
Defect Properties and Lithium Incorporation in Li2ZrO3


 and
and 
Abstract
:
1. Introduction
2. Computational Methods
3. Results and Discussion
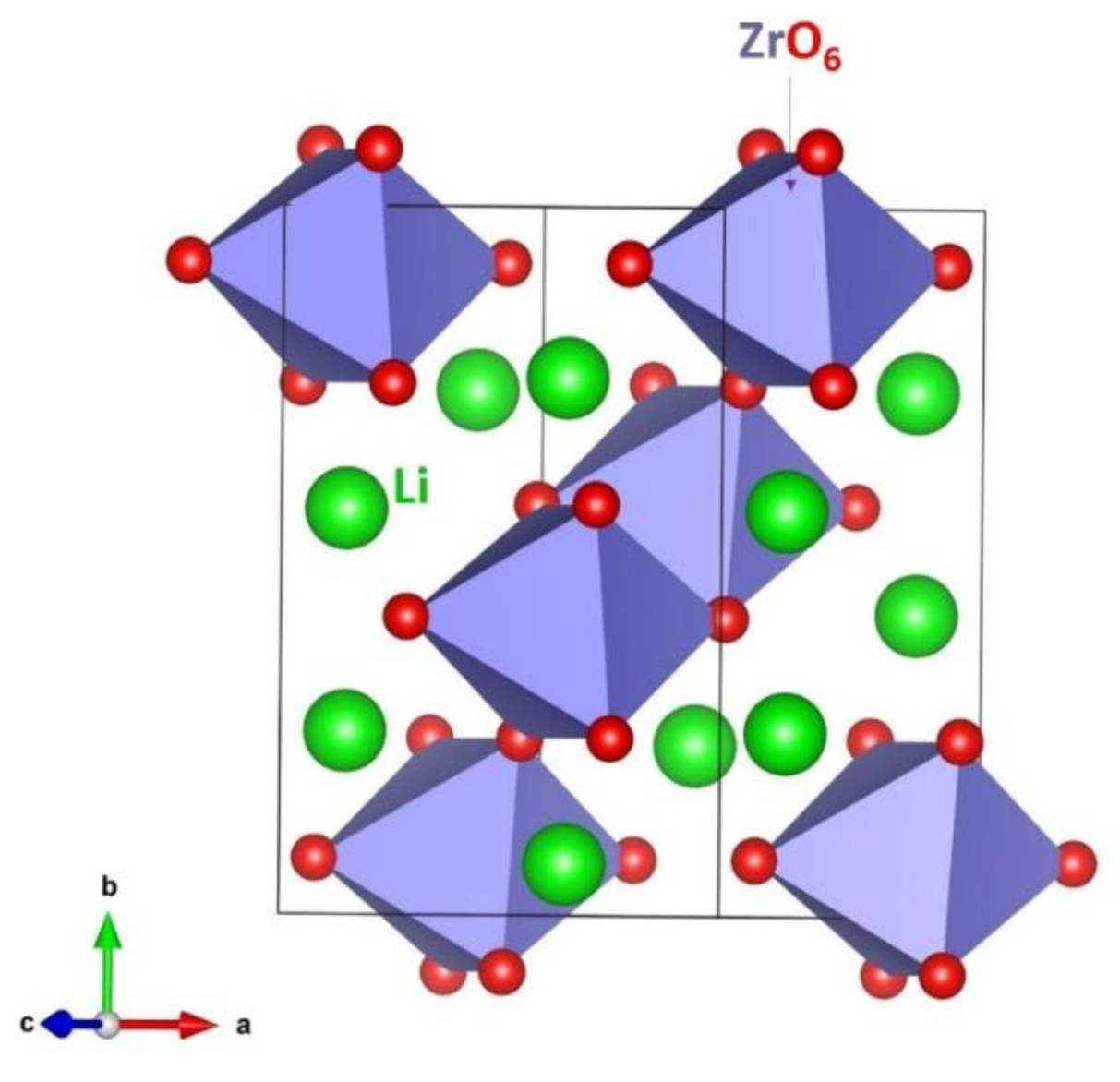
3.1. Structure of LZO
3.2. Defect Energetics
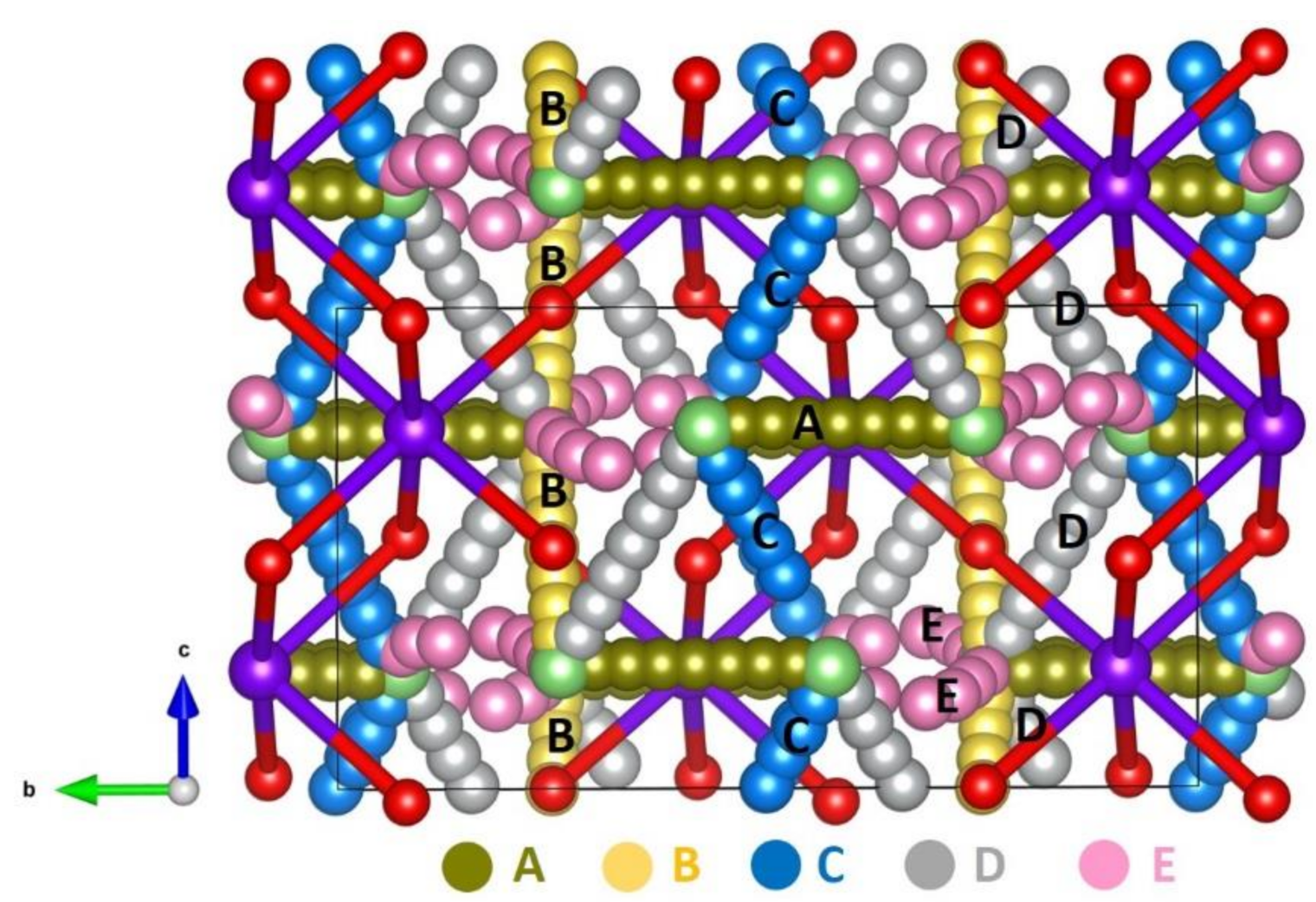
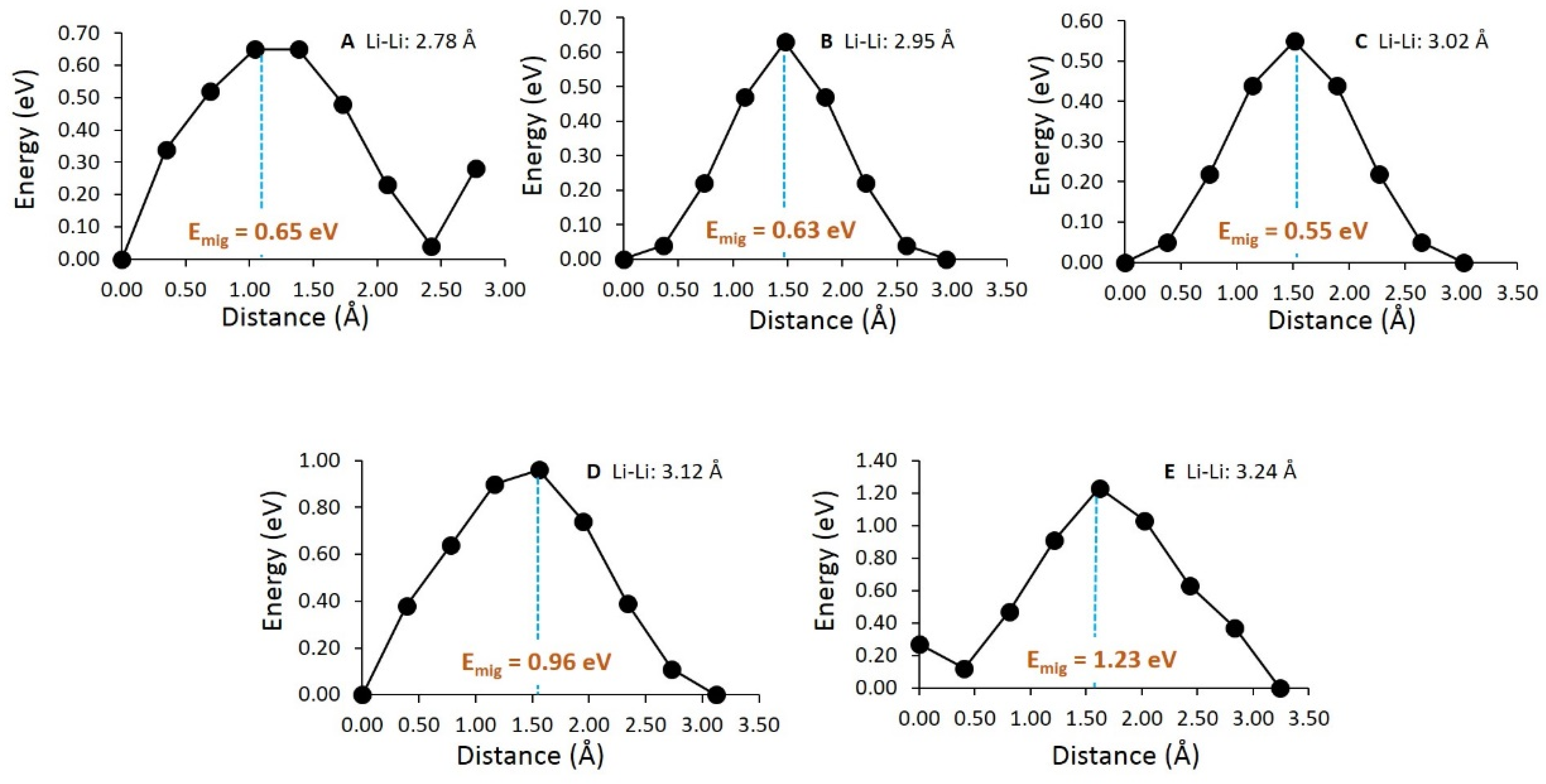
3.3. Diffusion of Li-Ions
3.4. Solution of Dopants
3.4.1. Isovalent Dopants
3.4.2. Aliovalent Dopants
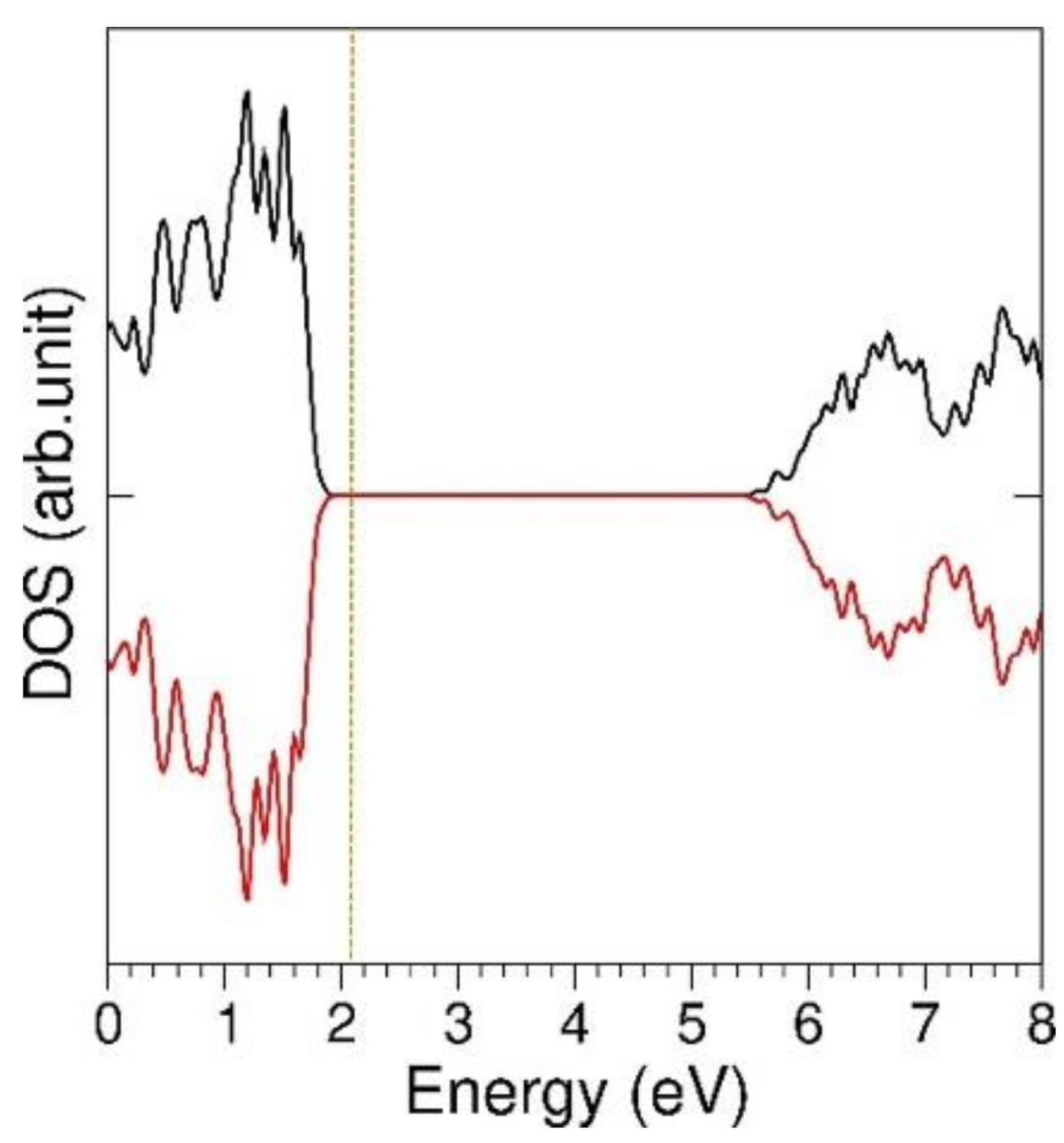
3.5. Li Incorporation in LZO
4. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, Y.; Zhao, Y.; Duan, H.; Huang, J. Electrochemical performance and lithium-ion insertion/extraction mechanism studies of the novel Li2ZrO3 anode materials. Electrochim. Acta 2015, 161, 219–225. [Google Scholar] [CrossRef]
- Thackeray, M.M.; Johnson, C.S.; Kim, J.S.; Lauzze, K.C.; Vaughey, J.T.; Dietz, N.; Abraham, D.; Hackney, S.; Zeltner, W.; Anderson, M. ZrO2- and Li2ZrO3-stabilized spinel and layered electrodes for lithium batteries. Electrochem. Commun. 2003, 5, 752–758. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, S.; Wu, Q.; Wan, N.; Pan, D.; Bai, Y. Improved electrochemical and thermal performances of layered Li[Li0.2Ni0.17Co0.07Mn0.56]O2 via Li2ZrO3 surface modification. J. Power Sources 2015, 282, 378–384. [Google Scholar] [CrossRef]
