
A Theoretical Evaluation of the Efficiencies of Metal-Free 1,3,4-Oxadiazole Dye-Sensitized Solar Cells: Insights from Electron–Hole Separation Distance Analysis


Abstract
1. Introduction
2. Methodology
Computational Details
3. Results and Discussion
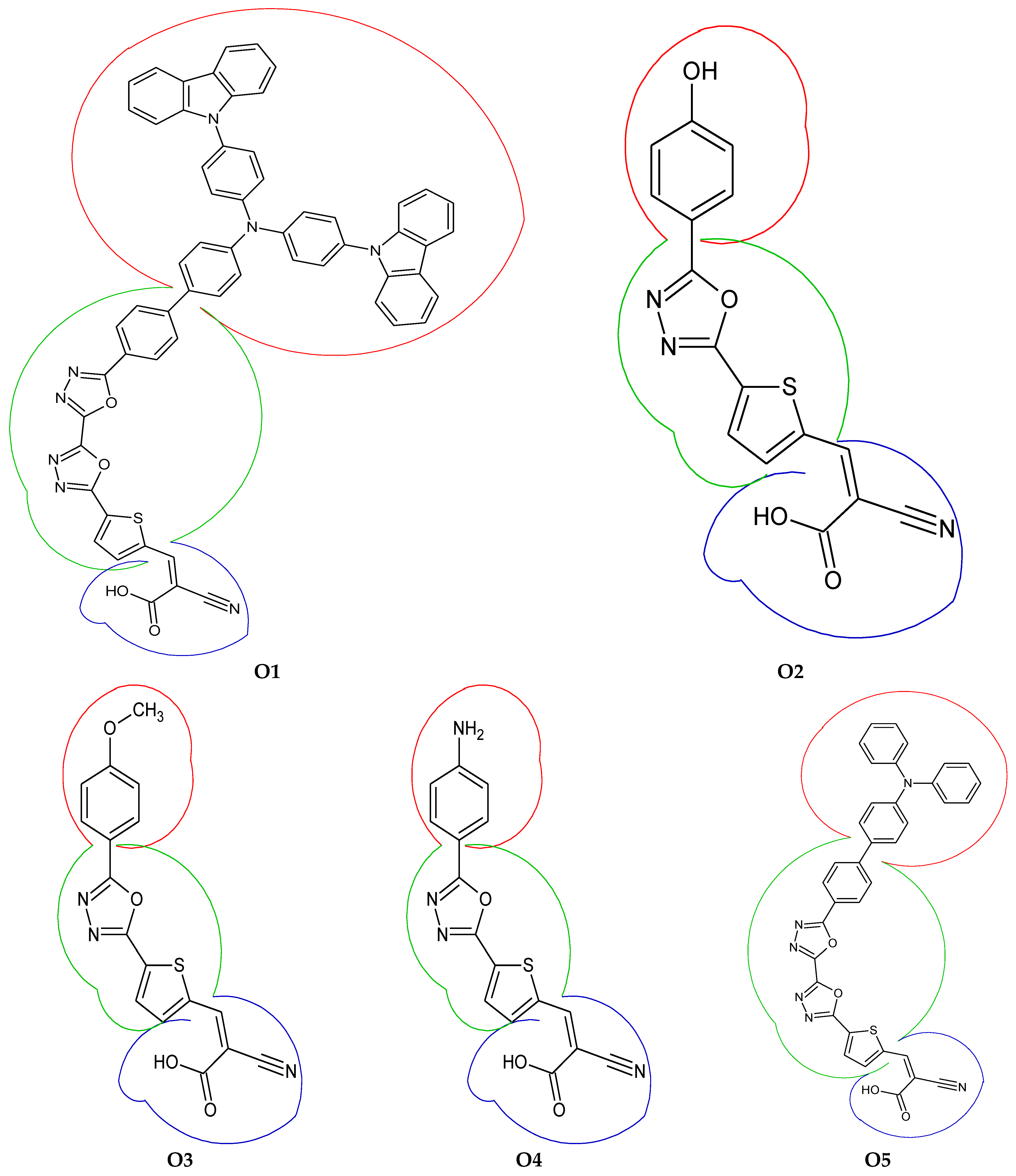
3.1. Optimized Geometry
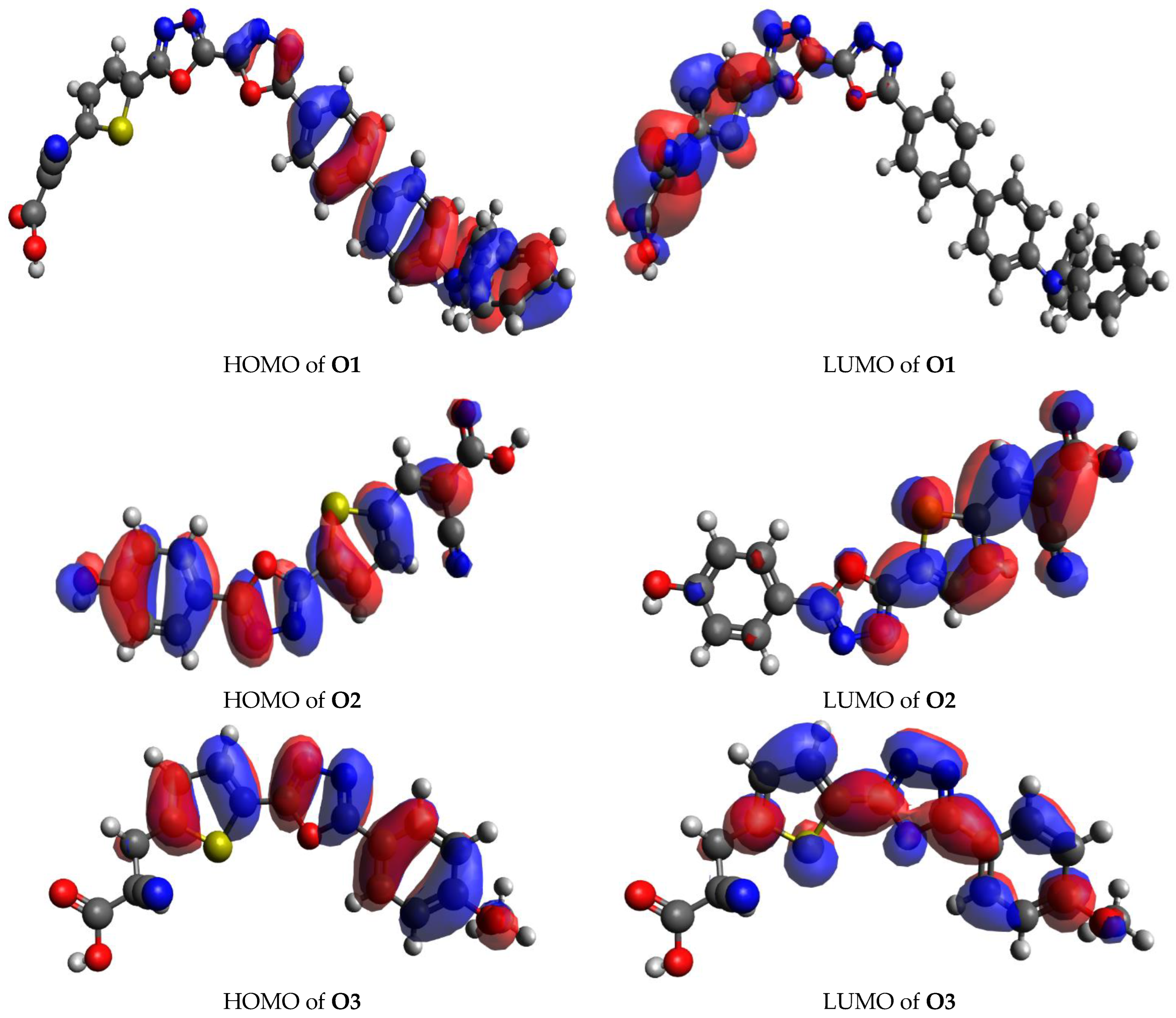
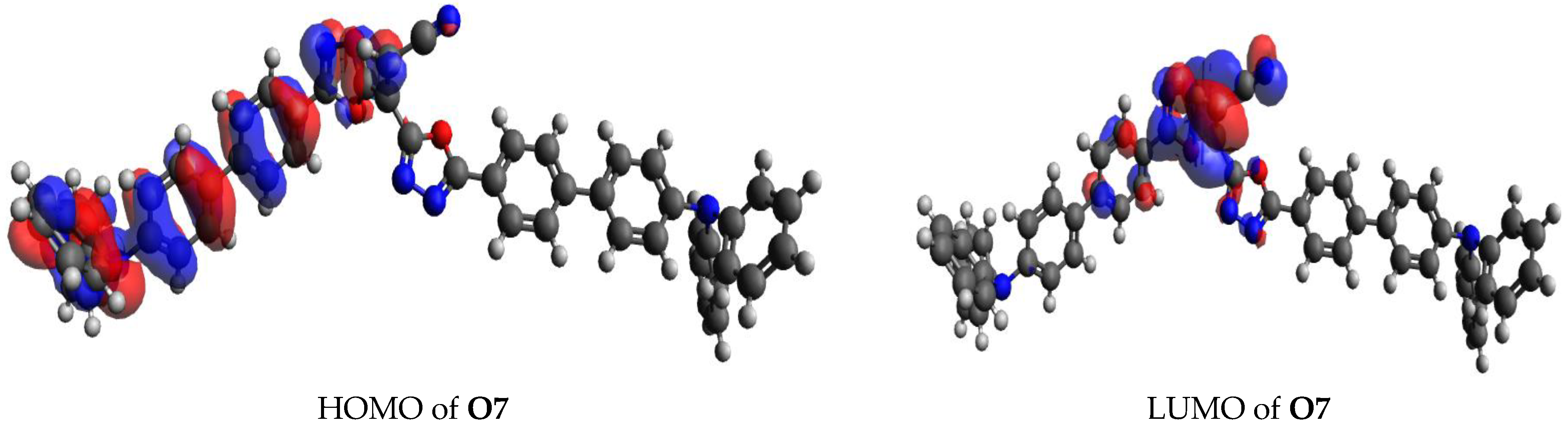
3.2. Coulomb Attractive Energy and Electron Excitation Analyses
Molecular Electrostatic Potential (MEP)
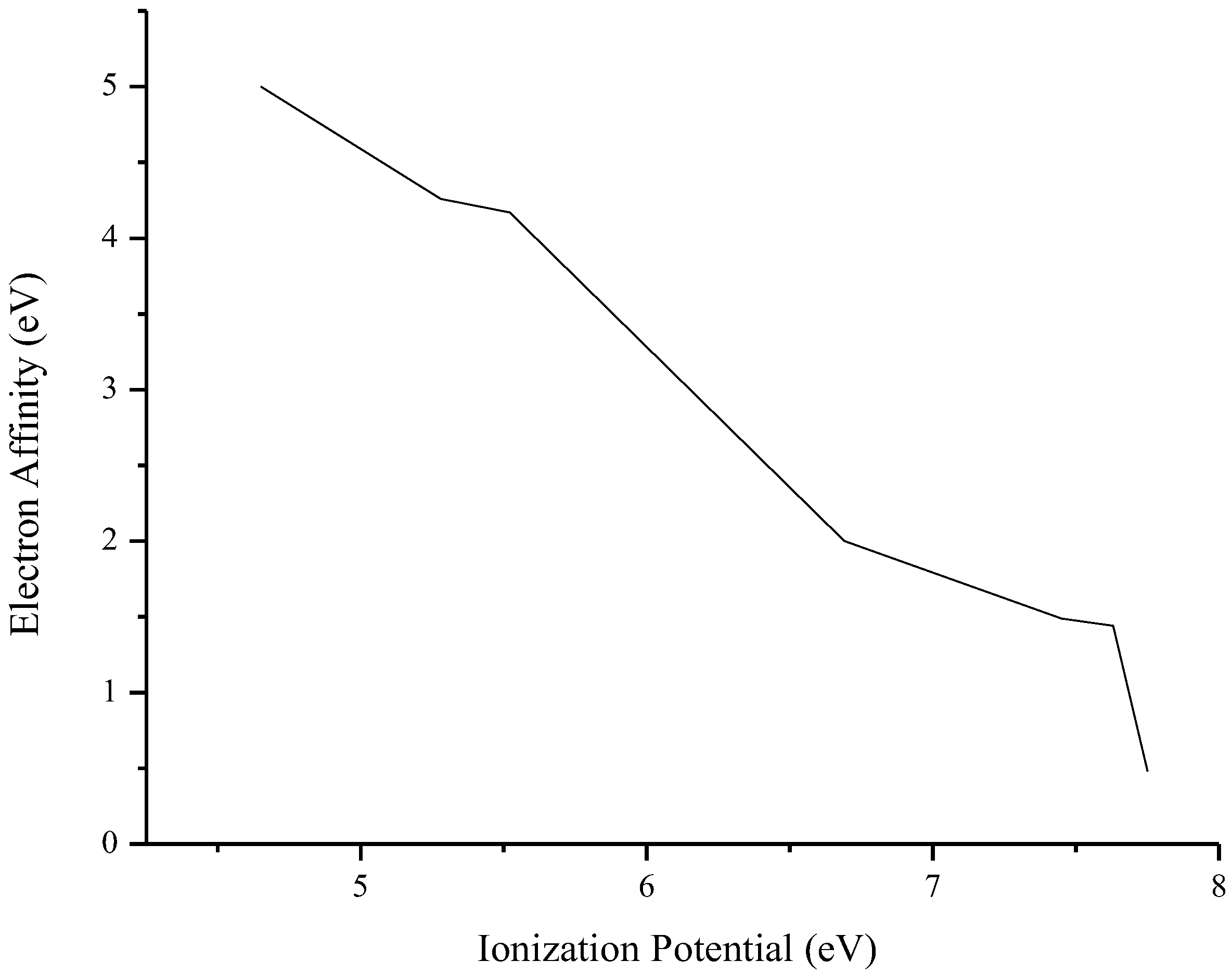
3.3. Photovoltaic Parameters
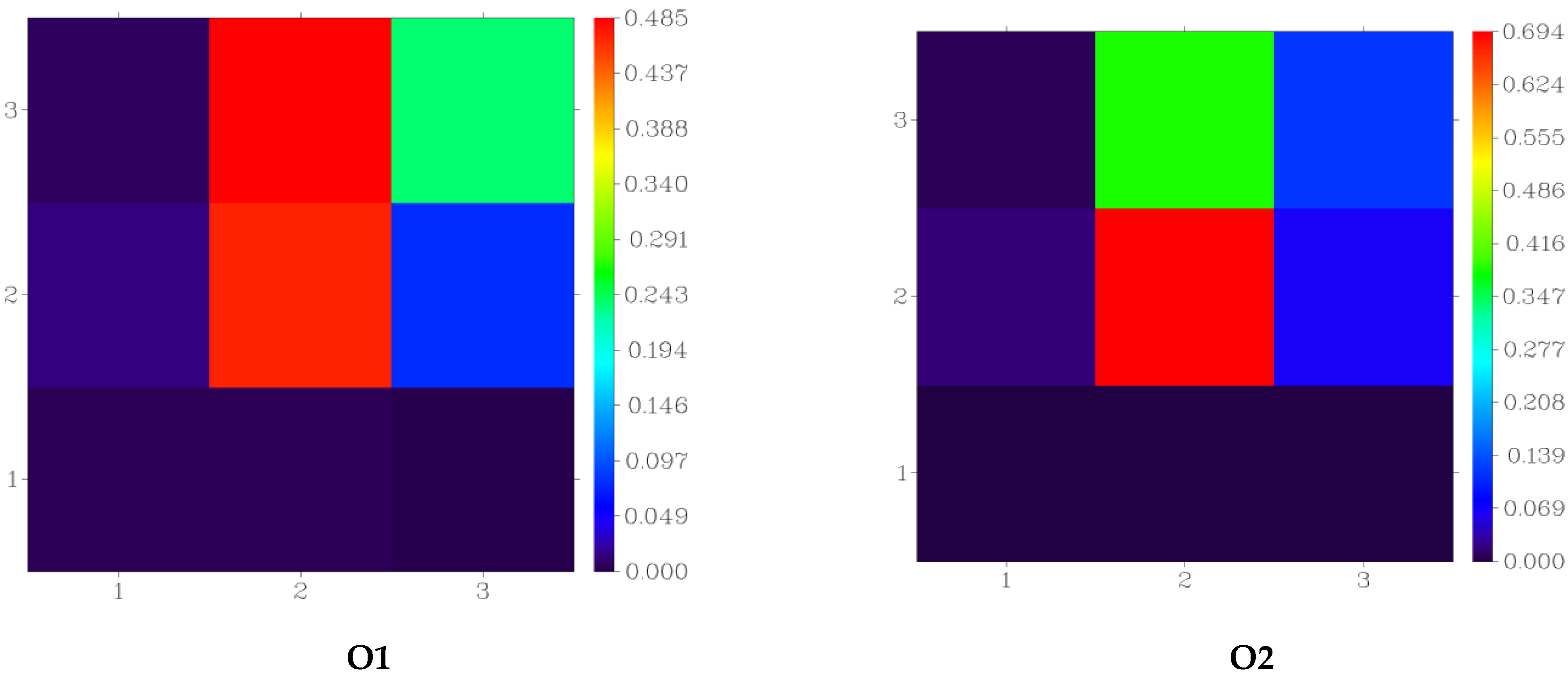
3.4. Fragment-Based Electron–Hole Analysis
3.5. Transition Density Matrix (TDM)
3.6. Intra-Fragment-Charge Transfer (IFCT) and Dihedral Angles
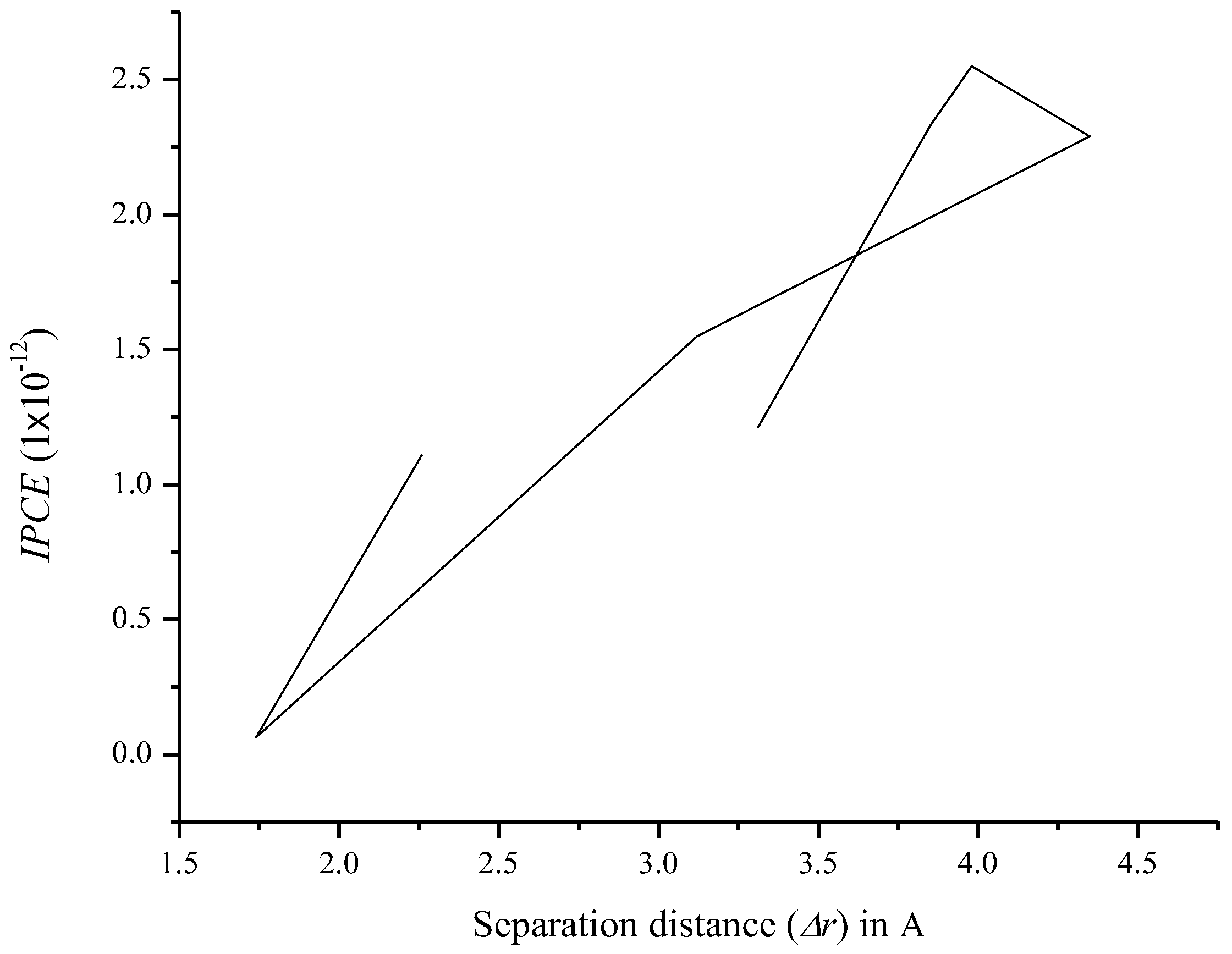
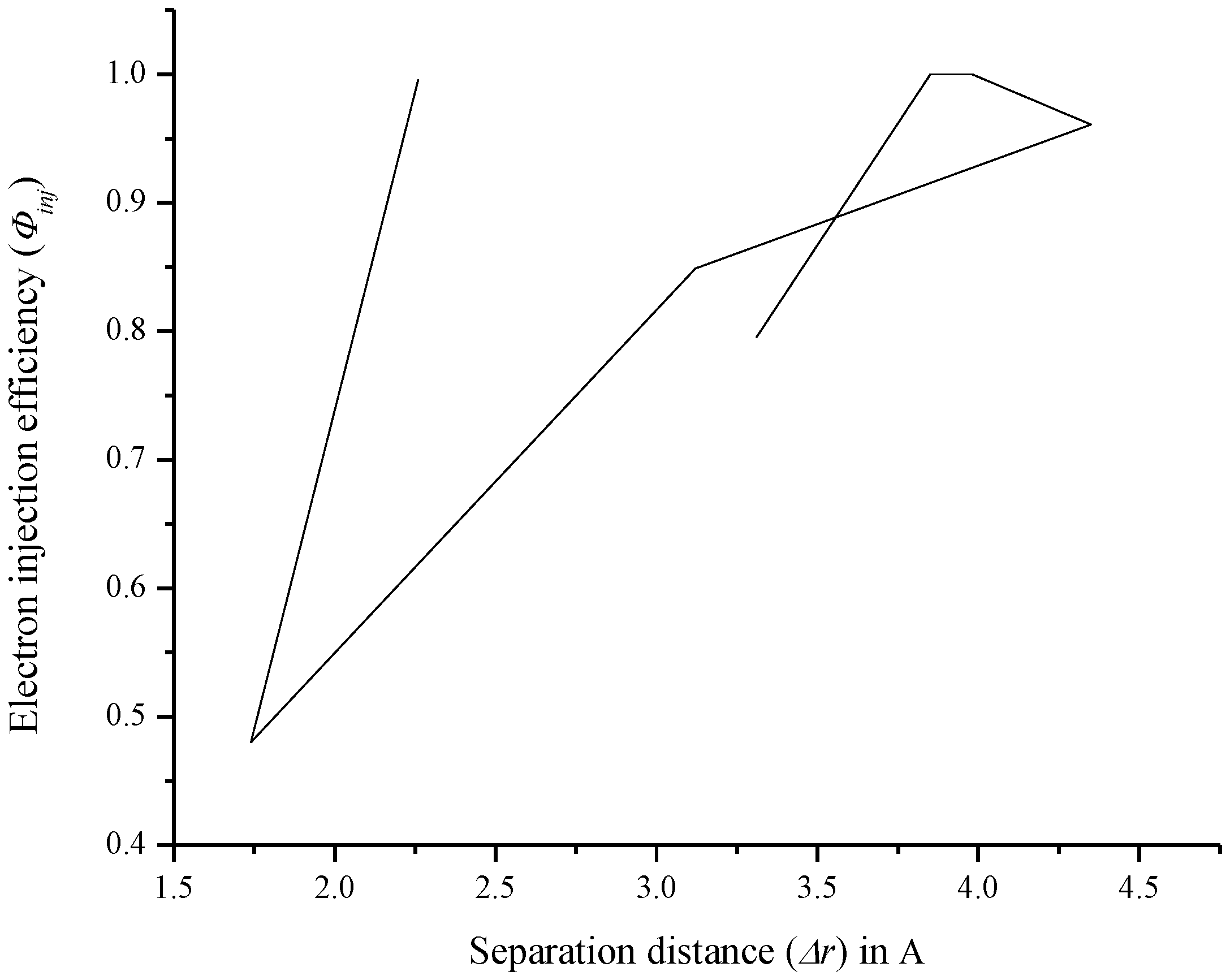
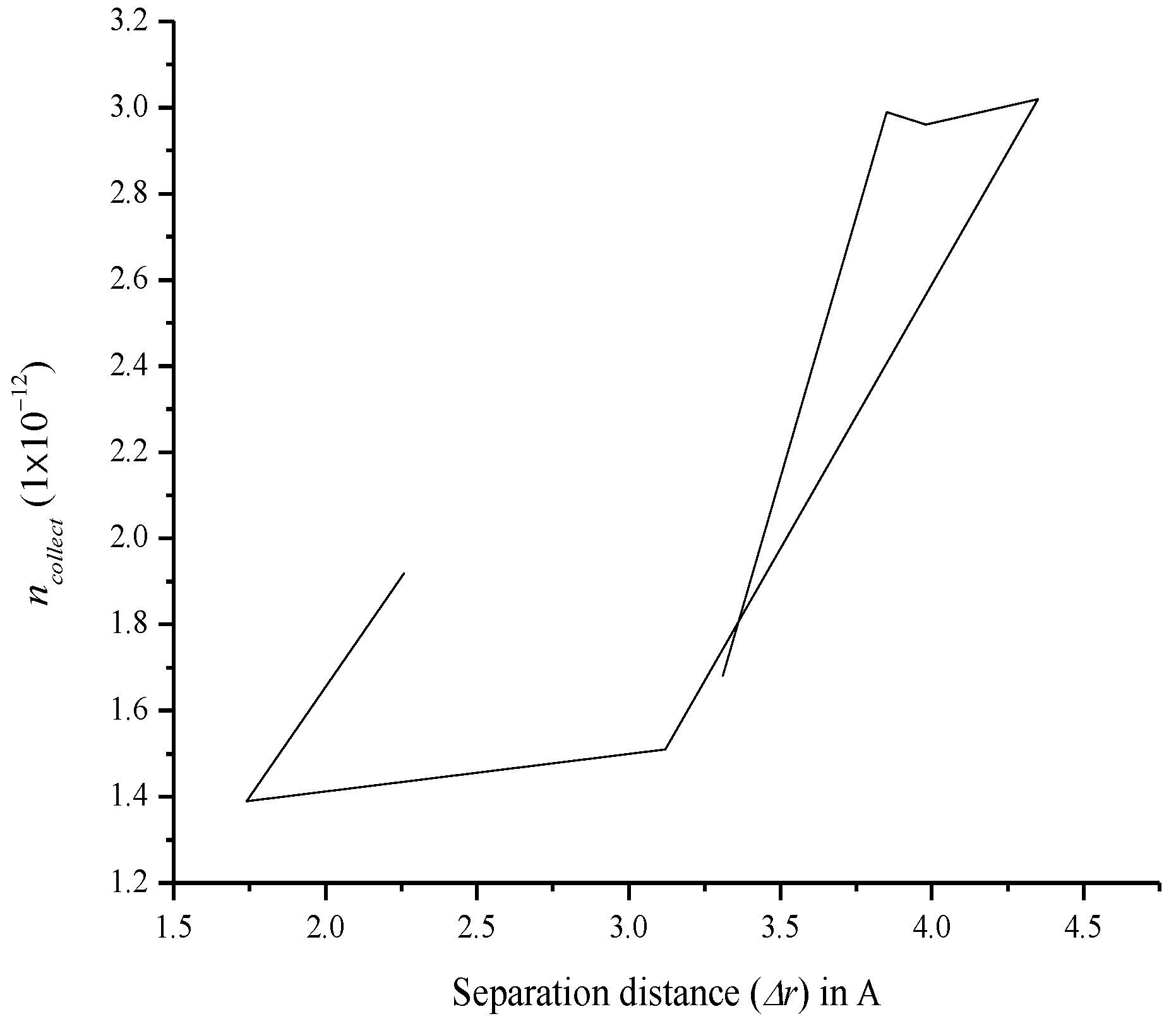
3.7. Relationship between Charge Separation Distance and Parameters Such as IPCE, Charge Regeneration, LHE, Electron Injection, and Charge Collection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abas, N.; Kalair, A.; Khan, N. Review of fossil fuels and future energy technologies. Futures 2015, 69, 31–49. [Google Scholar] [CrossRef]
- Robertson, N. Optimizing dyes for dye-sensitized solar cells. Angew. Chem. Int. Ed. 2006, 45, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- Tibebu, S.; Hailu, A. Design, Construction, and Evaluation of the Performance of Dual-Axis Sun Trucker Parabolic Solar Cooker and Comparison of Cooker. J. Renew. Energy 2021, 2021, 8944722. [Google Scholar] [CrossRef]
- Owusu, P.A.; Sarkodie, S.A. A review of renewable energy sources, sustainability issues and climate change mitigation. Cog. Eng. 2016, 3, 1167990. [Google Scholar] [CrossRef]
- Usman, Z.; Tah, J.; Abanda, H.; Nche, C. A critical appraisal of pv-systems’ performance. Buildings 2020, 10, 192. [Google Scholar] [CrossRef]
- Luceño-Sánchez, J.A.; Díez-Pascual, A.M.; Peña Capilla, R. Materials for photovoltaics: State of art and recent developments. Int. J. Mol. Sci. 2019, 20, 976. [Google Scholar] [CrossRef]
- Lee, J.K.; Yang, M. Progress in light harvesting and charge injection of dye-sensitized solar cells. Mat. Sci. Eng. B 2011, 176, 1142–1160. [Google Scholar] [CrossRef]
- Perera, I.R.; Hettiarachchi, C.V.; Ranatunga, R.J.K.U. Metal–organic frameworks in dye-sensitized solar cells. In Advances in Solar Energy Research; Springer: Singapore, 2019; pp. 175–219. [Google Scholar]
- Ahmad, S.; Liu, J.; Ji, W.; Sun, L. Metal–organic framework thin film-based dye sensitized solar cells with enhanced photocurrent. Materials 2018, 11, 1868. [Google Scholar] [CrossRef]
