovery lies in the fact that this element, like other alkaline earth metals, is not prone to complex formation. One of the few effective complexing agents for binding alkaline earth metal cations are crown ethers (CEs)—cyclic compounds consisting of oxoethyl fragments (-CH2-CH2-O-) [6]. The general pattern is that the closer the radius of the cation to the size of the cavity, the stronger the binding will be [7,8]. As a consequence, 18-crown-6 (1,4,7,10,13,16-hexaoxacyclooctadodecane, hereinafter 18C6) and its derivatives are the most suitable CEs for radium bonding [9,10]. A key aspect of developing methods for the selective recovery of radium by CEs is a detailed study of the properties of 18C6 and its derivatives. While a lot of work is devoted to 18C6 and dibenzo-18C6, other derivatives are little studied, although they have great potential for application. For example, 18C6 derivatives can be covalently bound to various nanoparticles to create materials for the selective sorption of different cations including Ra2+ [11,12,13].
A convenient tool for studying the behavior of organic compounds, especially hardly accessible ones, is the introduction of a tritium label into their molecules by the method of thermal activation of tritium [14,15] for their further detection by beta-radiation. This method makes it possible to obtain tritium-labeled compounds of various classes, including those containing oxoethyl groups, for example, pluronics [16,17]. At the same time, only one work on the labeling of CEs with tritium, published in 1984, is presented in the literature to date [18]. In this work tritium labeling of 15-crown-5, dibenzo-18-crown-6, and 22-crown-6-tetraon was carried out using a method characterized by high consumption of tritium. To achieve specific radioactivity of the studied CEs from 4 to 90 TBq/mol, 1.5 to 3.7 GBq of gaseous tritium was used in the experiments, while modern methods make it possible to carry out labeling with significantly lower consumption of tritium.
The aim of our work was to find the optimal conditions for the introduction of a tritium label into 18C6 and its derivatives, as well as to determine their phase distribution for further development of methods for radium recovery.
2. Materials and Methods
2.1. Studied CEs
In this work, 18C6 and its derivatives—benzo-18-crown-6, 2-hydroxymethyl-18-crown-6, and 4′-aminodibenzo-18-crown-6 (hereinafter B18C6, HM18C6, and ADB18C6)—were used. The structures of these compounds are shown in Figure 1.
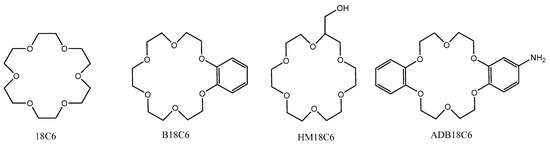
Figure 1.
Structures of the used CEs.
18C6, B18C6, and HM18C6 were obtained from Acros Organics (Belgium) with a purity of 97 to 99% according to the manufacturer data. 18C6 was colorless hygroscopic crystals melting at 39 °C, and B18C6 was white crystals melting at 43.1 °C. HM18C6 was a colorless liquid with a boiling point of 150 °C, well soluble in both water and organic solvents.
ADB18C6 was synthesized by us from dibenzo-18C6 (Acros Organics, 99%). For this purpose, a mixture of 0.01 mmol of dibenzo-18C6, 0.01 mmol of NaN3, and 10 g of polyphosphoric acid were heated at 90–100 °C for 3 h and then diluted with 100 mL of water. Then pH was adjusted to 8–9 by adding ammonia solution. The formed precipitate was filtered off. The mother liquor was extracted with three portions of 50 mL of CH2Cl2. The solvent was evaporated, and the residue was combined with the precipitate. The product was recrystallized from butanol resulting in 2.26 g (61%) of ADB18C6; m.p. 148–152 °C. The resulting precipitate was purified using a chromatography column with Al2O3; chloroform and a mixture of chloroform and isopropanol 100:1 were used as eluents. The yield of ADB18C6 (pale flesh-colored powder) was 1.1 g (29%), melting point of 158–163 °C (butanol). 1H NMR spectrum (using Bruker CXP-200, Bruker, Russia), DMSO-d6, ppm: 3.42 (2H, br. C, NH2), 4.02 (8H, m, OCH2); 4.10 (4H, m, OCH2); 4.16 (4H, m, OCH2); 6.21 (1H, dd, J = 8.4, J = 2.6, ArH); 6.29 (1H, d, J = 2.6 Hz, ArH); 6.88 (4H, m, ArH); 6.70 (1H, d, J = 8.4, ArH). By elemental analysis on a C,H,N-analyzer Carlo Erba Strumentazione (Italy) and an atomic-emission spectrometer with inductively coupled plasma IRIS Advantage ICP/OES-00-0311 (Thermo Scientific (Jarrell-Ash), Waltham, MA, USA) elemental composition was determined, %: C 64.14; H 6.67; N 3.69; formula C20H25NO6; and calculated from the gross formula, %: C 63.99; H 6.71; N 3.73.