- Kanazawa, T.; Nishikawa, M.; Yamasaki, H.; Katayama, K.; Kashimura, H.; Hanada, T.; Fukada, S. Study on Tritium Release Behavior from Li2ZrO3. Fusion Sci. Technol. 2011, 60, 1167–1170. [Google Scholar] [CrossRef]
- Hollenberg, G.W. Fast neutron irradiation results on Li2O, Li4SiO4, Li2ZrO3 and LiAlO2. J. Nucl. Mater. 1984, 123, 896–900. [Google Scholar] [CrossRef]
- Kawamura, Y.; Enoeda, M.; Okuno, K. Isotope exchange reaction in Li2ZrO3 packed bed. Fusion Eng. Des. 1998, 39, 713–721. [Google Scholar] [CrossRef]
- Wang, C.; Dou, B.; Song, Y.; Chen, H.; Xu, Y.; Xie, B. High Temperature CO2 Sorption on Li2ZrO3 Based Sorbents. Ind. Eng. Chem. Res. 2014, 53, 12744–12752. [Google Scholar] [CrossRef]
- Peltzer, D.; Mùnera, J.; Cornaglia, L.; Strumendo, M. Characterization of potassium doped Li2ZrO3 based CO2 sorbents: Stability properties and CO2 desorption kinetics. Chem. Eng. J. 2018, 336, 1–11. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, Y.; Pfeiffer, H.; Louis, B.; Sun, L.; O’Hare, D.; Wang, Q. Recent advances in lithium containing ceramic based sorbents for high-temperature CO2 capture. J. Mater. Chem. A 2019, 7, 7962–8005. [Google Scholar] [CrossRef]
- Baklanova, Y.V.; Arapova, I.Y.; Buzlukov, A.L.; Gerashenko, A.P.; Verkhovskii, S.V.; Mikhalev, K.N.; Denisova, T.; Shein, I.; Maksimova, L. Localization of vacancies and mobility of lithium ions in Li2ZrO3 as obtained by 6,7 Li NMR. J. Solid State Chem. 2013, 208, 43–49. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, Y.; Lu, Z.; Wang, H.; Sun, D.; Yang, G. The preparation and role of Li2ZrO3 surface coating LiNi0.5Co0.2Mn0.3O2 as cathode for lithium-ion batteries. Appl. Surf. Sci. 2016, 361, 150–156. [Google Scholar] [CrossRef]
- Ferreira, A.R.; Reuter, K.; Scheurer, C. DFT simulations of 7 Li solid state NMR spectral parameters and Li+ ion migration barriers in Li2ZrO3. RSC Adv. 2016, 6, 41015–41024. [Google Scholar] [CrossRef]
- Alexopoulos, K.; Varotsos, P. Calculation of diffusion coefficients at any temperature and pressure from a single measurement. II. Heterodiffusion. Phys. Rev. B 1981, 24, 3606–3609. [Google Scholar] [CrossRef]
- Varotsos, P.; Alexopoulos, K. Thermodynamics of Point Defects and their Relation with the Bulk Properties; Elsevier: Amsterdam, North-Holland, The Netherlands, 1986. [Google Scholar]
- Rupasov, D.; Chroneos, A.; Parfitt, D.; Kilner, J.A.; Grimes, R.W.; Istomin, S.Y.; Antipov, E.V. Oxygen diffusion in Sr0.75Y0.25CoO2.625: A molecular dynamics study. Phys. Rev. B 2009, 79, 172102. [Google Scholar] [CrossRef] [Green Version]
- Kordatos, A.; Christopoulos, S.R.G.; Kelaidis, N.; Chroneos, A. Defect processes in Li2ZrO3: Insights from atomistic modelling. J. Mater. Sci. Mater. Electron. 2017, 28, 11789–11793. [Google Scholar] [CrossRef] [Green Version]
