
High Dispersion of CeO2 on CeO2/MgO Prepared under Dry Conditions and Its Improved Redox Properties


Abstract
1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Catalyst Characterization
2.3. Dry Reforming Reaction
3. Results and Discussion
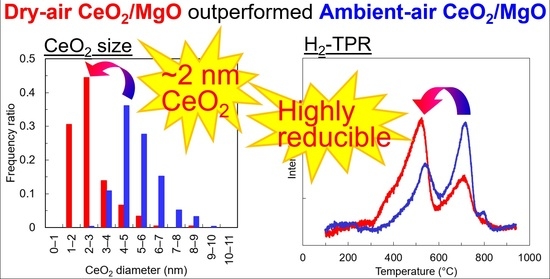
3.1. Characterization of the Catalysts
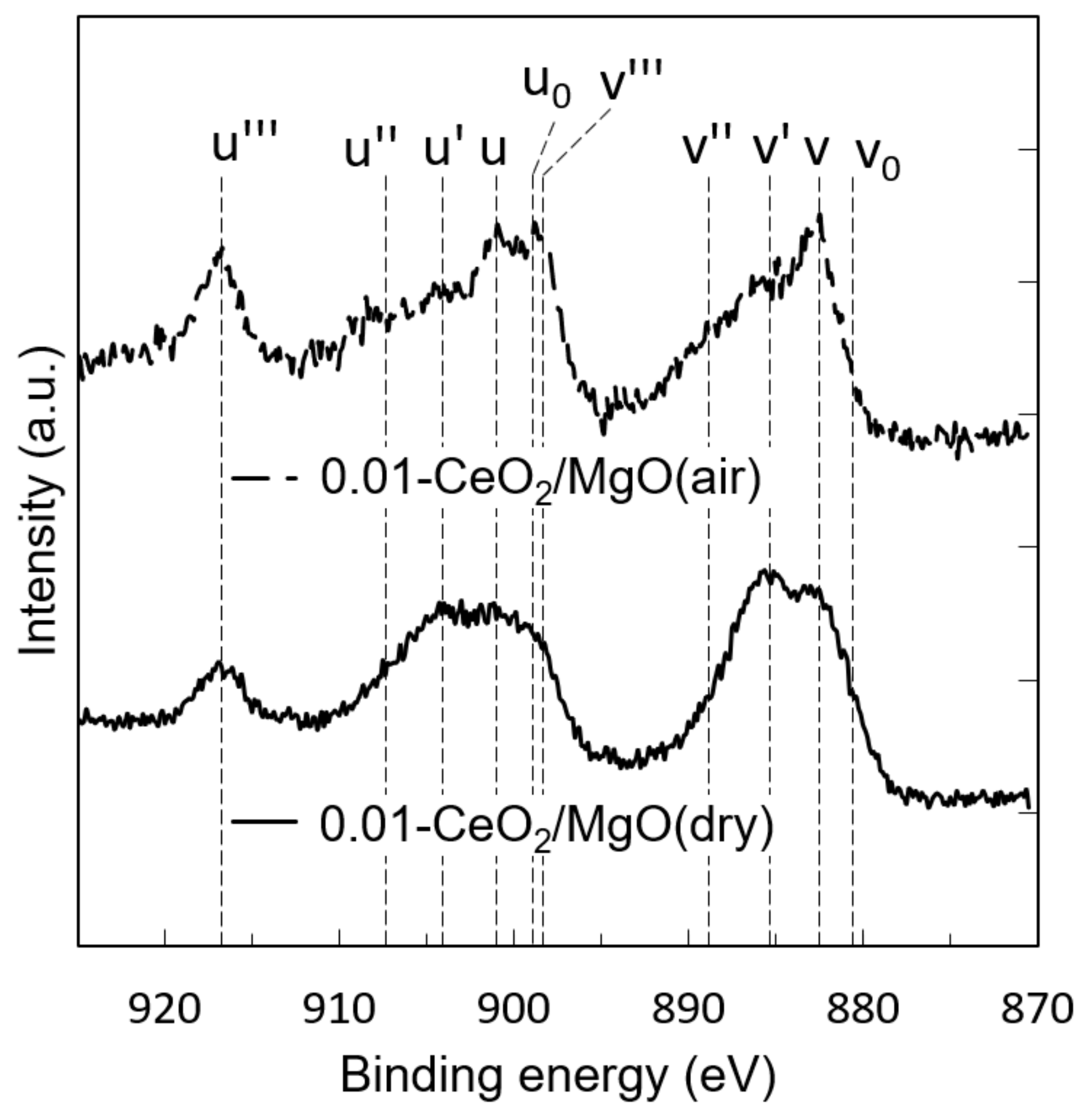
3.2. Mobility of Oxygen
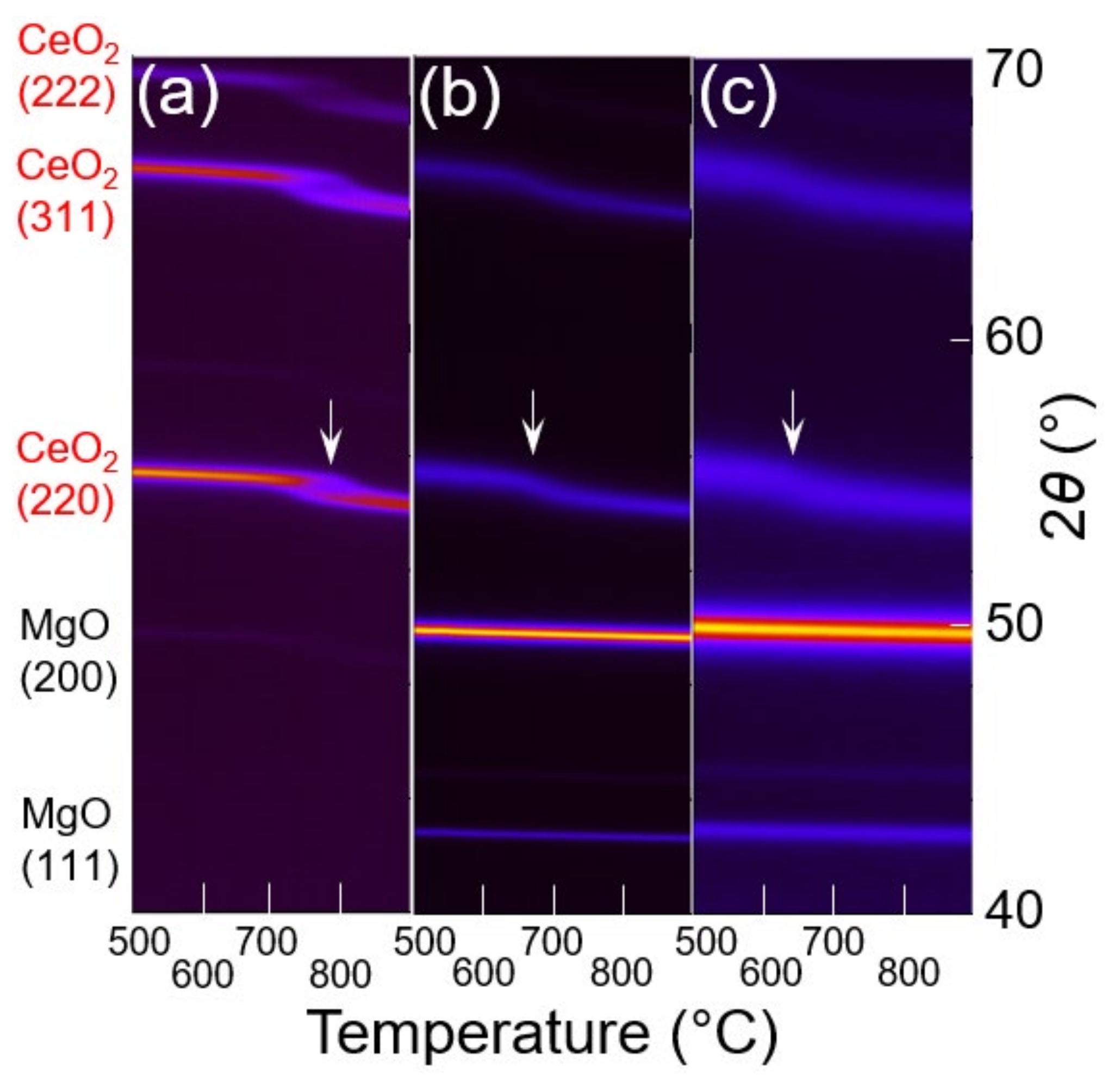
3.3. In Situ XRD Study under the Reducing Atmosphere
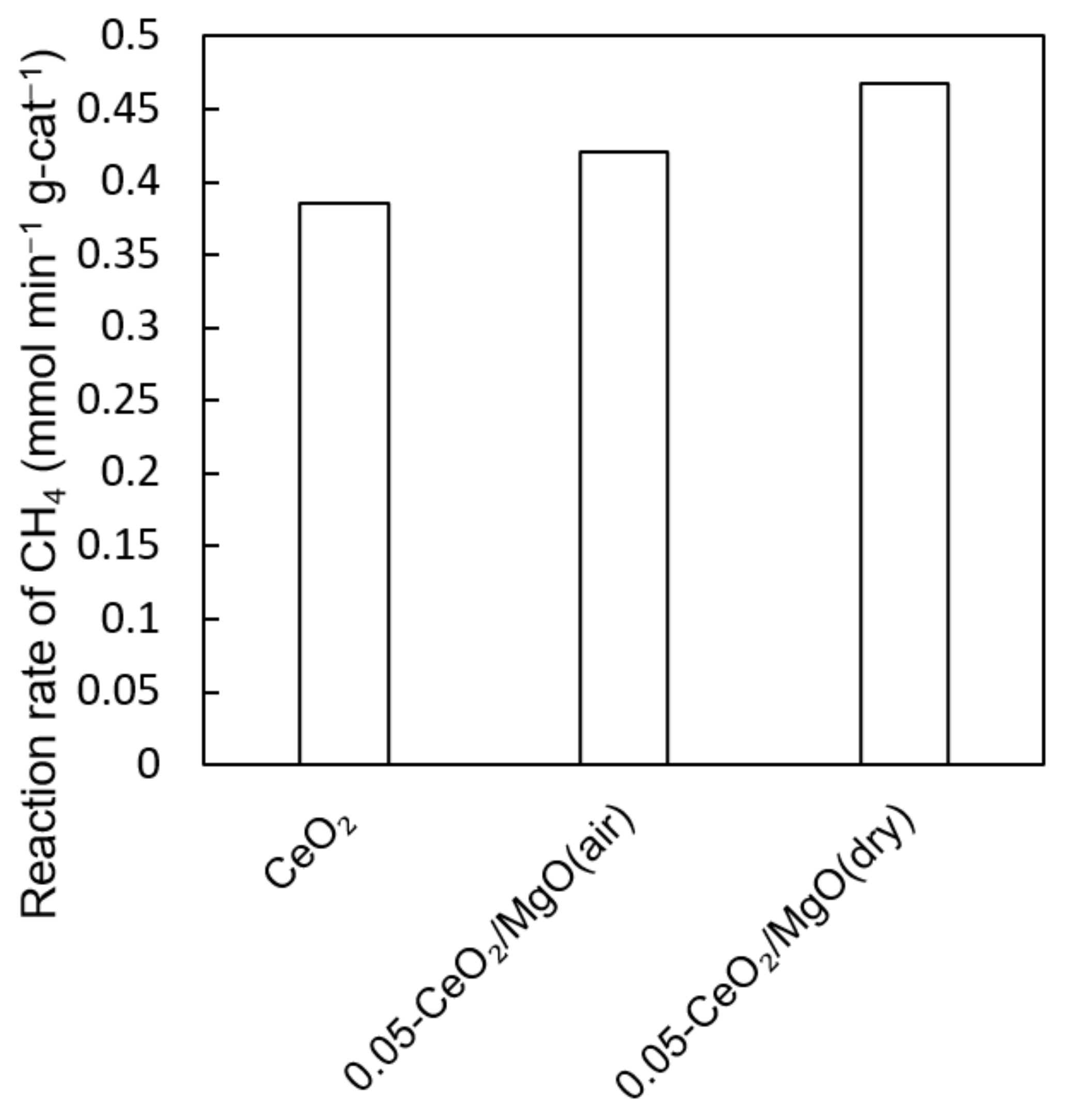
3.4. Dry Reforming Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Trovarelli, A. Catalytic properties of ceria and CeO2-Containing materials. Catal. Rev.-Sci. Eng. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Yang, X.; Da, J.; Yu, H.; Wang, H. Characterization and performance evaluation of Ni-based catalysts with Ce promoter for methane and hydrocarbons steam reforming process. Fuel 2016, 179, 353–361. [Google Scholar] [CrossRef]
- Savuto, E.; Navarro, R.M.; Mota, N.; Di Carlo, A.; Bocci, E.; Carlini, M.; Fierro, J.L.G. Steam reforming of tar model compounds over Ni/Mayenite catalysts: Effect of Ce addition. Fuel 2018, 224, 676–686. [Google Scholar] [CrossRef]
- Gac, W.; Zawadzki, W.; Rotko, M.; Słowik, G.; Greluk, M. CO2 Methanation in the Presence of Ce-Promoted Alumina Supported Nickel Catalysts: H2S Deactivation Studies. Top. Catal. 2019, 62, 524–534. [Google Scholar] [CrossRef]
