
Mechanistic Insights into Hydrodeoxygenation of Acetone over Mo/HZSM-5 Bifunctional Catalyst for the Production of Hydrocarbons


Abstract
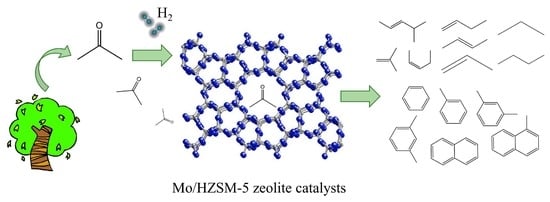
:
1. Introduction
2. Experiment Methods
2.1. Materials
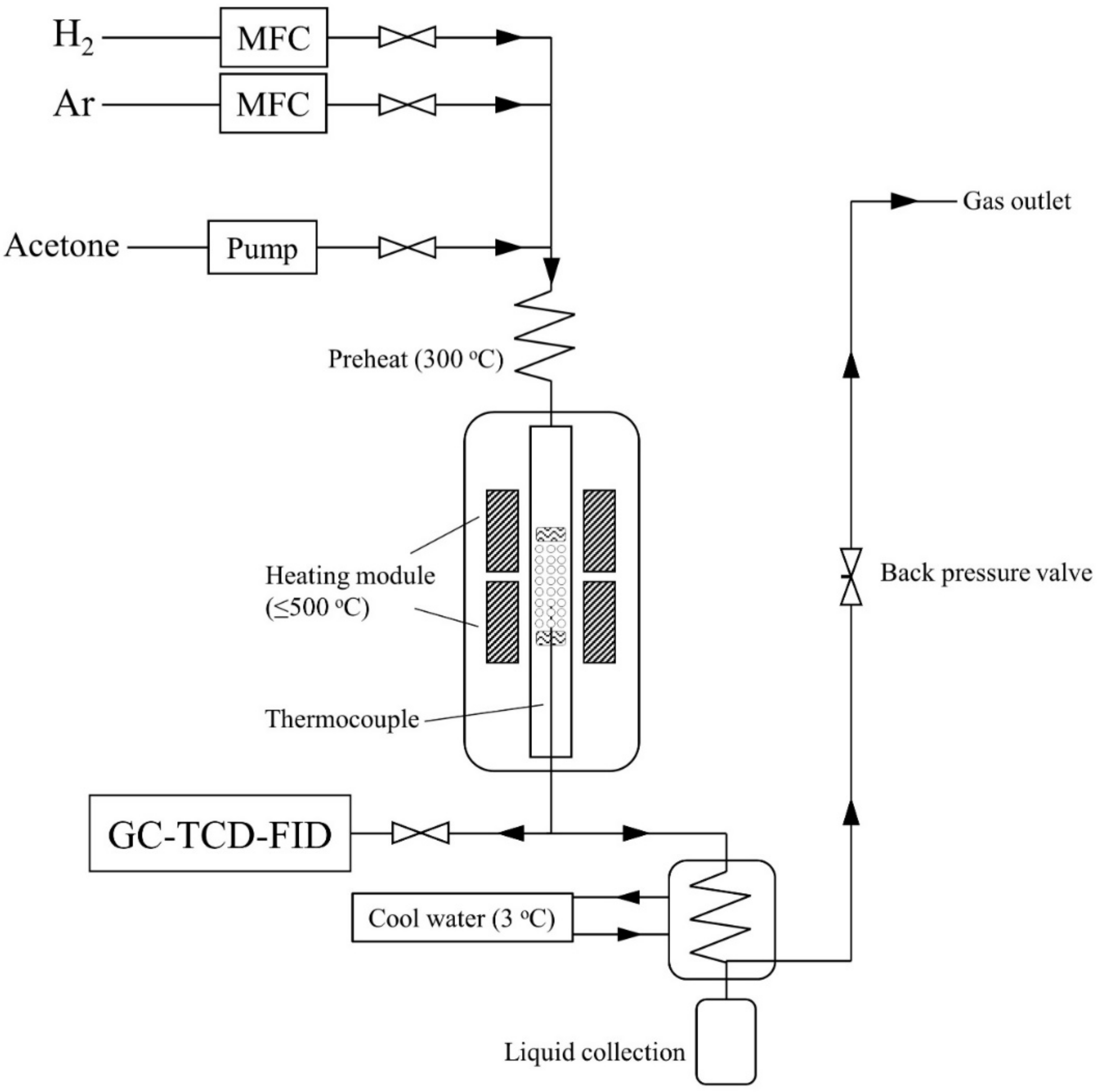
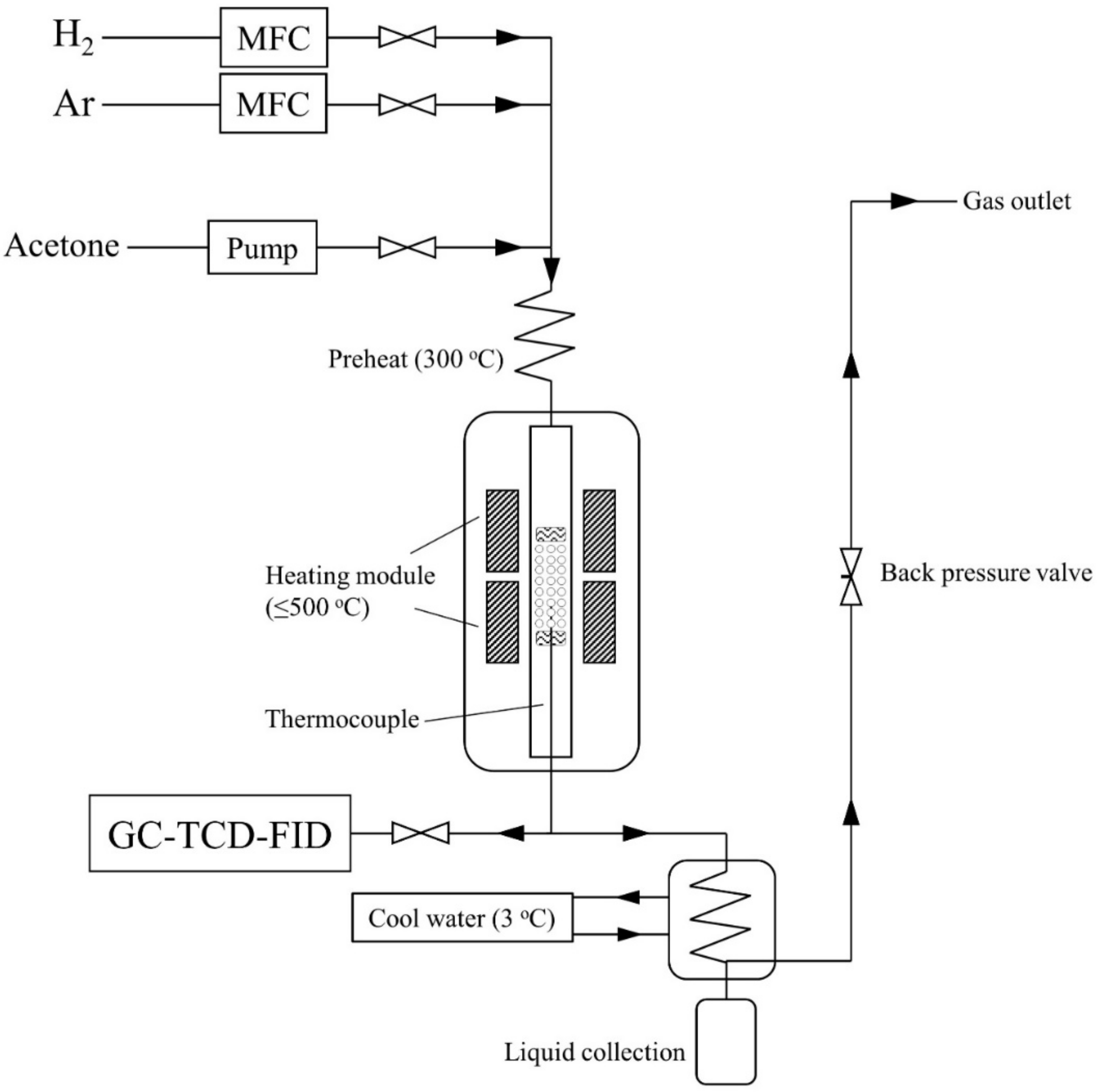
2.2. Experiment
2.3. Catalyst Characterization
3. Results and Discussion
3.1. Catalyst Characterization
3.2. Hydrodeoxygenation of Acetone
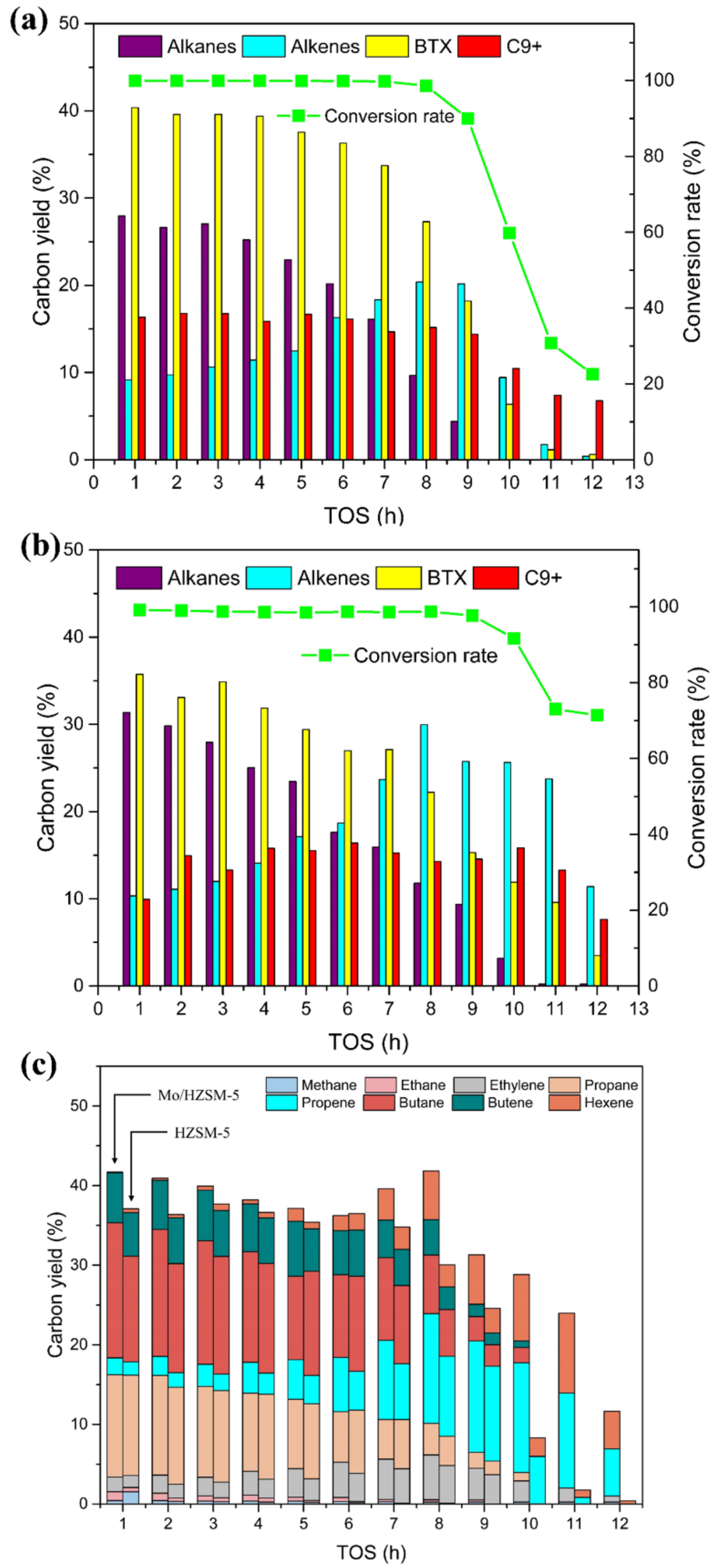
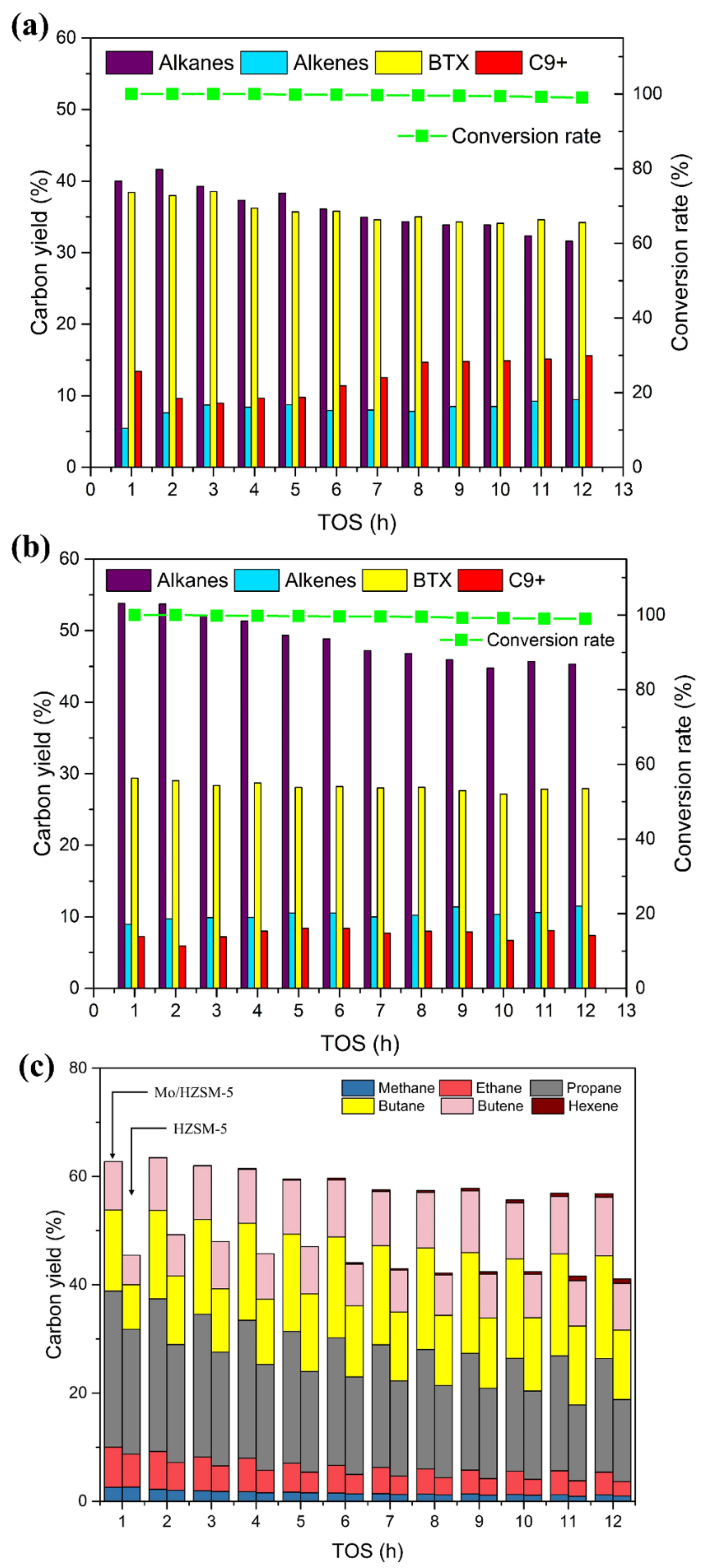
3.2.1. Atmospheric Hydrogen Pressure
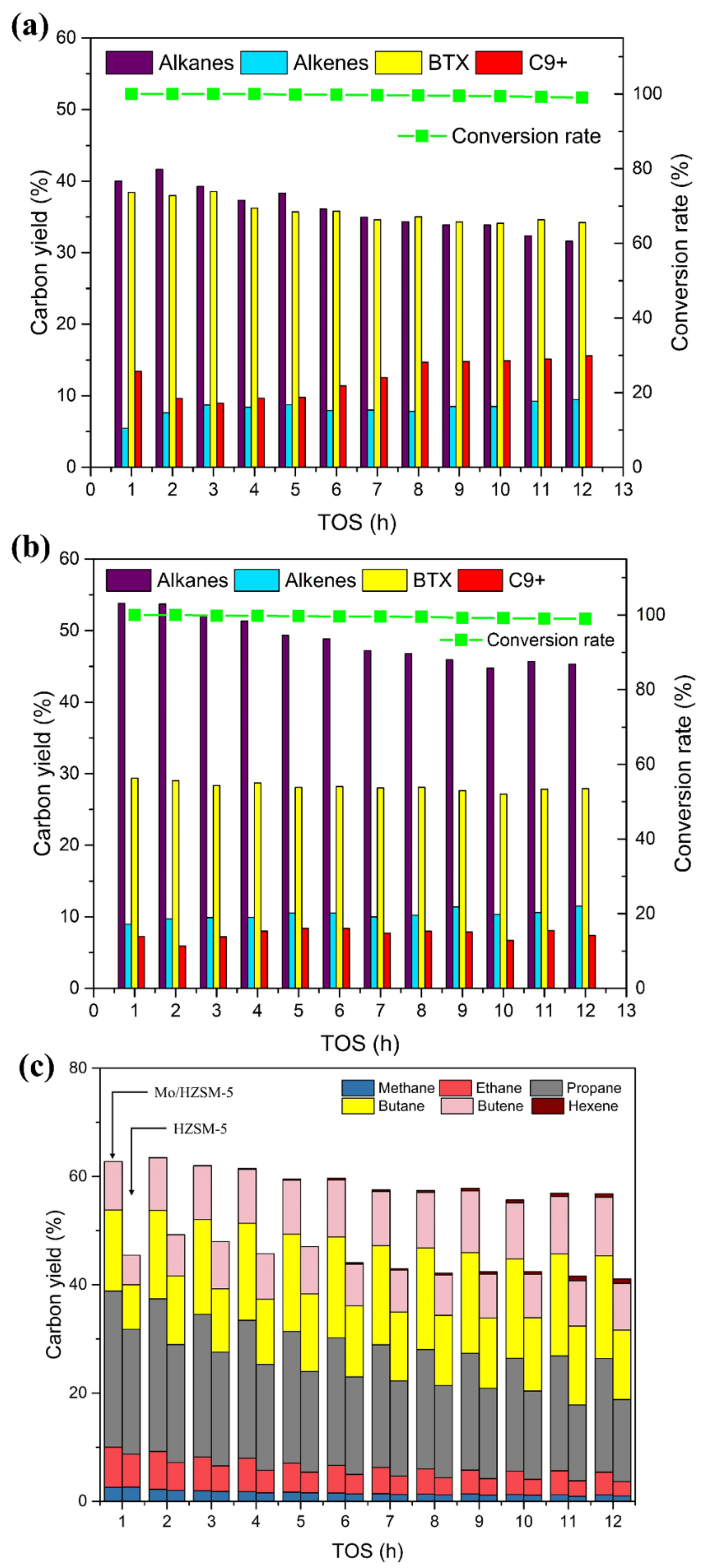
3.2.2. Influence of Hydrogen Pressure on Hydrodeoxygenation of Acetone
3.3. Characterization of the Spent Catalysts
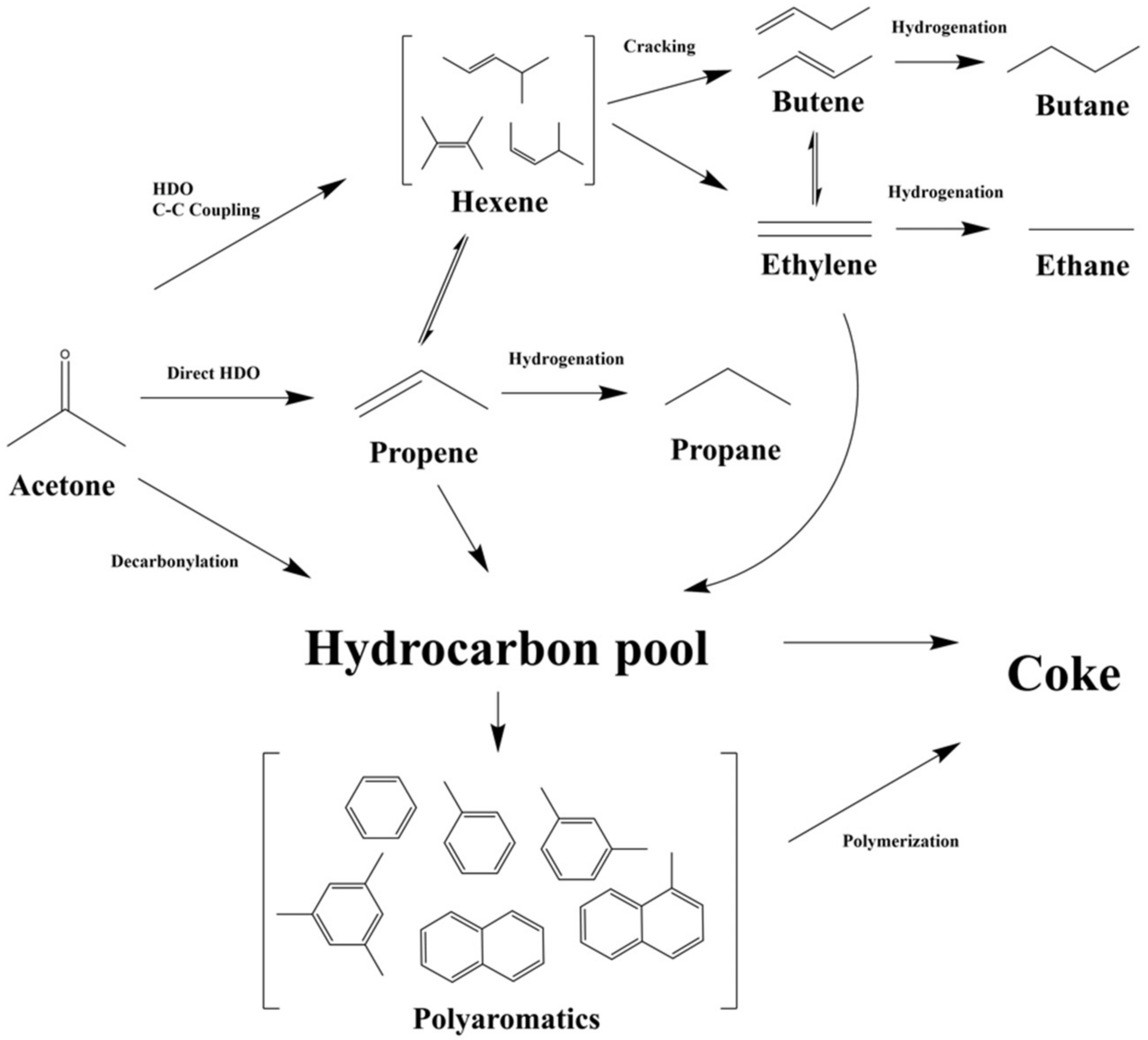
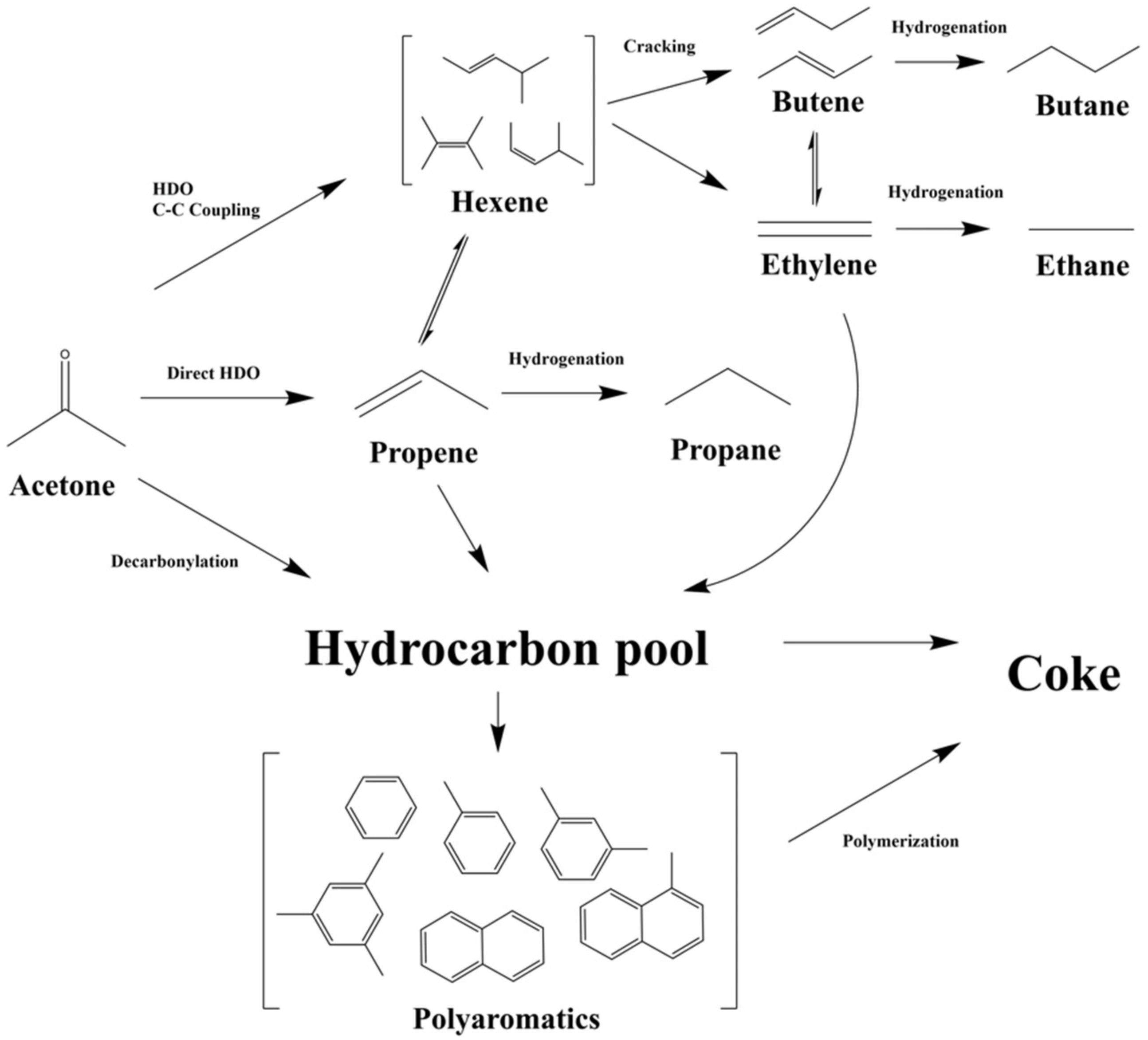
3.4. Reaction Network of Catalytic HDO of Acetone over Mo/HZSM-5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dabros, T.M.H.; Stummann, M.Z.; Høj, M.; Jensen, P.A.; Grunwaldt, J.-D.; Gabrielsen, J.; Mortensen, P.M.; Jensen, A.D. Transportation fuels from biomass fast pyrolysis, catalytic hydrodeoxygenation, and catalytic fast hydropyrolysis. Prog. Energy Combust. Sci. 2018, 68, 268–309. [Google Scholar] [CrossRef]
- Liu, Z.; Hughes, M.; Tong, Y.; Zhou, J.; Kreutter, W.; Lopez, H.C.; Singer, S.; Zitomer, D.; McNamara, P. Paper mill sludge biochar to enhance energy recovery from pyrolysis: A comprehensive evaluation and comparison. Energy 2022, 239, 121925. [Google Scholar] [CrossRef]