2.2. Tritium Labeling of CEs and Liquid Scintillation Counting
Tritium labeling of CEs was carried out via thermal activation of tritium according to the following procedure. 1 mL of a CE solution with a concentration of 0.6 mg/mL was applied to the walls of a cylindrical reaction vessel (6.5 cm in diameter), and the solvent was removed by evaporation, for which a gas flow (CH4 or N2) was supplied to the vessel. The following solvents were used for CEs: chloroform for 18C6 and B18C6, ethanol and chloroform mixture (1:1) for ADB18C6, and ethanol for HM18C6. The vessel with the obtained thin CE film (target) was attached to a special device for working with gaseous tritium. After removing air to a residual pressure of less than 0.01 Pa, the system was filled with molecular tritium. During the process of vacuuming, the walls of the reaction vessel were cooled to 77 K with liquid nitrogen. Molecular tritium was atomized by heating a tungsten wire located along the central axis of the reaction vessel to 1900 K, passing an alternating electric current through it. The tritium pressure was 0.5 Pa to ensure an acceptable atomization rate and preserve the initial energy of tritium atoms since the free path of tritium atoms was higher than the distance between the atomizer and the walls of the vessel. The heating time of the tungsten wire was 10 s. Such conditions resulted in the maximal isotopic exchange of hydrogen for tritium in a thin surface layer of the target and the minimal destruction of the substance.
After the isotope exchange reaction, the residual gas was pumped out, the system was filled with air, and the vessel was disconnected from the vacuum unit. The labeled substance was dissolved in the same solvent from which the target was prepared, the solution was transferred into a round bottom flask. After holding for several hours, the solution was evaporated using a rotary evaporator and dissolved in a new portion of the solvent. This procedure was repeated several times; afterward, chromatographic analysis was carried out using thin layer chromatography (see below).
Content of tritium in solutions was determined by liquid scintillation (LS) spectrometry (LS-spectrometers GreenStar, Moscow, Russia, and RackBeta 1215, LKB Instruments, Turku, Finland) using LS cocktail UltimaGold (PerkinElmer Inc., Waltham, MA, USA).
2.3. Purification of Labeled CEs via Thin-Layer and Column Chromatography
To control the degree of purification of labeled CEs (hereinafter LCEs), thin layer chromatography (TLC) was used. The selection of conditions was carried out using stable CEs, the position of which on the plates was visualized in an iodine chamber. Merck (Darmstadt, Germany) plates Silica gel 60 F254 and aluminum oxide 60 F254 were used. The eluents were ethyl acetate, heptane, dioxane, ethanol, chloroform, water, benzene, formic acid, or acetone, as well as their various combinations. The optimal conditions found for the TLC of each CE were then used to determine the purity of the LCEs.
Liquid column chromatography was used to purify LCEs from radioactive impurities that could be formed during the reaction of atomic tritium with the main compound or from possible impurities remaining in the product after its synthesis. Chromatographic columns, 4 cm high and 0.4 cm in diameter, were filled with Al2O3 were used. Elution was carried out with 95% ethanol under external pressure. The eluent was collected in 100 μL fractions, their radioactivity was measured. After purification, the fractions with LCEs were combined and used for further work.
2.4. Phase Distribution Study
For each organic substance, the counting coefficient was preliminarily determined. 1 mL of the organic phase was mixed with water for 5 min, after which 500 µL were taken from the organic phase, tritium-labeled 18C6 was added to it, then a scintillator was added, and the counting rate was determined. The counting coefficient was determined as the ratio of the counting rate of labeled 18C6 in the organic phase mixed with 1 mL of the scintillator to its counting rate in 500 µL of distilled water mixed with 1 mL of the scintillator. Substances of different classes were used as organic phases: alkanes, haloalkanes, aromatic hydrocarbons, alcohols, ketones, and esters. Organic phases, their properties, and the counting coefficients determined by us are listed in Table 1.