- Yildirim, O.; Bonomo, M.; Barbero, N.; Atzori, C.; Civalleri, B.; Bonino, F.; Viscardi, G.; Barolo, C. Application of metal-organic frameworks and covalent organic frameworks as (photo) active material in hybrid photovoltaic technologies. Energies 2020, 13, 5602. [Google Scholar] [CrossRef]
- He, Y.; Zhang, Z.; Wang, W.; Fu, L.J. Metal organic frameworks derived high performance photoanodes for DSSCs. J. Alloys Compd. 2020, 825, 154089. [Google Scholar] [CrossRef]
- Spoerke, E.D.; Small, L.J.; Foster, M.E.; Wheeler, J.; Ullman, A.M.; Stavila, V.; Rodriguez, M.; Allendorf, M.D. MOF-Sensitized Solar Cells Enabled by a Pillared Porphyrin Framework. J. Phys. Chem. C 2017, 121, 4816–4824. [Google Scholar] [CrossRef]
- Dou, J.; Li, Y.; Xie, F.; Ding, X.; Wei, M. Metal-Organic Framework Derived Hierarchical Porous Anatase TiO2 as a Photoanode for Dye-Sensitized Solar Cells. Cryst. Growth Des. 2016, 16, 121–125. [Google Scholar] [CrossRef]
- Khajavian, R.; Mirzaei, M.; Alizadeh, H. Current status and future prospects of metal–organic frameworks at the interface of dye-sensitized solar cells. Dalt. Trans. 2020, 49, 13936–13947. [Google Scholar] [CrossRef]
- Lee, C.P.; Li, C.T.; Ho, K.C. Use of organic materials in dye-sensitized solar cells. Mat. Tod. 2017, 20, 267–283. [Google Scholar] [CrossRef]
- Chauhan, R.; Kushwaha, R.; Bahadur, L. Study of Light Harvesting Properties of Different Classes of Metal-Free Organic Dyes in TiO2 Based Dye-Sensitized Solar Cells. J. Energy 2014, 2014, 517574. [Google Scholar] [CrossRef]
- Cao, D.; Peng, J.; Hong, Y.; Fang, X.; Wang, L.; Meier, H. Enhanced performance of the dye-sensitized solar cells with phenothiazine-based dyes containing double D−A branches. Org. Lett. 2011, 13, 1610–1613. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, S.; Anandan, S. Cyanovinyl substituted benzimidazole based (D–π–A) organic dyes for fabrication of dye sensitized solar cells. Dyes Pigment. 2014, 105, 223–231. [Google Scholar] [CrossRef]
- Castillo-Vallés, M.; Andrés-Castán, J.M.; Garín, J.; Orduna, J.; Villacampa, B.; Franco, S.; Blesa, M.J. Dye-sensitized-solar-cells based on calix[4]arene scaffolds. RSC Adv. 2015, 5, 90667–90670. [Google Scholar] [CrossRef][Green Version]
- Tan, L.L.; Liu, J.M.; Li, S.Y.; Xiao, L.M.; Kuang, D.B.; Su, C.Y. Dye-Sensitized Solar Cells with Improved Performance using Cone-Calix[4]Arene Based Dyes. ChemSusChem 2014, 8, 280–287. [Google Scholar] [CrossRef]