- Liu, C.; Wang, H.; Karim, A.M.; Sun, J.; Wang, Y. Catalytic fast pyrolysis of lignocellulosic biomass. Chem. Soc. Rev. 2014, 43, 7594–7623. [Google Scholar] [CrossRef]
- Zahra, H.; Sawada, D.; Kumagai, S.; Ogawa, Y.; Johansson, L.-S.; Ge, Y.; Guizani, C.; Yoshioka, T.; Hummel, M. Evolution of carbon nanostructure during pyrolysis of homogeneous chitosan-cellulose composite fibers. Carbon 2021, 185, 27–38. [Google Scholar] [CrossRef]
- Saraeian, A.; Burkhow, S.J.; Jing, D.; Smith, E.A.; Shanks, B.H. Catalyst Property Effects on Product Distribution during the Hydrodeoxygenation of Lignin Pyrolysis Vapors over MoO3/γ-Al2O3. ACS Sustain. Chem. Eng. 2021, 9, 6685–6696. [Google Scholar] [CrossRef]
- Williams, P.T.; Nugranad, N. Comparison of products from the pyrolysis and catalytic pyrolysis of rice husks. Energy 2000, 25, 493–513. [Google Scholar] [CrossRef]
- Mastral, J.F.; Berrueco, C.; Gea, M.; Ceamanos, J. Catalytic degradation of high density polyethylene over nanocrystalline HZSM-5 zeolite. Polym. Degrad. Stab. 2006, 91, 3330–3338. [Google Scholar] [CrossRef]
- Stefanidis, S.D.; Kalogiannis, K.G.; Iliopoulou, E.F.; Lappas, A.A.; Pilavachi, P.A. In-situ upgrading of biomass pyrolysis vapors: Catalyst screening on a fixed bed reactor. Bioresour. Technol. 2011, 102, 8261–8267. [Google Scholar] [CrossRef]