Table 1.
Used organic phases and their properties.
To study the phase distribution of CEs, a small amount of carrier was added to a solution of purified LCEs in ethanol so that its content in the experiment was no more than a few μg. 990 μL of distilled water were placed in 2 mL Eppendorf tubes, then 10 μL of the resulting CE solution with the carrier was added. After that, 1 mL of the organic phase was added to the tube and mixed on a shaker at 1400 rpm. After mixing, 500 μL of each phase was taken, to which 1 mL of the scintillator was then added, and the tritium content was determined. The distribution coefficient D was calculated as the ratio of the activity of tritium in the organic phase to the activity of tritium in the aqueous phase, taking into account the counting coefficient.
To study the effect of pH on the phase distribution of CEs, aliquots of HCl or NH3 solutions were preliminarily added to water, controlling the pH of the solution with a pH meter.
3. Results
3.1. Tritium Labeling of Studied CEs
All studied CEs (18C6, B18C6, HM18C6, ADB18C6) were successfully labeled with tritium. Despite the fact that the conditions for the introduction of tritium into these substances were the same, the specific radioactivity of the LCEs varied. The specific radioactivity of 89 TBq/mol was obtained for labeled 18C6. The specific radioactivity of B18C6 was lower and amounted to 55 TBq/mol. This could have happened because, under the chosen reaction conditions, the isotopic exchange of hydrogen for tritium in aromatic fragments of the molecule occurs less efficiently than in aliphatic ones. At the same time, it was found that the introduction of an amino group into the benzene ring increases the efficiency of isotope exchange; therefore, a sufficiently high specific activity of 108 TBq/mol was obtained for the ADB18C6 molecule. Finally, HM18C6 had the lowest specific activity and amounted to 18 TBq/mol. The reason for the decrease in the specific activity of HM18C6 could have been that in the process of preparing the substance for the introduction of tritium, the former did not become a thin film on the walls of the vessel, since G18C6 is a liquid at room temperature. During the evaporation of the solvent, HM18C6 accumulated on the glass surface in the form of microdroplets, and this reduced the target surface accessible to tritium atoms.
3.2. TLC and Column Chromatography of Studied CEs
The TLC of the studied CEs before the introduction of the tritium label was carried out on Al2O3 and SiO2 plates. It was established that 95% ethanol is the optimal eluent on Al2O3 plates; in this case, Rf from 0.8 to 0.9 was observed for all CEs. For LCEs, one peak in radioactivity was observed, which coincided with the position of the non-radioactive standards on the plates. When using SiO2 plates, it was not possible to select TLC conditions with a compact position of the non-radioactive standard on the plate; therefore, this option could not be used for the analysis of LCEs. Thus, the optimal conditions for TLC of LCEs were the use of Al2O3 plates and ethanol as eluent. TLC in 95% ethanol on Al2O3 plates showed that the purity of labeled 18C6, B18C6, HM18C6, and ADB18C6 was 96, 91, 97, and 99%, respectively.