- Yeo, T.Y.; Ashok, J.; Kawi, S. Recent developments in sulphur-resilient catalytic systems for syngas production. Renew. Sustain. Energy Rev. 2019, 100, 52–70. [Google Scholar] [CrossRef]
- Nair, M.M.; Abanades, S. Tailoring Hybrid Nonstoichiometric Ceria Redox Cycle for Combined Solar Methane Reforming and Thermochemical Conversion of H2O/CO2. Energy Fuels 2016, 30, 6050–6058. [Google Scholar] [CrossRef]
- Arifin, D.; Weimer, A.W. Kinetics and mechanism of solar-thermochemical H2 and CO production by oxidation of reduced CeO2. Sol. Energy 2018, 160, 178–185. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, K.; Wang, H.; Tian, D.; Wang, Y.; Zhu, X.; Wei, Y.; Zheng, M.; Luo, Y. Designed oxygen carriers from macroporous LaFeO3 supported CeO2 for chemical-looping reforming of methane. Appl. Catal. B Environ. 2017, 202, 51–63. [Google Scholar] [CrossRef]
- Galvita, V.V.; Poelman, H.; Bliznuk, V.; Detavernier, C.; Marin, G.B. CeO2-Modified Fe2O3 for CO2 Utilization via Chemical Looping. Ind. Eng. Chem. Res. 2013, 52, 8416–8426. [Google Scholar] [CrossRef]
- Zaimes, G.G.; Hubler, B.J.; Wang, S.; Khanna, V. Environmental life cycle perspective on rare earth oxide production. ACS Sustain. Chem. Eng. 2015, 3, 237–244. [Google Scholar] [CrossRef]
- Haque, N.; Hughes, A.; Lim, S.; Vernon, C. Rare earth elements: Overview of mining, mineralogy, uses, sustainability and environmental impact. Resources 2014, 3, 614–635. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Z.; Wen, J.; Poeppelmeier, K.R.; Marks, L.D. Imaging the atomic surface structures of CeO2 nanoparticles. Nano Lett. 2014, 14, 191–196. [Google Scholar] [CrossRef]