- Tan, L.L.; Huang, J.F.; Shen, Y.; Xiao, L.M.; Liu, J.M.; Kuang, D.B.; Su, C.Y. Highly efficient and stable organic sensitizers with duplex starburst triphenylamine and carbazole donors for liquid and quasi-solid-state dye-sensitized solar cells. J. Mat. Chem. A 2014, 2, 8988–8994. [Google Scholar] [CrossRef]
- Kungwan, N.; Khongpracha, P.; Namuangruk, S.; Meeprasert, J.; Chitpakdee, C.; Jungsuttiwong, S.; Promarak, V. Theoretical study of linker-type effect in carbazole–carbazole-based dyes on performances of dye-sensitized solar cells. Theor. Chem. Acc. 2014, 133, 1523. [Google Scholar] [CrossRef]
- Abdellah, I.M.; El-Shafei, A. Influence of carbonyl group on photocurrent density of novel fluorene based D-π-A photosensitizers: Synthesis, photophysical and photovoltaic studies. J. Photochem. Photobiol. A Chem. 2020, 387, 112133. [Google Scholar] [CrossRef]
- Gilbert, M.; Albinsson, B. Photoinduced charge and energy transfer in molecular wires. Chem. Soc. Rev. 2015, 44, 845–862. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Hua, J.; Wu, W.; Li, J.; Jin, Z.; Long, Y.; Tian, H. New starburst sensitizer with carbazole antennas for efficient and stable dye-sensitized solar cells. Energy Environ. Sci. 2010, 3, 1736–1745. [Google Scholar] [CrossRef]
- Ning, Z.; Zhang, Q.; Wu, W.; Pei, H.; Liu, B.O.; Tian, H.E. Starburst triarylamine based dyes for efficient dye-sensitized solar cells. J. Org. Chem. 2008, 73, 3791–3797. [Google Scholar] [CrossRef]
- Naik, P.; Elmorsy, M.R.; Su, R.; Babu, D.D.; El-Shafei, A.; Adhikari, A.V. New carbazole based metal-free organic dyes with D-π-A-π-A architecture for DSSCs: Synthesis, theoretical and cell performance studies. Solar Energy 2017, 153, 600–610. [Google Scholar] [CrossRef]
- Naik, P.; Su, R.; Elmorsy, M.R.; El-Shafei, A.; Adhikari, A.V. New di-anchoring A-π-D-π-A configured organic chromophores for DSSC application: Sensitization and co-sensitization studies. Photochem. Photobiol. Sci. 2018, 17, 302–314. [Google Scholar] [CrossRef]
- Sharma, K.; Sharma, V.; Sharma, S.S. Dye-sensitized solar cells: Fundamentals and current status. Nanoscale Res. Lett. 2018, 13, 381. [Google Scholar] [CrossRef]
- Zarate, X.; Schott-Verdugo, S.; Rodriguez-Serrano, A.; Schott, E. The nature of the donor motif in acceptor-bridge-donor dyes as an influence in the electron photo-injection mechanism in DSSCs. J. Phys. Chem. A 2016, 120, 1613–1624. [Google Scholar] [CrossRef]
- Koyama, Y.; Miki, T.; Wang, X.F.; Nagae, H. Dye-sensitized solar cells based on the principles and materials of photosynthesis: Mechanisms of suppression and enhancement of photocurrent and conversion efficiency. Int. J. Mol. Sci. 2009, 10, 4575–4622. [Google Scholar] [CrossRef]