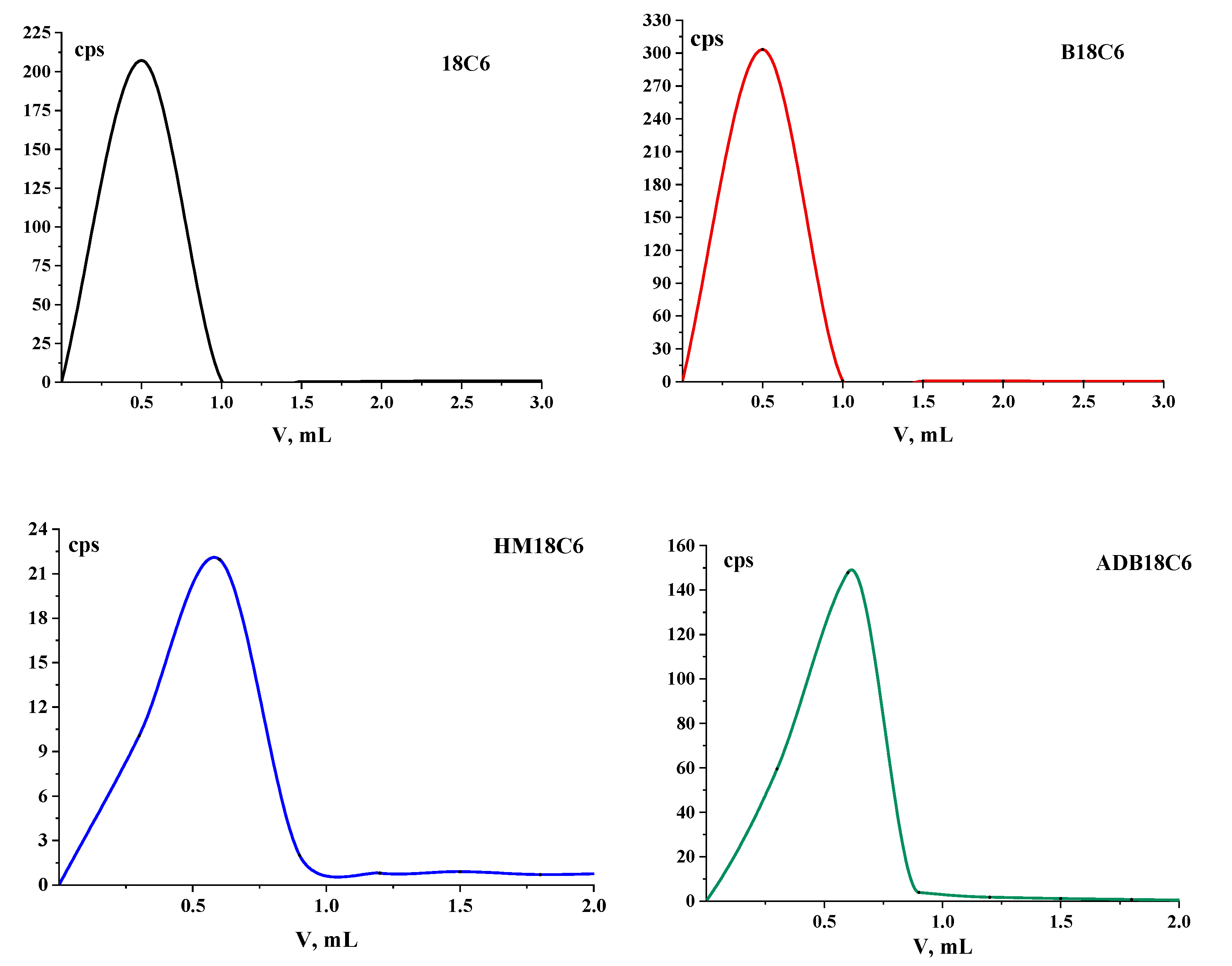
The elution profiles of the studied LCEs during further column chromatography on columns with Al2O3 are shown in Figure 2. It can be seen that the main activity left the column in the first fractions, after which, in each subsequent sample the activity slightly exceeded the background. No other peaks were found in the chromatograms, and, most likely, impurities remained on the column, since they remained at the start in TLC.
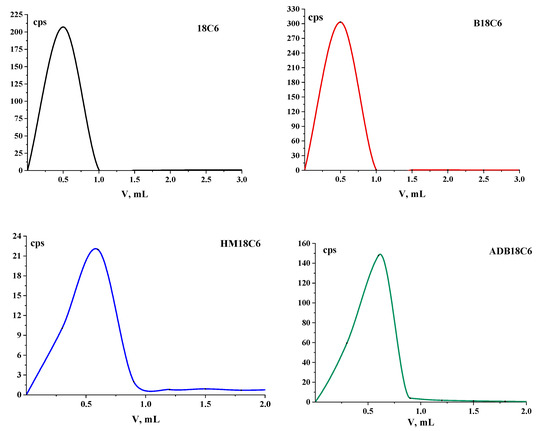
Figure 2.
Elution curves of the studied LCEs on columns with Al2O3.