- Paun, C.; Safonova, O.V.; Szlachetko, J.; Abdala, P.M.; Nachtegaal, M.; Sa, J.; Kleymenov, E.; Cervellino, A.; Krumeich, F.; Van Bokhoven, J.A. Polyhedral CeO2 nanoparticles: Size-dependent geometrical and electronic structure. J. Phys. Chem. C 2012, 116, 7312–7317. [Google Scholar] [CrossRef]
- Matei-Rutkovska, F.; Postole, G.; Rotaru, C.G.; Florea, M.; Pârvulescu, V.I.; Gelin, P. Synthesis of ceria nanopowders by microwave-assisted hydrothermal method for dry reforming of methane. Int. J. Hydrog. Energy 2016, 41, 2512–2525. [Google Scholar] [CrossRef]
- Laosiripojana, N.; Charojrochkul, S.; Kim-lohsoontorn, P.; Assabumrungrat, S. Role and advantages of H2S in catalytic steam reforming over nanoscale CeO2 -based catalysts. J. Catal. 2010, 276, 6–15. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, J.; Benfield, R.E.; Ding, Y.; Grandjean, D.; Zhang, Z.; Ju, X. Structure and Chemical Transformation in Cerium Oxide Nanoparticles Coated by Surfactant Cetyltrimethylammonium Bromide (CTAB): An X-ray Absorption Spectroscopic Study. J. Phys. Chem. B 2002, 106, 4569–4577. [Google Scholar] [CrossRef][Green Version]
- Zhang, F.; Chan, S.W.; Spanier, J.E.; Apak, E.; Jin, Q.; Robinson, R.D.; Herman, I.P. Cerium oxide nanoparticles: Size-selective formation and structure analysis. Appl. Phys. Lett. 2002, 80, 127–129. [Google Scholar] [CrossRef]