- Sanusi, K.; Fatomi, N.O.; Borisade, A.O.; Tilmaz, Y.; Ceylan, U.; Fashima, A. An approximate procedure for profiling dye molecules with potentials as sensitizers in solar cell application: A DFT/TD-DFT approach. Chem. Phys. Lett. 2019, 1–7. [Google Scholar] [CrossRef]
- Hamann, T.W.; Martinson, A.B.; Elam, J.W.; Pellin, M.J.; Hupp, J.T. Atomic layer deposition of TiO2 on aerogel templates: New photoanodes for dye-sensitized solar cells. J. Phys. Chem. C 2008, 112, 10303–10307. [Google Scholar] [CrossRef]
- Mehmood, U.; Hussein, I.A.; Daud, M. New 1,3,4-Oxadiazole Based Photosensitizers for Dye Sensitized Solar Cells. Int. J. Photoen. 2015, 2015, 637652. [Google Scholar] [CrossRef]
- Srinivas, K.; Sivakumar, G.; Chitumalla, R.K.; Reddy, M.A.; Bhanuprakash, K.; Vaidya, J.R.; Chen, C.W.; Hsu, Y.C.; Lin, J.T. Novel 1, 3, 4-oxadiazole derivatives as efficient sensitizers for dye-sensitized solar cells: A combined experimental and computational study. Synth. Met. 2011, 161, 1671–1681. [Google Scholar] [CrossRef]
- Singh, A.; Kociok-Köhn, G.; Trivedi, M.; Chauhan, R.; Kumar, A.; Gosavi, S.W.; Terashima, C.; Kujishima, A.N.J. Ferrocenylethenyl-substituted oxadiazoles with phenolic and nitro anchors as sensitizers in dye sensitized solar cells. New J. Chem. 2019, 43, 4745–4756. [Google Scholar] [CrossRef]
- Paun, A.; Hadade, N.D.; Paraschivescu, C.C.; Matache, M. 1,3,4-Oxadiazoles as luminescent materials for organic light emitting diodes via cross-coupling reactions. J. Mat. Chem. C 2016, 4, 8596–8610. [Google Scholar] [CrossRef]
- Friesh, M.J.; Truks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Pettersson, G. Gaussian 09; Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Account. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Atomic dipole moment corrected Hirshfeld population method. J. Theor. Comput. Chem. 2012, 11, 163–183. [Google Scholar] [CrossRef]
- Ning, Z.; Zhang, Q.; Pei, H.; Luan, J.; Lu, C.; Cui, Y.; Tian, H. Photovoltage improvement for dye-sensitized solar cells via cone-shaped structural design. J. Phys. Chem. C 2009, 113, 10307–10313. [Google Scholar] [CrossRef]
- Mubashar, U.; Farhat, A.; Khera, R.A.; Iqbal, N.; Saleem, R.; Iqbal, J. Designing and theoretical study of fluorinated small molecule donor materials for organic solar cells. J. Mol. Model. 2021, 27, 216. [Google Scholar] [CrossRef] [PubMed]
- Mathiyalagan, A.; Manimaran, K.; Muhu, K.; Rajakantham, M. Density functional theory study on the electronic structures and spectral properties of 3,5-Dimethylanisole dye sensitizer for solar cell applications. Res. Chem. 2021, 3, 1–38. [Google Scholar] [CrossRef]