3.3. Phase Distribution of LCEs
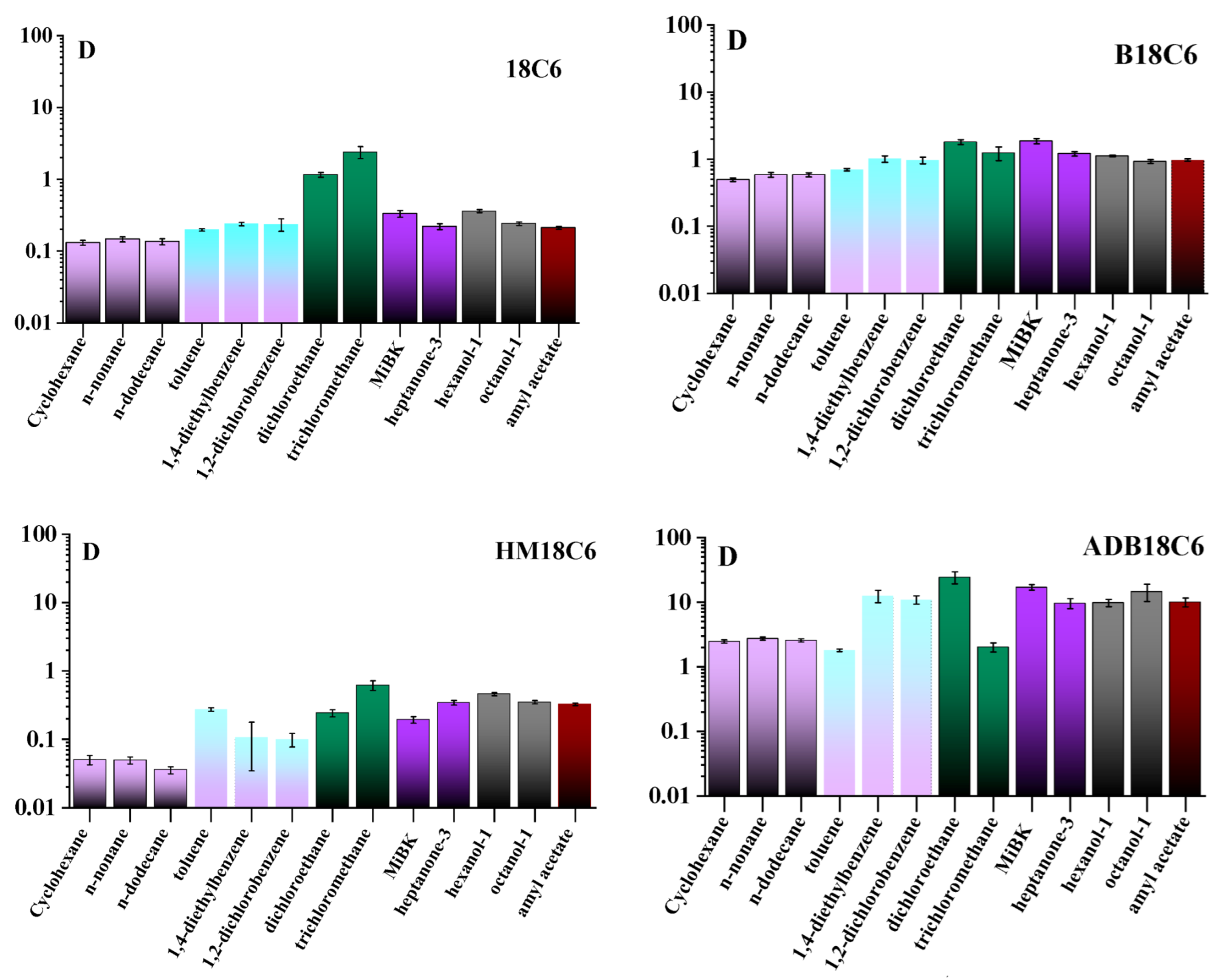
We have determined that changes in pH (from 5 to 9) and ionic strength (addition of up to 1 M NaClO4) of the aqueous phase did not significantly affect the phase distribution of the studied LCEs; therefore, the experiments were carried out in bidistilled water. The kinetics of LCEs distribution between the phases was studied from 5 min to 24 h; it was found that for all studied organic substances, with the exception of alkanes, equilibrium was reached in 1 h, while in the case of alkanes, D slowly increased up to 24 h. Figure 3 shows the values of D in the studied systems after 1 h of phase contact. It was found that D can vary significantly in different systems, as well as for different CEs in the same system. Thus, hydrophilic 18C6 does not tend to pass into the organic phase in all the studied systems (D was less than 0.4), except for dichloroethane and chloroform, for which the distribution of 18C6 between them and water is approximately the same (D was slightly more than 1). When a benzene ring appears in the 18C6 molecule, hydrophobicity increases, and, in the case of B18C6, D increases and ranges from 0.5 for alkanes to 2 for dichloroethane and MiBK. The addition of a hydroxo-group to 18C6 (HM18C6), on the contrary, increases hydrophilicity, and the D of the studied solvents does not exceed 0.7. Finally, for ADB18C6, D increases strongly in comparison with other studied LCEs due to the appearance of two benzene rings and an amino group in the structure. D for the studied solvents varies from 2 in the case of toluene and chloroform to 30 for dichloroethane. Among organic homologs, as a rule, differences in D of studied CEs are low, however, there are exceptions for ADB18C6. Thus, D for the toluene-water system turned out to be an order of magnitude lower than for diethylbenzene, as well as D for the chloroform-water system in comparison with dichloroethane-water.