- Gale, J.D. GULP: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef]
- Fisher, C.A.J.; Kuganathan, N.; Islam, M.S. Defect chemistry and lithium-ion migration in polymorphs of the cathode material Li2MnSiO4. J. Mater. Chem. A 2013, 1, 4207–4214. [Google Scholar] [CrossRef] [Green Version]
- Minervini, L.; Grimes, R.W.; Sickafus, K.E. Disorder in Pyrochlore Oxides. J. Am. Ceram. Soc. 2000, 83, 1873–1878. [Google Scholar] [CrossRef]
- Cherry, M.; Islam, M.S.; Catlow, C.R.A. Oxygen Ion Migration in Perovskite-Type Oxides. J. Solid State Chem. 1995, 118, 125–132. [Google Scholar] [CrossRef]
- Mott, N.F.; Littleton, M.J. Conduction in polar crystals. I. Electrolytic conduction in solid salts. Trans. Faraday Soc. 1938, 34, 485–499. [Google Scholar] [CrossRef]
- Kuganathan, N.; Ganeshalingam, S.; Chroneos, A. Defects, Diffusion, and Dopants in Li2Ti6O13: Atomistic Simulation Study. Materials 2019, 12, 2851. [Google Scholar] [CrossRef] [Green Version]
- Kuganathan, N.; Kordatos, A.; Anurakavan, S.; Iyngaran, P.; Chroneos, A. Li3SbO4 lithium-ion battery material: Defects, lithium ion diffusion and tetravalent dopants. Mater. Chem. Phys. 2019, 225, 34–41. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Kelaidis, N.; Chroneos, A. Defects, Lithium Mobility and Tetravalent Dopants in the Li3NbO4 Cathode Material. Sci. Rep. 2019, 9, 2192. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. Numerical Recipes in C: The Art of Scientific Computing, 2nd ed.; Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Heiba, Z.K.; El-Sayed, K. Structural and anisotropic thermal expansion correlation of Li2ZrO3 at different temperatures. J. Appl. Crystallogr. 2002, 35, 634–636. [Google Scholar] [CrossRef] [Green Version]
- Oberkampf, W.L.; Barone, M.F. Measures of agreement between computation and experiment: Validation metrics. J. Comput. Phys. 2006, 217, 5–36. [Google Scholar] [CrossRef] [Green Version]
- Kröger, F.A.; Vink, H.J. Relations between the Concentrations of Imperfections in Crystalline Solids. In Solid State Physics; Seitz, F., Turnbull, D., Eds.; Academic Press: New York, NY, USA, 1956; Volume 3, pp. 307–435. [Google Scholar]
- Armstrong, A.R.; Kuganathan, N.; Islam, M.S.; Bruce, P.G. Structure and Lithium Transport Pathways in Li2FeSiO4 Cathodes for Lithium Batteries. J. Am. Chem. Soc. 2011, 133, 13031–13035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nytén, A.; Kamali, S.; Häggström, L.; Gustafsson, T.; Thomas, J.O. The lithium extraction/insertion mechanism in Li2FeSiO4. J. Mater. Chem. 2006, 16, 2266–2272. [Google Scholar] [CrossRef]
