Modelling Carbon Corrosion during a PEMFC Startup: Simulation of Mitigation Strategies


Abstract
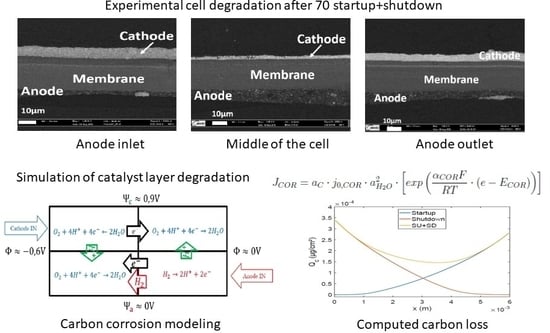
1. Introduction
- controlling the cathode electrode potential by using a current leak through a dummy load consisting of a parallel resistance [24];
- changing the anodic rib/channel design in order to slow down the kinetic of the degradation of the cathode facing the rib. To our knowledge, no simulation studies were performed so far on the design of the bipolar plate as a way to mitigate the carbon corrosion during startup/shutdown.
2. Dynamic Model of Carbon Support Corrosion during Startup
3. Simulation of the Transient Potentials and the Resulting Carbon Support Corrosion during Startup
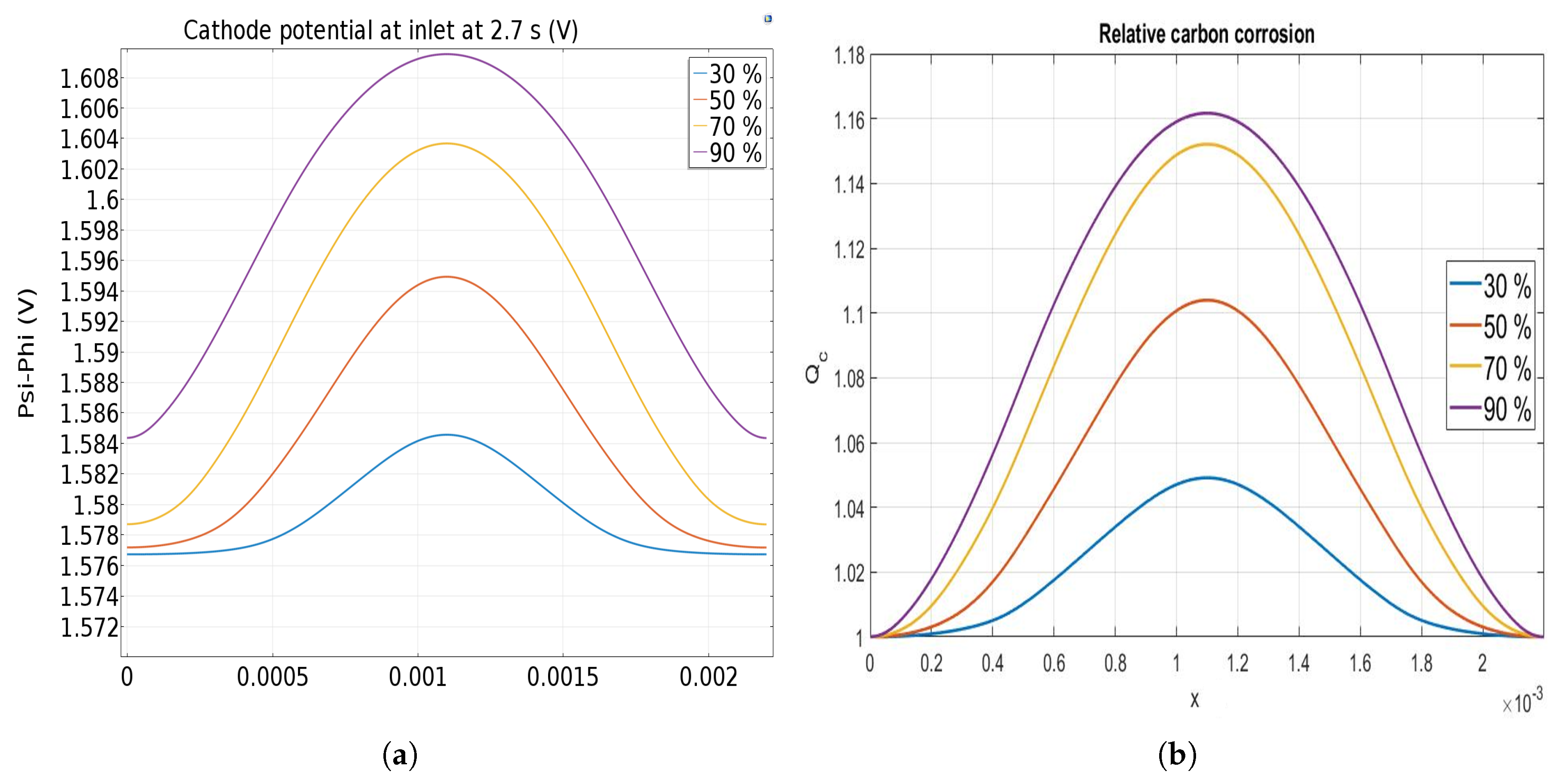
3.1. Along the Channel
3.2. Rib/Channel
4. Mitigation Strategies Simulation
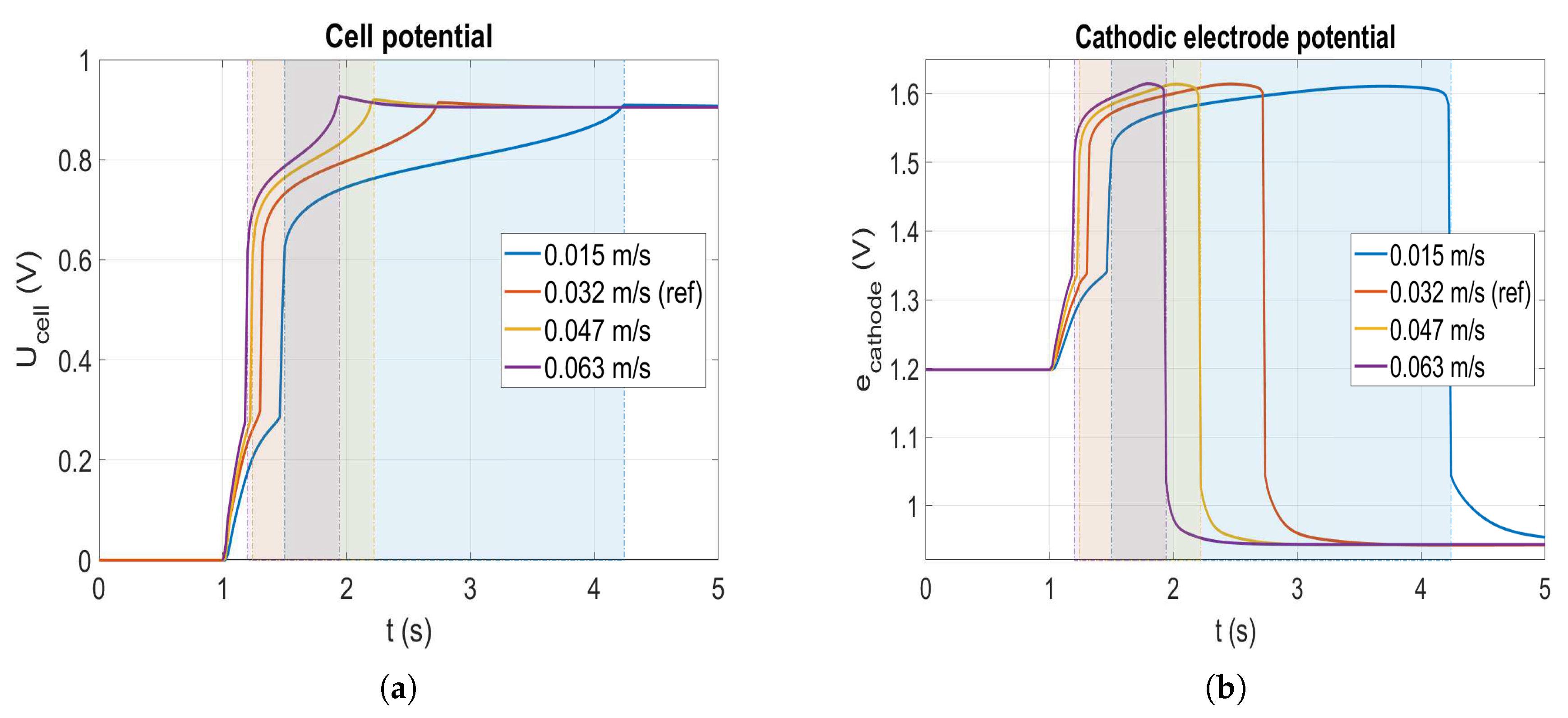
4.1. Gas Flow Velocity
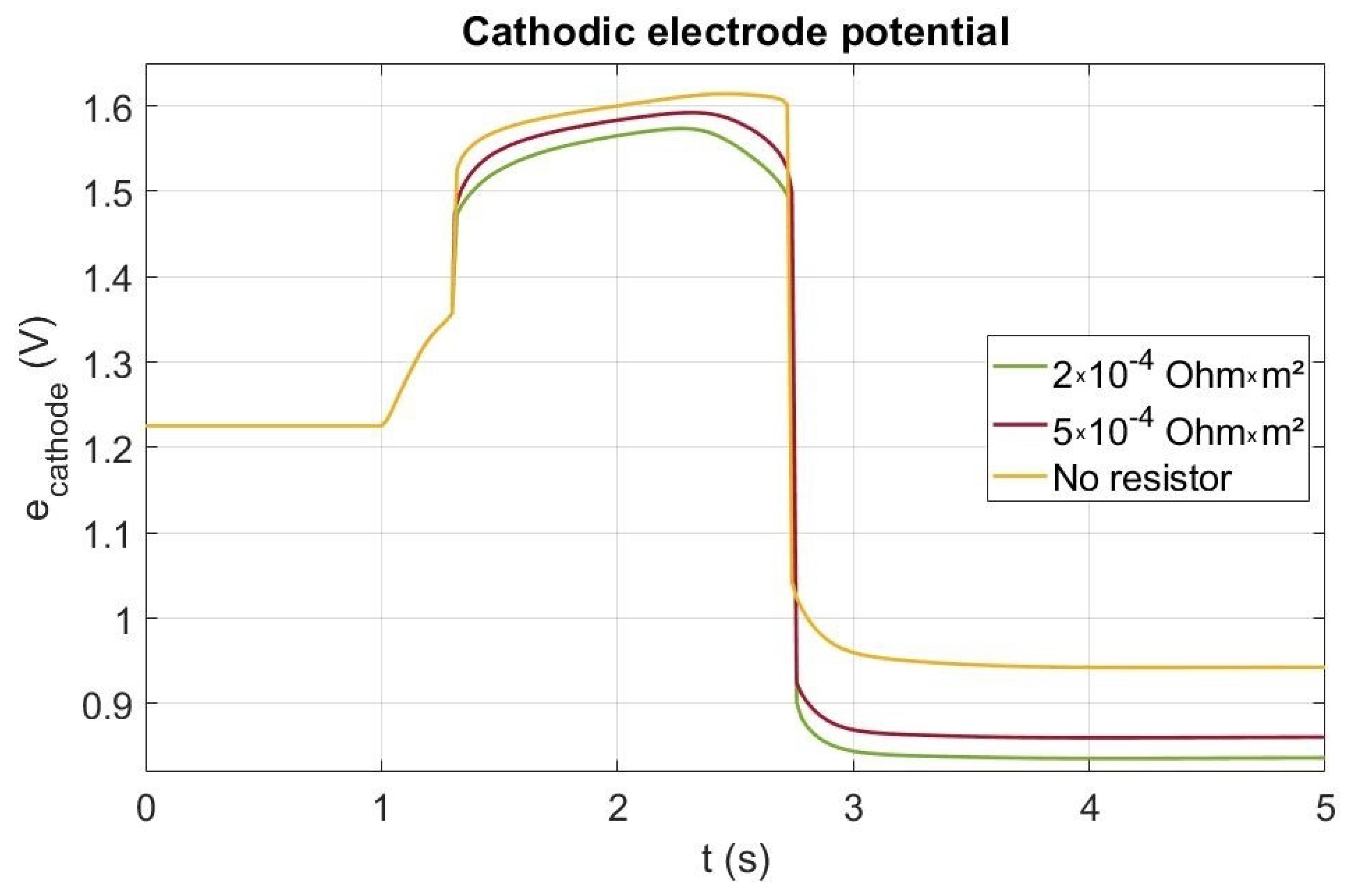
4.2. Startup with a Current Leak
4.3. Impact of Rib-Channel Design
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ionic potential | |
| Electronic potential | |
| Stoichiometry coefficient of the specie | |
| Standard molar enthalpy | |
| Standard molar entropy | |
| Enthalpy of formation of the activated complex (in the way of oxidation) | |
| Entropy of formation of the activated complex (in the way of oxidation) | |