- Natile, M.M.; Boccaletti, G.; Glisenti, A. Properties and reactivity of nanostructured CeO2 powders: Comparison among two synthesis procedures. Chem. Mater. 2005, 17, 6272–6286. [Google Scholar] [CrossRef]
- Spanier, J.E.; Robinson, R.D.; Zhang, F.; Chan, S.; Herman, I.P. Size-dependent properties of CeO2-y nanoparticles as studied by Raman scattering. Phys. Rev. B 2001, 64, 245407. [Google Scholar] [CrossRef]
- Hailstone, R.K.; DiFrancesco, A.G.; Leong, J.G.; Allston, T.D.; Reed, K.J. A study of lattice expansion in CeO2 Nanoparticles by Transmission Electron Microscopy. J. Phys. Chem. C 2009, 113, 15155–15159. [Google Scholar] [CrossRef]
- Han, W.Q.; Wen, W.; Hanson, J.C.; Teng, X.; Marinkovic, N.; Rodriguez, J.A. One-dimensional ceria as catalyst for the low-temperature water-gas shift reaction. J. Phys. Chem. C 2009, 113, 21949–21955. [Google Scholar] [CrossRef]
- Zhang, J.; Kumagai, H.; Yamamura, K.; Ohara, S.; Takami, S.; Morikawa, A.; Shinjoh, H.; Kaneko, K.; Adschiri, T.; Suda, A. Extra-Low-Temperature Oxygen Storage Capacity of CeO2 Nanocrystals with Cubic Facets. Nano Lett. 2011, 11, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Aneggi, E.; Wiater, D.; de Leitenburg, C.; Llorca, J.; Trovarelli, A. Shape-Dependent Activity of Ceria in Soot Combustion. ACS Catal. 2014, 4, 172–181. [Google Scholar] [CrossRef]
- Liu, Z.; Sorrell, C.C.; Koshy, P.; Hart, J.N. DFT Study of Methanol Adsorption on Defect-Free CeO2 Low-Index Surfaces. ChemPhysChem 2019, 20, 2074–2081. [Google Scholar] [CrossRef]