- Puertolas, B.; Veses, A.; Callen, M.S.; Mitchell, S.; Garcia, T.; Perez-Ramirez, J. Porosity-Acidity Interplay in Hierarchical ZSM-5 Zeolites for Pyrolysis Oil Valorization to Aromatics. ChemSusChem 2015, 8, 3283–3293. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Zhang, J.; Wang, J. Parametric study of two-stage hydropyrolysis of lignocellulosic biomass for production of gaseous and light aromatic hydrocarbons. Bioresour. Technol. 2017, 244 Pt 1, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Carlson, T.R.; Tompsett, G.A.; Conner, W.C.; Huber, G.W. Aromatic Production from Catalytic Fast Pyrolysis of Biomass-Derived Feedstocks. Top. Catal. 2009, 52, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Xiao, R.; Huang, H.; Xiao, G. Comparison of non-catalytic and catalytic fast pyrolysis of corncob in a fluidized bed reactor. Bioresour. Technol. 2009, 100, 1428–1434. [Google Scholar] [CrossRef]
- Cheng, S.; Wei, L.; Julson, J.; Rabnawaz, M. Upgrading pyrolysis bio-oil through hydrodeoxygenation (HDO) using non-sulfided Fe-Co/SiO2 catalyst. Energy Convers. Manag. 2017, 150, 331–342. [Google Scholar] [CrossRef]
- Prasomsri, T.; Shetty, M.; Murugappan, K.; Román-Leshkov, Y. Insights into the catalytic activity and surface modification of MoO3 during the hydrodeoxygenation of lignin-derived model compounds into aromatic hydrocarbons under low hydrogen pressures. Energy Environ. Sci. 2014, 7, 2660–2669. [Google Scholar] [CrossRef]