- Politaev, V.V.; Petrenko, A.A.; Nalbandyan, V.B.; Medvedev, B.S.; Shvetsova, E.S. Crystal structure, phase relations and electrochemical properties of monoclinic Li2MnSiO4. J. Solid State Chem. 2007, 180, 1045–1050. [Google Scholar] [CrossRef]
- Pizzagalli, L. Classical Atomistic Simulations in Materials Sciences: An Introduction. Mater. Sci. 2004, 10, 125. [Google Scholar]
- Islam, M.S.; Fisher, C.A.J. Lithium and sodium battery cathode materials: Computational insights into voltage, diffusion and nanostructural properties. Chem. Soc. Rev. 2014, 43, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Kuganathan, N.; Chroneos, A. Na3V(PO4)2 cathode material for Na ion batteries: Defects, dopants and Na diffusion. Solid State Ion. 2019, 336, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Fisher, C.A.J.; Hart Prieto, V.M.; Islam, M.S. Lithium Battery Materials LiMPO4 (M = Mn, Fe, Co, and Ni): Insights into Defect Association, Transport Mechanisms, and Doping Behavior. Chem. Mater. 2008, 20, 5907–5915. [Google Scholar] [CrossRef]
- Gladysz, G.M.; Chawla, K.K. 2—Intrinsic voids in crystalline materials: Ideal materials and real materials. In Voids in Materials, 2nd ed.; Gladysz, G.M., Chawla, K.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 17–39. [Google Scholar]
- Duan, Y. Electronic structural and electrochemical properties of lithium zirconates and their capabilities of CO2 capture: A first-principles density-functional theory and phonon dynamics approach. J. Renew. Sustain. Energy 2011, 3, 013102. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]