| GDL | Gas diffusion layer |
| MPL | Micro-porous layer |
| CL | Catalyst layer |
| MB | Membrane |
| BP | Bipolar plates |
| ORR | Oxygen Reduction Reaction |
| OER | Oxygen Evolution Reaction |
| HER | Hydrogen Evolution Reaction |
| HOR | Hydrogen Oxidation Reaction |
| COR | Carbon Oxidation Reaction |
| Molar mass of specie | |
| e | Electrode potential |
| Constant reaction rate | |
| F | Faraday constant |
| n | Number of electrons or number of moles |
| Activity of specie | |
| h | Planck constant |
| the Boltzmann constant | |
| Avogadro constant | |
| Carbon specific surface | |
| Current density of the reaction | |
| Exchange current density |
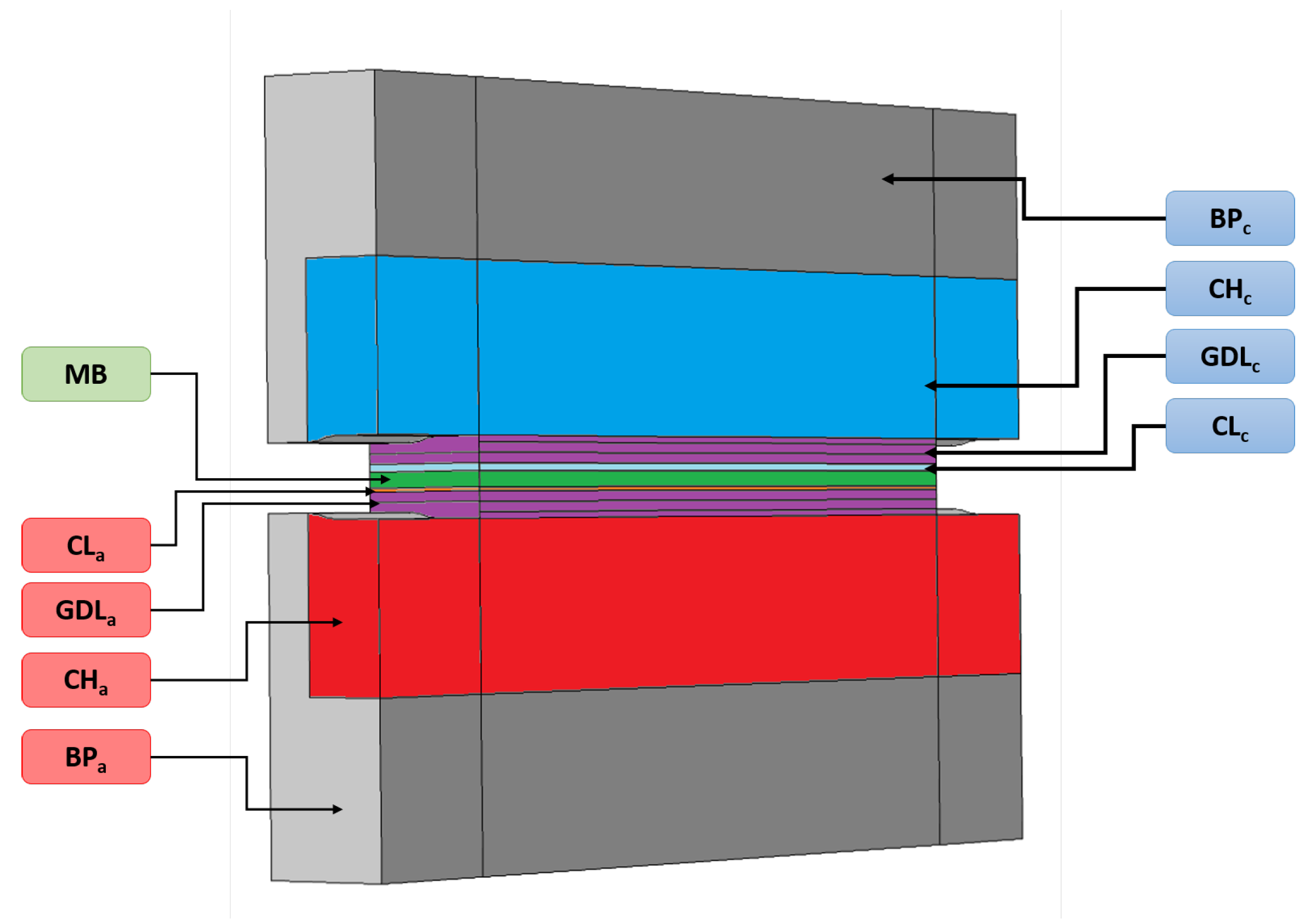