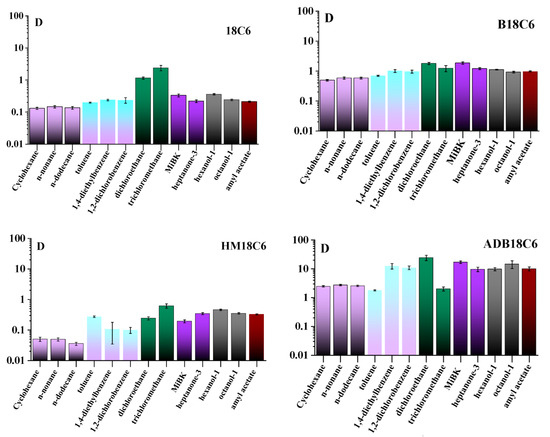
Figure 3.
Distribution coefficients (D) of the studied LCEs between water and organic phases for 1 h of contact.
4. Discussion
4.1. Labeling of CEs and Purification
As we wrote in the Introduction, the only attempt to apply the method of thermal activation of tritium to obtain LCEs was made in the work [18], where labeling of 15-crown-5, dibenzo-18-crown-6, and 22-crown-6-tetraon was studied. In this work, 2 mL of CEs solutions (from 5 to 18 mg of CEs) in organic diluents were preliminarily applied to filter paper 220 × 100 mm that was then placed into the reaction vessel. To atomize tritium, a tungsten wire was heated to 2200 K for 30 s. Purification of LCEs was carried out by gas-liquid chromatography on columns 2 m long, where the columns were heated to 200 °C for 14 min each. The eluent was methanol, which, after purification, was removed on a rotary evaporator. In our work, substances were applied directly to the walls of the reaction vessel, and we also used milder reaction conditions: we reduced the atomizer temperature from 2200 to 1900 K, reduced gas pressure not less than twofold, and lowered reaction time threefold. We also made the purification of LCEs to be much faster and simpler. As a result, by reducing the consumption of tritium (from twofold to fourfold) and increasing the yield of the labeled product, comparable levels of specific radioactivity (from 18 to 108 TBq/mol in our work and from 4 to 90 TBq/mol in [18]) were obtained, which are quite sufficient for the described experiments. Thus, for the first time, tritium-labeled 18C6, B18C6, HM18C6, and ADB18C6 were obtained and high specific activity was achieved, and a method for monitoring the purity of LCEs and a method for their rapid purification from impurities were developed.
4.2. Phase Distribution of LCEs
We determined that the D of the studied LCEs does not correlate with the dielectric constant and dipole moment of organic phases (see Supplementary Materials). However, the change in these parameters among substances of the same class allows us to make assumptions about the effect of various functional groups on D.