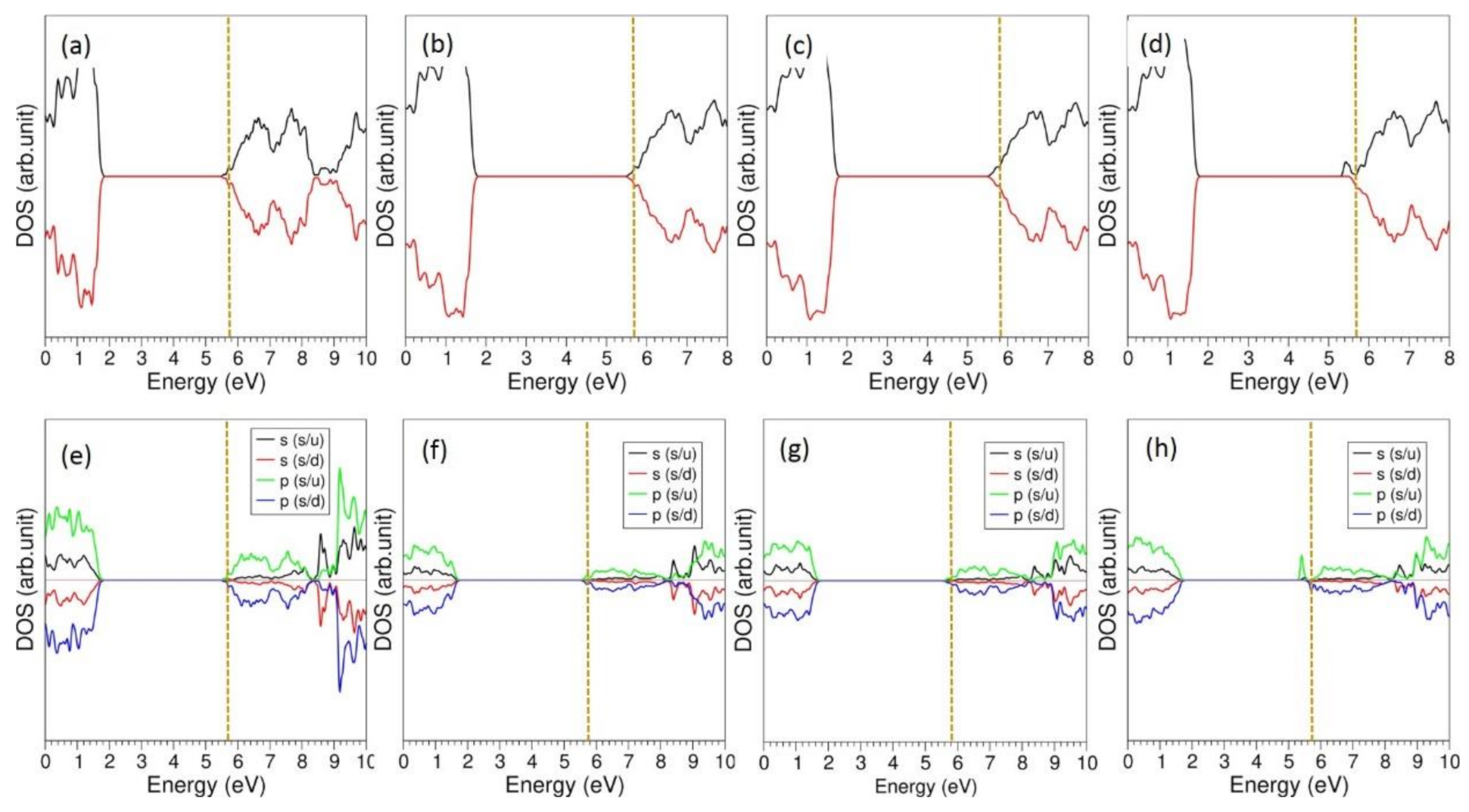

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction | A/eV | ρ/Å | C/eV·Å6 | Y/e | K/eV·Å–2 |
|---|---|---|---|---|---|
| Li+ ‒ O2− | 632.1018 | 0.2906 | 0.00 | 1.00 | 99,999 |
| Zr4+ ‒ O2− | 1502.11 | 0.3477 | 5.10 | 1.35 | 169.617 |
| O2− ‒ O2− | 22,764.0 | 0.1490 | 43.0 | −2.80 | 31.0 |
| Parameter | Calculated | Experiment [31] | |∆|(%) | ||
|---|---|---|---|---|---|
| Force Field | DFT | Force Field | DFT | ||
| a (Å) | 5.5912 | 5.3411 | 5.4089 | 3.37 | 1.25 |
| b (Å) | 8.8458 | 9.0737 | 9.0309 | 2.05 | 0.47 |
| c (Å) | 5.4787 | 5.3874 | 5.4144 | 1.19 | 0.50 |
| α = γ (°) | 90.00 | 90.00 | 90.00 | 0.00 | 0.00 |
| β (°) | 115.66 | 111.38 | 112.50 | 2.81 | 0.99 |
| V (Å3) | 244.25 | 243.12 | 244.35 | 0.04 | 0.50 |
| Defect Process | Equation Number | Defect Energy (eV) | Defect Energy (eV)/Defect |
|---|---|---|---|
| Li Frenkel | (1) | 2.18 | 1.09 |
| Zr Frenkel | (2) | 13.64 | 6.82 |
| O Frenkel | (3) | 6.46 | 3.23 |
| Schottky | (4) | 14.6 | 2.45 |
| Li2O Schottky | (5) | 6.74 | 2.25 |
| ZrO2 Schottky | (6) | 9.97 | 3.32 |
| Li-Zr anti-site (isolated) | (7) | 5.76 | 2.88 |
| Li-Zr anti-site (cluster) | (8) | 2.92 | 1.46 |