- Bulat, F.A.; Murray, J.S.; Politzer, P. Identifying the most energetic electrons in a molecule: The highest occupied molecular orbital and the average local ionization energy. Comp. Theory Chem. 2021, 1199, 1–6. [Google Scholar] [CrossRef]
- Coetzee, L.C.C.; Muller, A.J.; Adeyinka, A.S.; Sonopo, M.S.; Williams, D.B.G. Synthesis, characterisation and DFT studies of [3, 5-bis (2-hydroxyphenyl)-1H-1, 2, 4-triazol-1-yl](phenyl) methanone derivatives. Results Chem. 2021, 3, 100165. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Q.; Liu, D.; Su, R.; Liu, J.; Li, Y. Computational Prediction of Electronic and Photovoltaic Properties of Anthracene-Based Organic Dyes for Dye-Sensitized Solar Cells. Int. J. Photoen. 2018, 2018, 4764830. [Google Scholar] [CrossRef]
- Sharmoukh, W.; Cong, J.; Ali, B.A.; Alam, N.K.; Kloo, L. Comparison between Benzothiadizole–Thiophene- and Benzothiadizole–Furan-Based D–A−π–A Dyes Applied in Dye-Sensitized Solar Cells: Experimental and Theoretical Insights. ACS Omega 2020, 5, 16856–16864. [Google Scholar] [CrossRef]
- Guido, C.A.; Cortona, P.; Mennucci, B.; Adamo, C. On the Metric of Charge Transfer Molecular Excitations: A Simple Chemical Descriptor. J. Chem. Theory Comput. 2013, 9, 3118–31141. [Google Scholar] [CrossRef]
- le Bahers, T.; Adamo, C.; Ciofini, I. A Qualitative Index of Spatial Extent in Charge-Transfer Excitations. J. Chem. Theory Comput. 2011, 7, 2498–2506. [Google Scholar] [CrossRef]
- Tretiak, S.; Mukamel, S. Density Matrix Analysis and Simulation of Electronic Excitations in Conjugated and Aggregated Molecules. Chem. Rev. 2002, 102, 3171–3212. [Google Scholar] [CrossRef]
- Preat, J.; Jacquemin, D.; Michaux, C.; Perpète, E.A. Improvement of the efficiency of thiophene-bridged compounds for dye-sensitized solar cells. Chem. Phys. 2010, 376, 56–68. [Google Scholar] [CrossRef]
- Kurumisawa, Y.; Higashino, T.; Nimura, S.; Tsuji, Y.; Liyama, H.; Imahori, H. Renaissance of fused porphyrins: Substituted methylene-bridged thiophene-fused strategy for high-performance dye-sensitized solar cells. J. Am. Chem. Soc. 2019, 141, 9910–9919. [Google Scholar] [CrossRef] [PubMed]
- Arrechea, S.; Clifford, J.N.; Pelleja, L.; Aljarilla, A.; de la Cruz, P.; Palomares, E.; Langa, F. Charge recombination losses in thiophene-substituted porphyrin dye-sensitized solar cells. Dye. Pigment. 2016, 126, 147–153. [Google Scholar] [CrossRef]
- Samae, R.; Surawatanawong, P.; Eiamprasert, U.; Pramjit, S.; Saengdee, L.; Tangboriboonrat, P.; Kiatisevi, S. Effect of Thiophene Spacer Position in Carbazole-Based Dye-Sensitized Solar Cells on Photophysical, Electrochemical and Photovoltaic Properties. Eur. J. Chem. 2016, 21, 3536–3549. [Google Scholar] [CrossRef]