The hydrophilicity of the studied CEs increases in the series ADB18C6, B18C6, 18C6, and HM18C6. Considering the distribution between water and saturated hydrocarbons, which are nonpolar, hydrophobic, and have the simplest structures among the studied substances, it can be seen that D depends only on the hydrophilicity of CEs (D decreases from ADB18C6 to B18C6). The length of the hydrocarbon chain does not affect D, except for coefficients for HM18C6 and dodecane, which are lower than for cyclohexane and nonane. This could happen because of the error of the LS method for low D values. The long kinetics of the transition of CEs to hydrocarbons can be explained by the steric factor. Aromatic hydrocarbons in our study are represented by toluene, diethylbenzene, and dichlorobenzene, which have different polarities. For 18C6 and B18C6 molecules, the polarity of the solvent does not have a significant effect, and the D values for aromatic and saturated hydrocarbons are approximately the same. In contrast, the polar HM18C6 molecule has an affinity for polar toluene, which can be seen from the increase in D. The reverse situation is observed for nonpolar ADB18C6, for which D remains the same in toluene as in hydrocarbons. D of HM18C6 and ADB18C6 in nonpolar diethylbenzene and polar dichlorobenzene are the same, which is unusual and requires further study. D of all studied CEs in haloalkanes are higher than in other solvents; however, it is impossible to correlate the polarity of the CE molecule, the solvent, and the obtained D. Probably, the D value, in this case, is affected by the size of solvent molecules, being smaller than ones of other studied solvents, which allows CEs to pass into the organic phase without steric hindrance. In the case of ketones, alcohols, and esters, D for all studied CEs are higher than in hydrocarbons, but the correlation between D and solvent properties is also difficult to draw. An interesting fact is that the position of the keto group and the branching of the chain in the ketone can either increase D or decrease it. To conclude, further detailed study of the interactions of CEs molecules with organic solvents of various classes is an interesting and complicated task.
As we wrote, 18C6 and its derivatives are promising complexing agents for 226Ra for its recovery and reprocessing. At least two methods can be used for these purposes: extraction of radium complexes with CEs into the organic phase and selective sorption of radium by nanomaterials with covalently bound CEs. We determined that, for the transition of CEs into the organic phase of a two-phase system, the presence of hydrophobic fragments in the composition of CE’s molecule is necessary. In particular, the appearance of a benzene ring in B18C6 increases D for most of the studied systems by several times compared to 18C6, as well as the appearance of a second benzene ring in ADB18C6 compared to B18C6. It should be taken into account that benzene rings increase the rigidity of the CEs structure, which may affect the binding of radium cations to them. It was shown that the studied CEs have an affinity for haloalkanes. Thus, to develop methods for radium extraction with 18C6 derivatives, it is recommended to use hydrophobized derivatives, and choose haloalkanes as the organic phase. In the case of covalent binding of the studied CEs (with the exception of ADB18C6) with nanomaterials of various nature and further suspension, the surface of the nanomaterials is likely to be sufficiently hydrophilic and, as a result, the suspension will be stable, which usually has a positive effect on the recovery process.
5. Conclusions
We developed an efficient method for introducing a tritium label into 18C6 and its derivatives, which made it possible to achieve high values of specific activity under mild labeling conditions. The conditions for monitoring the purity of LCEs by thin-layer chromatography were determined, and a method for their purification by column chromatography was developed. The distribution of obtained LCEs between water and organic substances of various classes was studied. It was established that the distribution coefficients depend on the chosen organic solvent in a non-trivial way, and further study of the mechanism of interaction of CEs with solvent molecules is necessary. As a result, the data obtained are the foundation for the development of methods for 226Ra reprocessing, which is an urgent and important task today.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/en15196867/s1, Figure S1. Distribution coefficients of the studied CEs depending on the dielectric constants of the organic phase molecules. Figure S2. Distribution coefficients of the studied CEs depending on the dipole moments of the organic phase molecules.
Author Contributions
Conceptualization, A.G.K. and G.A.B.; methodology, A.G.K. and G.A.B.; validation, G.A.B.; investigation, T.Y.E., J.S.B. and G.A.B.; resources, A.G.K., S.E.V. and G.A.B.; writing—original draft preparation, A.G.K., T.Y.E. and G.A.B.; writing—review and editing, S.E.V. and G.A.B.; visualization, A.G.K. and J.S.B.; supervision, A.G.K. and S.E.V.; project administration, A.G.K.; funding acquisition, A.G.K. All authors have read and agreed to the published version of the manuscript.