- Aqsha, A.; Katta, L.; Tijani, M.M.; Oliveira, C.F.; Mahinpey, N. Investigation of catalytic hydrodeoxygenation of anisole as bio-oil model compound over Ni-Mo/TiO2 and Ni-V/TiO2 catalysts: Synthesis, kinetic, and reaction pathways studies. Can. J. Chem. Eng. 2020, 99, 1094–1106. [Google Scholar] [CrossRef]
- Marker, T.L.; Felix, L.G.; Linck, M.B.; Roberts, M.J.; Ortiz-Toral, P.; Wangerow, J. Integrated hydropyrolysis and hydroconversion (IH2®) for the direct production of gasoline and diesel fuels or blending components from biomass, Part 2: Continuous testing. Environ. Prog. Sustain. Energy 2014, 33, 762–768. [Google Scholar] [CrossRef]
- Marker, T.L.; Felix, L.G.; Linck, M.B.; Roberts, M.J. Integrated hydropyrolysis and hydroconversion (IH2) for the direct production of gasoline and diesel fuels or blending components from biomass, Part 1: Proof of principle testing. Environ. Prog. Sustain. Energy 2012, 31, 191–199. [Google Scholar] [CrossRef]
- Rasmussen, M.J.; Medlin, J.W. Role of tungsten modifiers in bimetallic catalysts for enhanced hydrodeoxygenation activity and selectivity. Catal. Sci. Technol. 2020, 10, 414–423. [Google Scholar] [CrossRef]
- Eschenbacher, A.; Saraeian, A.; Shanks, B.H.; Mentzel, U.V.; Jensen, P.A.; Henriksen, U.B.; Ahrenfeldt, J.; Jensen, A.D. Performance-screening of metal-impregnated industrial HZSM-5/γ-Al2O3 extrudates for deoxygenation and hydrodeoxygenation of fast pyrolysis vapors. J. Anal. Appl. Pyrolysis 2020, 150, 104892. [Google Scholar] [CrossRef]