- Gao, P.; Tsao, H.N.; Grätzel, M.; Nazeeruddin, M.K. Fine-tuning the electronic structure of organic dyes for dye-sensitized solar cells. Org. Lett. 2012, 14, 4330–4333. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Wu, H.; Dong, G.; Shen, Z.; Li, S.; Zhang, M. Benzothiadiazole–ethynylthiophenezoic acid as an acceptor of photosensitizer for efficient organic dye-sensitized solar cells. J. Mater. Chem. A 2018, 6, 21493–21500. [Google Scholar] [CrossRef]
- Wu, Z.; Li, X.; Agren, H.; Hua, J.; Tian, H. Pyrimidine-2-carboxylic acid as an electron-accepting and anchoring group for dye-sensitized solar cells. Appl. Mater. Interfac. 2015, 7, 26355–26359. [Google Scholar] [CrossRef]
- Zhang, L.; Cole, J.M.; Waddell, P.G.; Low, K.S.; Liu, X. Relating Electron Donor and Carboxylic Acid Anchoring Substitution Effects in Azo Dyes to Dye-Sensitized Solar Cell Performance. ACS Sustain. Chem. Eng. 2013, 1, 1440–1452. [Google Scholar] [CrossRef]
- Cecconi, B.; Mordini, A.; Reginato, G.; Zani, L.; Taddei, M.; de Biani, F.F.; de Angelis, F.; Marotta, G.; Salvatori, P.; Calamante, M. Pyridine-N-Oxide 2-Carboxylic Acid: An Acceptor Group for Organic Sensitizers with Enhanced Anchoring Stability in Dye-Sensitized Solar Cells. Asian J. Org. Chem. 2014, 3, 140–152. [Google Scholar] [CrossRef]
- Ganesan, P.; Chandiran, A.; Gao, P.; Rajalingam, R.; Grätzel, M.; Nazeeruddin, M.K. Molecular engineering of 2-quinolinone based anchoring groups for dye-sensitized solar cells. J. Phys. Chem. C 2014, 118, 16896–16903. [Google Scholar] [CrossRef]
- Ompang, D.; Singh, J. Diffusion length and Langevin recombination of singlet and triplet excitons in organic heterojunction solar cells. Chem. Phys. Chem. 2015, 16, 1281–1285. [Google Scholar] [CrossRef]
- Pivrikas, A.; Juška, G.; Österbacka, R.; Westerling, M.; Viliūnas, M.; Arlauskas, K.; Stubb, H. Langevin recombination and space-charge-perturbed current transients in regiorandom poly(3-hexylthiophene). Phys. Rev. B 2005, 71, 125205. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
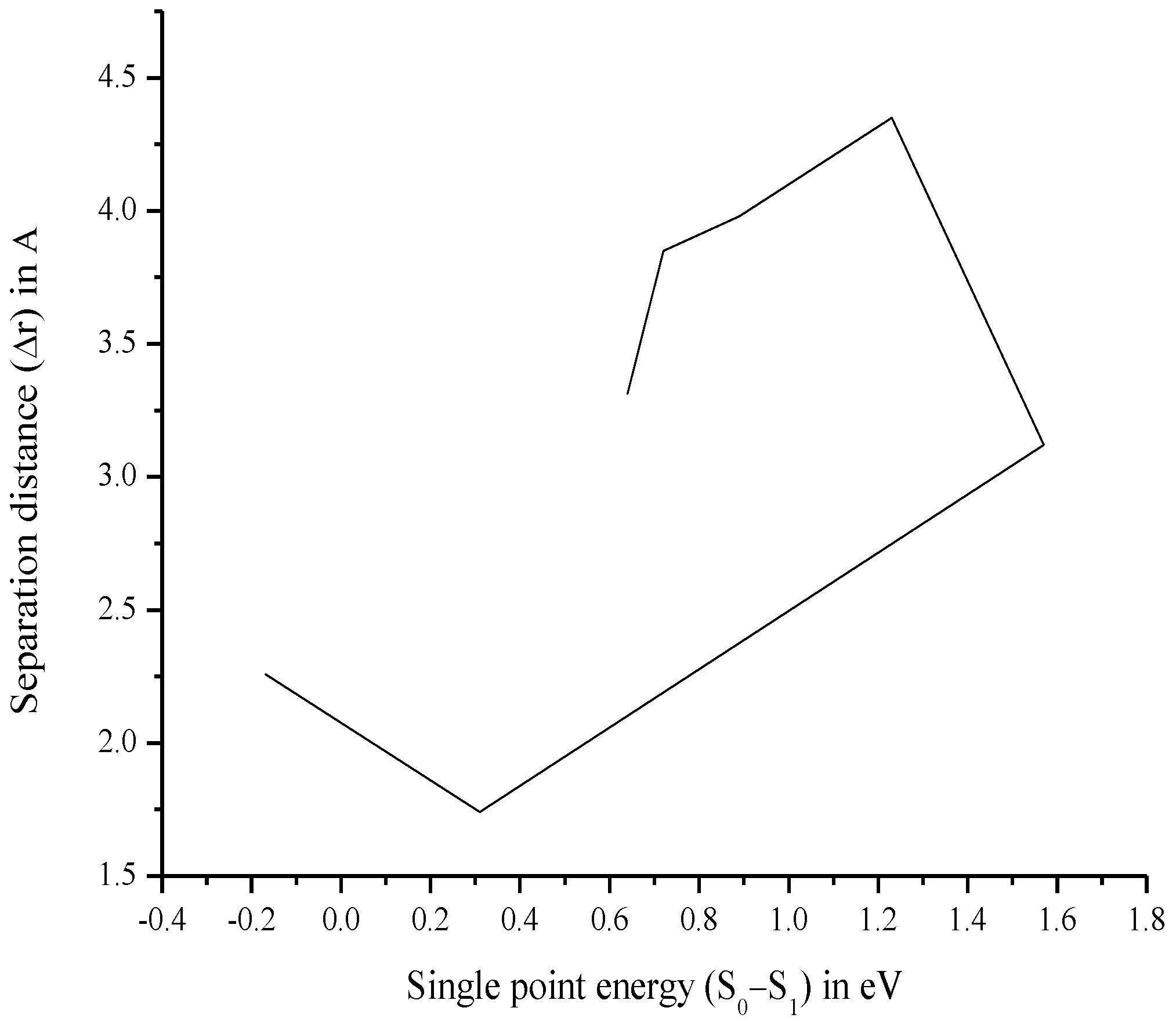{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | HOMO (eV) | LUMO (eV) | E(LUMO-HOMO) (eV) | Eb (eV) | E(S1-S0) (eV) | μ (Debye) | f | εabs (L·mol−1·cm−1) | λmax (nm) | IP (eV) | EA (eV) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| O1 | −6.95 | −2.59 | 4.36 | 3.72 | 0.64 | 7.48 | 0.545 | 27,692 | 337 | 6.69 | 2.00 |
| O2 | −7.37 | −2.15 | 5.22 | 4.50 | 0.72 | 2.81 | 1.030 | 41,649 | 356 | 7.45 | 1.49 |