- Branda, M.M.; Ferullo, R.M.; Causá, M.; Illas, F. Relative stabilities of low index and stepped CeO2 surfaces from hybrid and GGA + U implementations of density functional theory. J. Phys. Chem. C 2011, 115, 3716–3721. [Google Scholar] [CrossRef]
- Damyanova, S.; Bueno, J.M.C. Effect of CeO2 loading on the surface and catalytic behaviors of CeO2-Al2O3-supported Pt catalysts. Appl. Catal. A Gen. 2003, 253, 135–150. [Google Scholar] [CrossRef]
- Reddy, B.M.; Rao, K.N.; Reddy, G.K.; Khan, A.; Park, S.E. Structural characterization and oxidehydrogenation activity of CeO 2/Al2O3 and V2O5/ CeO2ZAl2O3 catalysts. J. Phys. Chem. C 2007, 111, 18751–18758. [Google Scholar] [CrossRef]
- Shyu, J.Z.; Weber, W.H.; Gandhi, H.S. Surface characterization of alumina-supported ceria. J. Phys. Chem. 1988, 92, 4964–4970. [Google Scholar] [CrossRef]
- Beniya, A.; Isomura, N.; Hirata, H.; Watanabe, Y. Morphology and chemical states of size-selected Pt n clusters on an aluminium oxide film on NiAl(110). Phys. Chem. Chem. Phys. 2014, 16, 26485–26492. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ma, Y.; Li, J.; Li, Q.; Hu, X.; Ye, Z.; Wu, X.Y.; Buckley, C.E.; Dong, D. CeO2-promotion of NiAl2O4 reduction via CeAlO3 formation for efficient methane reforming. J. Energy Inst. 2020, 93, 991–999. [Google Scholar] [CrossRef]
- Nataliya Bochvar, N.; Liberov, Y.; Fabrichnaya, O.; MSIT®. Ce-Mg-O Ternary Phase Diagram Evaluation. Available online: https://materials.springer.com/msi/docs/sm_msi_r_10_012253_02 (accessed on 26 November 2020).