- Prasomsri, T.; Nimmanwudipong, T.; Román-Leshkov, Y. Effective hydrodeoxygenation of biomass-derived oxygenates into unsaturated hydrocarbons by MoO3 using low H2 pressures. Energy Environ. Sci. 2013, 6, 1732–1738. [Google Scholar] [CrossRef]
- Nolte, M.W.; Saraeian, A.; Shanks, B.H. Hydrodeoxygenation of cellulose pyrolysis model compounds using molybdenum oxide and low pressure hydrogen. Green Chem. 2017, 19, 3654–3664. [Google Scholar] [CrossRef]
- Murugappan, K.; Mukarakate, C.; Budhi, S.; Shetty, M.; Nimlos, M.R.; Román-Leshkov, Y. Supported molybdenum oxides as effective catalysts for the catalytic fast pyrolysis of lignocellulosic biomass. Green Chem. 2016, 18, 5548–5557. [Google Scholar] [CrossRef] [Green Version]
- Shetty, M.; Murugappan, K.; Green, W.H.; Román-Leshkov, Y. Structural Properties and Reactivity Trends of Molybdenum Oxide Catalysts Supported on Zirconia for the Hydrodeoxygenation of Anisole. ACS Sustain. Chem. Eng. 2017, 5, 5293–5301. [Google Scholar] [CrossRef]
- Xue, Y.; Sharma, A.; Huo, J.; Qu, W.; Bai, X. Low-pressure two-stage catalytic hydropyrolysis of lignin and lignin-derived phenolic monomers using zeolite-based bifunctional catalysts. J. Anal. Appl. Pyrolysis 2020, 146, 104779. [Google Scholar] [CrossRef]
- Thangalazhy-Gopakumar, S.; Adhikari, S.; Gupta, R.B. Catalytic Pyrolysis of Biomass over H+ZSM-5 under Hydrogen Pressure. Energy Fuels 2012, 26, 5300–5306. [Google Scholar] [CrossRef]