| O3 | −7.23 | −2.17 | 5.06 | 4.17 | 0.89 | 5.73 | 0.853 | 34,505 | 360 | 7.75 | 0.48 |
| O4 | −7.59 | −2.12 | 5.47 | 4.23 | 1.23 | 0.91 | 0.675 | 28,404 | 325 | 7.63 | 1.44 |
| O5 | −7.39 | −2.45 | 4.94 | 3.37 | 1.57 | 6.66 | 0.782 | 37,278 | 325 | 5.52 | 4.17 |
| O6 | −6.69 | −2.95 | 3.74 | 3.43 | 0.31 | 3.18 | 0.042 | 3522 | 429 | 5.28 | 4.26 |
| O7 | −7.06 | −2.58 | 4.48 | 4.65 | −0.17 | 5.12 | 0.380 | 15,470 | 496 | 4.65 | 5.00 |
| Atoms | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | N15 | O17 | O18 | N25 | N27 | N28 | O27 | O28 | O29 | O30 | O31 | N53 | N182 | O184 | O185 |
| O1 | 235 | 349 | 414 | ||||||||||||
| O2 | 246 | 364 | 362 | ||||||||||||
| O3 | 251 | 308 | 411 | ||||||||||||
| O4 | 229 | 327 | 400 | ||||||||||||
| O5 | 233 | 341 | 402 | ||||||||||||
| O6 | 206 | 288 | 412 | ||||||||||||
| O7 | 86.6 | ||||||||||||||
| Compound | rdye (m) (1 × 10−7) | Dπ (1 × 10−11) | δp | ncollect (1 × 10−12) | LHE | t (s) (1 × 10−38) | Φinj | IPCE (1 × 10−12) | |
|---|---|---|---|---|---|---|---|---|---|
| O1 | 1.62 | 6.73 | −6.08 | 1.68 | 0.715 | 0.212 | 0.795 | 1.21 | 2.35 |
| O2 | 1.15 | 9.51 | −5.64 | 2.99 | 0.907 | 0.120 | 1.00 | 2.33 | 2.77 |
| O3 | 1.16 | 9.47 | −5.66 | 2.96 | 0.860 | 0.148 | 1.00 | 2.55 | 2.63 |
| O4 | 1.15 | 9.51 | −5.61 | 3.02 | 0.789 | 0.154 | 0.961 | 2.29 | 2.99 |
| O5 | 1.41 | 7.73 | −5.94 | 1.51 | 0.835 | 0.136 | 0.849 | 1.55 | 2.79 |
| O6 | 1.89 | 5.76 | −6.44 | 1.39 | 0.0922 | 4.56 | 0.480 | 0.0615 | 2.09 |
| O7 | 1.54 | 7.07 | −6.07 | 1.92 | 0.583 | 0.633 | 0.996 | 1.11 | 2.46 |
| Fragment | Hole (%) | Electron (%) | Overlap (%) | Difference (%) |
|---|---|---|---|---|
| Donor | 1.38 | 0.26 | 0.60 | −1.12 |
| π-spacer | 75.52 | 39.84 | 54.85 | −35.68 |
| Acceptor | 23.38 | 59.90 | 37.42 | 36.52 |
| Fragment | Hole (%) | Electron (%) | Overlap (%) | Difference (%) |
|---|---|---|---|---|
| Donor | 1.18 | 0.19 | 0.47 | −0.99 |
| π-spacer | 86.04 | 56.91 | 69.98 | −29.12 |
| Acceptor | 12.79 | 42.90 | 23.42 | 30.12 |
| Fragment | Hole (%) | Electron (%) | Overlap (%) | Difference (%) |
|---|---|---|---|---|
| Donor | 2.58 | 0.23 | 0.77 | −2.36 |
| π-spacer | 80.66 | 58.70 | 68.81 | −21.97 |
| Acceptor | 16.32 | 40.89 | 25.83 | 24.57 |
| Fragment | Hole (%) | Electron (%) | Overlap (%) | Difference (%) |
|---|---|---|---|---|
| Donor | 1.32 | 0.10 | 0.35 | −1.23 |
| π-spacer | 85.29 | 47.41 | 63.59 | −37.88 |
| Acceptor | 13.39 | 52.50 | 26.51 | 39.11 |
| Fragment | Hole (%) | Electron (%) | Overlap (%) | Difference (%) |
|---|---|---|---|---|
| Donor | 3.44 | 0.87 | 1.74 | −2.57 |
| π-spacer | 74.77 | 48.71 | 60.35 | −26.07 |