- Tinoco, M.; Sanchez, J.J.; Yeste, M.P.; Lopez-Haro, M.; Trasobares, S.; Hungria, A.B.; Bayle-guillemaud, P.; Blanco, G.; Pintado, J.M.; Calvino, J.J. Low-Lanthanide-Content CeO2/MgO Catalysts with Outstandingly Stable Oxygen Storage Capacities: An In-Depth Structural Characterization by Advanced STEM Techniques. ChemCatChem 2015, 7, 3763–3778. [Google Scholar] [CrossRef]
- Pérez Casero, R.; Gómez San Román, R.; Perrière, J.; Laurent, A.; Seiler, W.; Gergaud, P.; Keller, D. Epitaxial growth of CeO2 on MgO by pulsed laser deposition. Appl. Surf. Sci. 1997, 109–110, 341–344. [Google Scholar] [CrossRef]
- Paunović, V.; Zichittella, G.; Mitchell, S.; Hauert, R.; Pérez-Ramírez, J. Selective Methane Oxybromination over Nanostructured Ceria Catalysts. ACS Catal. 2018, 8, 291–303. [Google Scholar] [CrossRef]
- Plewa, J.; Steindor, J. Kinetics of reduction of magnesium sulfate by carbon oxide. J. Therm. Anal. 1987, 32, 1809–1820. [Google Scholar] [CrossRef]
- Taira, K. Dry reforming reactions of CH4 over CeO2/MgO catalysts at high concentrations of H2S. 2021, J. Cat. Under revision. J. Cat. Under revision.. 2021. [Google Scholar]
- Ferreira, V.J.; Tavares, P.; Figueiredo, J.L.; Faria, J.L. Effect of Mg, Ca, and Sr on CeO2 based catalysts for the oxidative coupling of methane: Investigation on the oxygen species responsible for catalytic performance. Ind. Eng. Chem. Res. 2012, 51, 10535–10541. [Google Scholar] [CrossRef]
- Geysermans, P.; Finocchi, F.; Goniakowski, J.; Hacquart, R.; Jupille, J. Combination of (100), (110) and (111) facets in MgO crystals shapes from dry to wet environment. Phys. Chem. Chem. Phys. 2009, 11, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.A.; Jones, C.F.; Watson, G.S.; Crossley, A.; Johnston, C. Surface modification of MgO substrates from aqueous exposure: An atomic force microscopy study. Thin Solid Film. 1997, 292, 96–102. [Google Scholar] [CrossRef]
- Eastman, P.F.; Culter, I.B. Effect of Water Vapor on Initial Sintering of Magnesia. J. Am. Ceram. Soc. 1966, 49, 526–530. [Google Scholar] [CrossRef]
- Ito, T.; Fujita, M.; Watanabe, M.; Tokuda, T. The initial sintering of alkaline earth oxides in water vapor and hydrogen gas. Bull. Cemical Soc. Japan 1981, 54, 2412–2419. [Google Scholar] [CrossRef]
- Green, J. Calcination of precipitated Mg(OH)2 to active MgO in the production of refractory and chemical grade MgO. J. Mater. Sci. 1983, 18, 637–651. [Google Scholar] [CrossRef]
- Peng, C.; Zhang, Z. Nitrate–citrate combustion synthesis of Ce1−xGdxO2−x/2 powder and its characterization. Ceram. Int. 2007, 33, 1133–1136. [Google Scholar] [CrossRef]