- Li, Z.; Zhong, Z.; Yang, Q.; Ben, H.; Seufitelli, G.V.S.; Resende, F.L.P. Parametric study of catalytic hydropyrolysis of rice husk over a hierarchical micro-mesoporous composite catalyst for production of light alkanes, alkenes, and liquid aromatic hydrocarbons. Fuel 2021, 310, 122475. [Google Scholar] [CrossRef]
- Venkatakrishnan, V.K.; Degenstein, J.C.; Smeltz, A.D.; Delgass, W.N.; Agrawal, R.; Ribeiro, F.H. High-pressure fast-pyrolysis, fast-hydropyrolysis and catalytic hydrodeoxygenation of cellulose: Production of liquid fuel from biomass. Green Chem. 2014, 16, 792–802. [Google Scholar] [CrossRef]
- Iisa, K.; Kim, Y.; Orton, K.A.; Robichaud, D.J.; Katahira, R.; Watson, M.J.; Wegener, E.C.; Nimlos, M.R.; Schaidle, J.A.; Mukarakate, C.; et al. Ga/ZSM-5 catalyst improves hydrocarbon yields and increases alkene selectivity during catalytic fast pyrolysis of biomass with co-fed hydrogen. Green Chem. 2020, 22, 2403–2418. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Z.; Chen, L.; Yang, S.; Xie, X.; Zhao, B.; Si, H.; Li, J.; Hua, D. Improving the monocyclic aromatic hydrocarbons production from fast pyrolysis of biomass over Fe-modified ZSM-5 catalysts. Int. J. Energy Res. 2020, 45, 6032–6040. [Google Scholar] [CrossRef]