Funding
All experiments with 18C6 and B18C6 were funded by RFBR and Moscow city Government according to project No. 21-33-70030. All experiments with HM18C6 and ADB18C6 were supported by the Russian Science Foundation grant No. 21-73-00279, https://rscf.ru/project/21-73-00279, (accessed on 15 September 2022).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Deng, D.; Zhang, L.; Dong, M.; Samuel, R.E.; Ofori-Boadu, A.; Lamssali, M. Radioactive waste: A review. Water Environ. Res. 2020, 92, 1818–1825. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, A.G.; Ekatova, T.Y.; Babenya, J.S. Photonuclear production of medical radiometals: A review of experimental studies. J. Radioanal. Nucl. Chem. 2021, 328, 493–505. [Google Scholar] [CrossRef]
- Apostolidis, C.; Molinet, R.; McGinley, J.; Abbas, K.; Möllenbeck, J.; Morgenstern, A. Cyclotron production of Ac-225 for targeted alpha therapy. Appl. Radiat. Isot. 2005, 62, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, K.; Suzuki, H.; Fukada, M.; Ito, T.; Ichinose, J.; Honda, Y.; Minegishi, K.; Higashi, T.; Zhang, M.R. Cyclotron production of 225Ac from an electroplated 226Ra target. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 279–289. [Google Scholar] [CrossRef] [PubMed]
- IAEA-Tecdoc-886. 1996. Available online: https://www.iaea.org/publications/5535/conditioning-and-interim-storage-of-spent-radium-sources (accessed on 15 September 2022).
- Yoshio, M.; Noguchi, H. Crown ethers for chemical analysis: A review. Anal. Lett. 1982, 15, 1197–1276. [Google Scholar] [CrossRef]
- Pedersen, C.J. Cyclic polyethers and their complexes. J. Am. Chem. Soc. 1967, 2, 7017–7036. [Google Scholar] [CrossRef]
- Abramov, A.A. Extraction of cations by crown-ethers. Moscow Univ. Bull. Ser. 2 Chem. 2000, 41, 3–15. (In Russian) [Google Scholar]
- Strzelbicki, J.; Strzelbicka, B.; Schugerl, K.; Bartsch, R. Crown ethers in separation of alkali metal cations. Process Metall. 1992, 7B, 1523–1528. [Google Scholar]
- Shiokawa, Y.; Kido, T.; Suzuki, S. Radiopolarographic determination of the stability constant of 18-crown-6 complex for Ra2+. J. Radioanal. Nucl. Chem. Lett. 1985, 96, 249–255. [Google Scholar] [CrossRef]
- Basiuk, V.A.; Henao-holguín, L.V.; Meza-laguna, V.; Basiuk, E.V. Solvent-free derivatization of oxidized single-walled carbon nanotubes and nanodiamond with aminobenzo-crown ethers. Fuller. Nanotub. Carbon Nanostruct. 2016, 24, 653–661. [Google Scholar] [CrossRef]
- Kawamura, M.; Sato, K. Magnetic nanoparticle-supported crown ethers. Chem. Commun. 2007, 3404–3405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Xiao, C.; Liu, Y.; Hu, Q.; Chen, C.; Kuraoka, E. Preparation of macroporous silica-based crown ether materials for strontium separation. J. Porous Mater. 2010, 17, 153–161. [Google Scholar] [CrossRef]
- Shishkov, A.V.; Filatov, E.S.; Unukovich, M.S.; Gol’danskii, V.I. Preparation of tritium-labeled biologically active compounds. Dokl. Akad. Nauk. SSSR 1976, 228, 1237. (In Russian) [Google Scholar] [PubMed]
- Shishkov, A.V.; Neiman, L.; Smolyakov, V. The preparation of labelled organic compounds by treatment with atomic tritium. Russ. Chem. Rev. 1984, 53, 656. (In Russian) [Google Scholar] [CrossRef]
- Badun, G.A.; Chernysheva, M.G.; Ksenofontov, A.L. Increase in the specific radioactivity of tritium-labeled compounds obtained by tritium thermal activation method. Radiochim. Acta 2012, 100, 401–408. [Google Scholar] [CrossRef]
- Chernysheva, M.G.; Shnitko, A.V.; Soboleva, O.A.; Badun, G.A. Competitive adsorption of lysozyme and non-ionic surfactants (Brij-35 and pluronic P123) from a mixed solution at water-air and water-xylene interfaces. Colloid Polym. Sci. 2018, 296, 223–232. [Google Scholar] [CrossRef]
- Neiman, L.A.; Antropova, L.P.; Konur, I.P.; Chepelev, V.M.; Nazarov, E.I. Tritium labeled macrocyclic polyethers. Sov. J. Bioorg. Chem. 1984, 10, 121–123. (In Russian) [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).