- Taira, K.; Nakao, K.; Suzuki, K.; Einaga, H. SOx tolerant Pt/TiO2 catalysts for CO oxidation and the effect of TiO2 supports on catalytic activity. Environ. Sci. Technol. 2016, 50, 9773–9780. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, P.; Hamnett, A.; Orchard, A.F.; Thornton, G. Satellite structure in the X-ray photoelectron spectra of some binary and mixed oxides of lanthanum and cerium. J. Chem. Soc. Dalt. Trans. 1976, 1686. [Google Scholar] [CrossRef]
- Romeo, M.; Bak, K.; El Fallah, J.; Le Normand, F.; Hilaire, L. XPS Study of the reduction of cerium dioxide. Surf. Interface Anal. 1993, 20, 508–512. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology. NIST X-Ray Photoelectron Spectroscopy Database; NIST Standard Reference Database Number 20; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2000. [Google Scholar] [CrossRef]
- Della Mea, G.B.; Matte, L.P.; Thill, A.S.; Lobato, F.O.; Benvenutti, E.V.; Arenas, L.T.; Jürgensen, A.; Hergenröder, R.; Poletto, F.; Bernardi, F. Tuning the oxygen vacancy population of cerium oxide (CeO2−x, 0 <x <0.5) nanoparticles. Appl. Surf. Sci. 2017, 422, 1102–1112. [Google Scholar] [CrossRef]
- Taira, K.; Sugiyama, T.; Einaga, H.; Nakao, K.; Suzuki, K. Promoting effect of 2000 ppm H2S on the dry reforming reaction of CH4 over pure CeO2, and in situ observation of the behavior of sulfur during the reaction. J. Catal. 2020, 389, 611–622. [Google Scholar] [CrossRef]
- Beruto, D.; Botter, R.; Searcy, A.W. H2O-Catalyzed Sintering of ~2-nm-Cross-Section Particles of MgO. J. Am. Ceram. Soc. 1987, 70, 155–159. [Google Scholar] [CrossRef]
- Cadigan, C.A.; Corpuz, A.R.; Lin, F.; Caskey, C.M.; Finch, K.B.H.; Wang, X.; Richards, R.M. Nanoscale (111) faceted rock-salt metal oxides in catalysis. Catal. Sci. Technol. 2013, 3, 900–911. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 3263, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Sangwal, K. Mechanism of dissolution of MgO crystals in acids. J. Mater. Sci. 1980, 15, 237–246. [Google Scholar] [CrossRef]
- Filley, J.; Ibrahim, M.A.; Nimlos, M.R.; Watt, A.S.; Blake, D.M. Magnesium and calcium chelation by a bis-spiropyran. J. Photochem. Photobiol. A Chem. 1998, 117, 193–198. [Google Scholar] [CrossRef]
- Hu, Y.; Lee, B.; Bell, C.; Jun, Y.-S. Environmentally Abundant Anions Influence the Nucleation, Growth, Ostwald Ripening, and Aggregation of Hydrous Fe(III) Oxides. Langmuir 2012, 28, 7737–7746. [Google Scholar] [CrossRef]
- Karen, P.; Kjekshus, A.; Huang, Q.; Karen, V.L. The crystal structure of magnesium dicarbide. J. Alloys Compd. 1999, 282, 72–75. [Google Scholar] [CrossRef]
- McBride, J.R.; Hass, K.C.; Poindexter, B.D.; Weber, W.H. Raman and X-ray studies of Ce1-xRExO2-y, where RE=La, Pr, Nd, Eu, Gd, and Tb. J. Appl. Phys. 1994, 76, 2435–2441. [Google Scholar] [CrossRef]
- Holgado, J.P.; Alvarez, R.; Munuera, G. Study of CeO2 XPS spectra by factor analysis: Reduction of CeO2. Appl. Surf. Sci. 2000, 161, 301–315. [Google Scholar] [CrossRef]