- Mukarakate, C.; Watson, M.J.; ten Dam, J.; Baucherel, X.; Budhi, S.; Yung, M.M.; Ben, H.; Iisa, K.; Baldwin, R.M.; Nimlos, M.R. Upgrading biomass pyrolysis vapors over β-zeolites: Role of silica-to-alumina ratio. Green Chem. 2014, 16, 4891–4905. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Kang, M.-Y.; Ihm, S.-K. Deactivation by coke deposition on the HZSM-5 catalysts in the methanol-to-hydrocarbon conversion. J. Phys. Chem. Solids 2012, 73, 1542–1545. [Google Scholar] [CrossRef]
- Zhang, H.; Shao, S.; Xiao, R.; Shen, D.; Zeng, J. Characterization of Coke Deposition in the Catalytic Fast Pyrolysis of Biomass Derivates. Energy Fuels 2013, 28, 52–57. [Google Scholar] [CrossRef]
- Jae, J.; Tompsett, G.A.; Foster, A.J.; Hammond, K.D.; Auerbach, S.M.; Lobo, R.F.; Huber, G.W. Investigation into the shape selectivity of zeolite catalysts for biomass conversion. J. Catal. 2011, 279, 257–268. [Google Scholar] [CrossRef]
- Johansson, R.; Hruby, S.L.; Rass-Hansen, J.; Christensen, C.H. The Hydrocarbon Pool in Ethanol-to-Gasoline over HZSM-5 Catalysts. Catal. Lett. 2008, 127, 1–6. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | HZSM-5 | 5% Mo |
|---|---|---|
| BET surface area (m2/g) | 397 | 367 |
| a Vmicro (m3/g) | 0.122 | 0.113 |
| b Vmeso (m3/g) | 0.084 | 0.075 |
| c Vtotal (m3/g) | 0.206 | 0.188 |
| Lewis acid | ||
| Density (mmol/g) | 1.158 | 1.223 |
| Peak position (°C) | 137.4 | 141.9 |
| Bronsted acid | ||
| Density (mmol/g) | 0.406 | 0.497 |
| Peak position (°C) | 445.8 | 418.3 |
| Spent Catalyst | HZSM-5 | Mo/HZSM-5 | ||
|---|---|---|---|---|
| Catalysis pressure/bar | 0.1 | 30 | 0.1 | 30 |
| Coke (C%) | 17.2 | 5.8 | 13.4 | 2.3 |