| Acceptor | 21.78 | 50.42 | 33.14 | 28.64 |
| Fragment | Hole (%) | Electron (%) | Overlap (%) | Difference (%) |
|---|---|---|---|---|
| Donor | 12.71 | −0.05 | 0.00 | −12.75 |
| π-spacer | 60.42 | 9.38 | 23.80 | −51.05 |
| Acceptor | 26.87 | 90.67 | 49.36 | 63.80 |
| Fragment | Hole (%) | Electron (%) | Overlap (%) | Difference (%) |
|---|---|---|---|---|
| Donor | 4.71 | 1.27 | 2.45 | −3.44 |
| π-spacer | 71.84 | 52.24 | 61.26 | −19.60 |
| Acceptor | 23.45 | 46.49 | 33.02 | 23.04 |
| Compound | Δr (Å) | Orbital Pair |
|---|---|---|
| O1 | 3.31 | 242 → 251 |
| O2 | 3.85 | 87 → 88 |
| O3 | 3.98 | 91 → 92 |
| O4 | 4.35 | 87 → 88 |
| O5 | 3.12 | 159 → 165 |
| 3.38 | 161 → 165 | |
| O6 | 1.74 | 402 → 404 |
| O7 | 2.26 | 213 → 217 |
| 2.89 | 216 → 217 |
| Compound | Λ | Orbital Pair |
|---|---|---|
| O1 | 0.324 | 242 → 251 |
| O2 | 0.506 | 87 → 88 |
| O3 | 0.471 | 91 → 92 |
| O4 | 0.280 | 87 → 88 |
| O5 | 0.150 | 159 → 165 |
| 0.118 | 161 → 165 | |
| O6 | 0.0476 | 367 → 404 |
| O7 | 0.271 | 213 → 217 |
| 0.143 | 216 → 217 |
| IFCT | Dihedral Angles (°) | |||
|---|---|---|---|---|
| Compound | (D-π-A) | (D-π) | (π-A) | (D-A) |
| O1 | 0.35923 | 86.04 | 54.25 | 81.91 |
| O2 | 0.29634 | 18.06 | 10.02 | 10.93 |
| O3 | 0.23401 | 89.97 | 0 | 89.97 |
| O4 | 0.38426 | 14.64 | 14.35 | 8.74 |
| O5 | 0.27091 | 88.35 | 53.68 | 86.59 |
| O6 | 0.52267 | 83.11 | 86.07 | 64.25 |
| O7 | 0.21145 | 65.74 | 40.10 | 37.49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coetzee, L.-C.C.; Adeyinka, A.S.; Magwa, N. A Theoretical Evaluation of the Efficiencies of Metal-Free 1,3,4-Oxadiazole Dye-Sensitized Solar Cells: Insights from Electron–Hole Separation Distance Analysis. Energies 2022, 15, 4913. https://doi.org/10.3390/en15134913
Coetzee L-CC, Adeyinka AS, Magwa N. A Theoretical Evaluation of the Efficiencies of Metal-Free 1,3,4-Oxadiazole Dye-Sensitized Solar Cells: Insights from Electron–Hole Separation Distance Analysis. Energies. 2022; 15(13):4913. https://doi.org/10.3390/en15134913
Chicago/Turabian StyleCoetzee, Louis-Charl Cloete, Adedapo Sunday Adeyinka, and Nomampondo Magwa. 2022. "A Theoretical Evaluation of the Efficiencies of Metal-Free 1,3,4-Oxadiazole Dye-Sensitized Solar Cells: Insights from Electron–Hole Separation Distance Analysis" Energies 15, no. 13: 4913. https://doi.org/10.3390/en15134913
APA StyleCoetzee, L.-C. C., Adeyinka, A. S., & Magwa, N. (2022). A Theoretical Evaluation of the Efficiencies of Metal-Free 1,3,4-Oxadiazole Dye-Sensitized Solar Cells: Insights from Electron–Hole Separation Distance Analysis. Energies, 15(13), 4913. https://doi.org/10.3390/en15134913