- Galvita, V.V.; Poelman, H.; Rampelberg, G.; De Schutter, B.; Detavernier, C.; Marin, G.B. Structural and kinetic study of the reduction of CuO-CeO2/Al2O3 by time-resolved X-ray diffraction. Catal. Lett. 2012, 142, 959–968. [Google Scholar] [CrossRef]
- Perrichon, V.; Laachir, A.; Bergeret, G.; Fréty, R.; Tournayan, L.; Touret, O. Reduction of cerias with different textures by hydrogen and their reoxidation by oxygen. J. Chem. Soc. Faraday Trans. 1994, 90, 773–781. [Google Scholar] [CrossRef]
- Stan, M.; Zhu, Y.T.; Jiang, H.; Butt, D.P. Kinetics of oxygen removal from ceria. J. Appl. Phys. 2004, 95, 3358–3361. [Google Scholar] [CrossRef]
- Al-Madfaa, H.A.; Khader, M.M. Reduction kinetics of ceria surface by hydrogen. Mater. Chem. Phys. 2004, 86, 180–188. [Google Scholar] [CrossRef]
- Goguet, A.; Meunier, F.C.; Tibiletti, D.; Breen, J.P.; Burch, R. Spectrokinetic Investigation of Reverse Water-Gas-Shift Reaction Intermediates over a Pt/CeO2 Catalyst. J. Phys. Chem. B 2004, 108, 20240–20246. [Google Scholar] [CrossRef]
- Warren, K.J.; Scheffe, J.R. Role of Surface Oxygen Vacancy Concentration on the Dissociation of Methane over Nonstoichiometric Ceria. J. Phys. Chem. C 2019, 123, 13208–13218. [Google Scholar] [CrossRef]
- Kumar, G.; Lau, S.L.J.; Krcha, M.D.; Janik, M.J. Correlation of Methane Activation and Oxide Catalyst Reducibility and Its Implications for Oxidative Coupling. ACS Catal. 2016, 6, 1812–1821. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nominal CeO2 Content (wt%) | CeO2 Content (ICP-OES) * (wt%) | BET Area (m2/g) | CeO2 Diameter (nm) ** | CeO2 Area (m2/g) *** | |
|---|---|---|---|---|---|
| MgO(air) | 0 | 0 | 33.1 | - | - |
| 0.01-CeO2/MgO(air) | 4.1 | 4.20 | 30.4 | 5.3 ± 1.3 | 3.8 |
| 0.05-CeO2/MgO(air) | 18.3 | 18.31 | 27.9 | 6.9 ± 2.0 | 12.5 |
| MgO(dry) | 0 | 0 | 97.7 | - | - |
| 0.01-CeO2/MgO(dry) | 4.1 | 4.15 | 58.9 | 2.6 ± 1.1 | 6.2 |
| 0.05-CeO2/MgO(dry) | 18.3 | 18.33 | 38.5 | 4.5 ± 1.9 | 15.8 |
| CeO2 | 100.0 | 100.0 | 13.5 | 61.6 | 13.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taira, K.; Murao, R. High Dispersion of CeO2 on CeO2/MgO Prepared under Dry Conditions and Its Improved Redox Properties. Energies 2021, 14, 7922. https://doi.org/10.3390/en14237922
Taira K, Murao R. High Dispersion of CeO2 on CeO2/MgO Prepared under Dry Conditions and Its Improved Redox Properties. Energies. 2021; 14(23):7922. https://doi.org/10.3390/en14237922
Chicago/Turabian StyleTaira, Kenji, and Reiko Murao. 2021. "High Dispersion of CeO2 on CeO2/MgO Prepared under Dry Conditions and Its Improved Redox Properties" Energies 14, no. 23: 7922. https://doi.org/10.3390/en14237922
APA StyleTaira, K., & Murao, R. (2021). High Dispersion of CeO2 on CeO2/MgO Prepared under Dry Conditions and Its Improved Redox Properties. Energies, 14(23), 7922. https://doi.org/10.3390/en14237922