| Surface area (m2/g) | 14 | 142 | 41 | 230 |
| a Vmicro (m3/g) | 0.001 | 0.044 | 0.011 | 0.084 |
| b Vmeso (m3/g) | 0.033 | 0.043 | 0.033 | 0.045 |
| c Vtotal (m3/g) | 0.034 | 0.087 | 0.044 | 0.129 |
| Lewis acid | ||||
| Density (mmol/g) | 0.470 | 0.594 | 0.654 | 0.813 |
| Peak position (°C) | 131.8 | 140.0 | 136.4 | 144.1 |
| Bronsted acid | ||||
| Density (mmol/g) | 0.76 | 0.163 | ||
| Peak position (°C) | 394.6 | 402.3 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, K.; Li, T.; Su, J.; Wang, C.; Wang, K. Mechanistic Insights into Hydrodeoxygenation of Acetone over Mo/HZSM-5 Bifunctional Catalyst for the Production of Hydrocarbons. Energies 2022, 15, 53. https://doi.org/10.3390/en15010053
Miao K, Li T, Su J, Wang C, Wang K. Mechanistic Insights into Hydrodeoxygenation of Acetone over Mo/HZSM-5 Bifunctional Catalyst for the Production of Hydrocarbons. Energies. 2022; 15(1):53. https://doi.org/10.3390/en15010053
Chicago/Turabian StyleMiao, Kai, Tan Li, Jing Su, Cong Wang, and Kaige Wang. 2022. "Mechanistic Insights into Hydrodeoxygenation of Acetone over Mo/HZSM-5 Bifunctional Catalyst for the Production of Hydrocarbons" Energies 15, no. 1: 53. https://doi.org/10.3390/en15010053
APA StyleMiao, K., Li, T., Su, J., Wang, C., & Wang, K. (2022). Mechanistic Insights into Hydrodeoxygenation of Acetone over Mo/HZSM-5 Bifunctional Catalyst for the Production of Hydrocarbons. Energies, 15(1), 53. https://doi.org/10.3390/en15010053