| Diffusion Path | Li-Li Separation (Å) | Activation Energy (eV) |
|---|---|---|
| A | 2.78 | 0.65 |
| B | 2.95 | 0.63 |
| C | 3.02 | 0.55 |
| D | 3.12 | 0.96 |
| E | 3.24 | 1.23 |
| Reaction | Incorporation Energy (eV) | Bader Charge (|e|) | ∆V (%) |
|---|---|---|---|
| Li + Li2ZrO3 → Li.Li2ZrO3 | −1.15 eV (0.52 eV) | +1.00 | 1.21 |
| Li + Li.Li2ZrO3 → 2Li.Li2ZrO3 | −1.12 eV (0.55 eV) | +1.00 (2) | 2.16 |
| Li + 2Li.Li2ZrO3 → 3Li.Li2ZrO3 | −0.60 eV (1.07 eV) | +1.00 (3) | 2.37 |
| Li + 3Li.Li2ZrO3 → 4Li.Li2ZrO3 | −0.85 eV (0.82 eV) | +1.00 (4) | 2.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rex, K.A.; Iyngaran, P.; Kuganathan, N.; Chroneos, A. Defect Properties and Lithium Incorporation in Li2ZrO3. Energies 2021, 14, 3963. https://doi.org/10.3390/en14133963
Rex KA, Iyngaran P, Kuganathan N, Chroneos A. Defect Properties and Lithium Incorporation in Li2ZrO3. Energies. 2021; 14(13):3963. https://doi.org/10.3390/en14133963
Chicago/Turabian StyleRex, Kobiny Antony, Poobalasuntharam Iyngaran, Navaratnarajah Kuganathan, and Alexander Chroneos. 2021. "Defect Properties and Lithium Incorporation in Li2ZrO3" Energies 14, no. 13: 3963. https://doi.org/10.3390/en14133963
APA StyleRex, K. A., Iyngaran, P., Kuganathan, N., & Chroneos, A. (2021). Defect Properties and Lithium Incorporation in Li2ZrO3. Energies, 14(13), 3963. https://doi.org/10.3390/en14133963