Appendix A. Summary of the Physics Solved in the Performance Model
References
- FCH2JU. 2018 Annual Work Plan and Budget; Technical Report; FCH2JU: Brussels, Belgium, 2018. [Google Scholar]
- Barbir, F. PEM Fuel Cells (Second Edition); Academic Press, Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar] [CrossRef]
- Shen, Q.; Hou, M.; Liang, D.; Zhou, Z.; Li, X.; Shao, Z.; Yi, B. Study on the processes of start-up and shutdown in proton exchange membrane fuel cells. J. Power Sources 2009, 189, 1114–1119. [Google Scholar] [CrossRef]
- Reiser, C.A.; Bregoli, L.; Patterson, T.W.; Yi, J.S.; Yang, J.D.; Perry, M.L.; Jarvi, T.D. A Reverse-Current Decay Mechanism for Fuel Cells. Electrochem. Solid-State Lett. 2005, 8, A273–A276. [Google Scholar] [CrossRef]
- Meyers, J.P.; Darling, R.M. Model of Carbon Corrosion in PEM Fuel Cells. J. Electrochem. Soc. 2006, 153, A1432–A1442. [Google Scholar] [CrossRef]
- Ohs, J.H.; Sauter, U.; Maass, S.; Stolten, D. Modeling hydrogen starvation conditions in proton-exchange membrane fuel cells. J. Power Sources 2011, 196, 255–263. [Google Scholar] [CrossRef]
- Maranzana, G.; Lamibrac, A.; Dillet, J.; Abbou, S.; Didierjean, S.; Lottin, O. Startup (and Shutdown) Model for Polymer Electrolyte Membrane Fuel Cells. J. Electrochem. Soc. 2015, 162, F694–F706. [Google Scholar] [CrossRef]
- Durst, J.; Lamibrac, A.; Charlot, F.; Dillet, J.; Castanheira, L.F.; Maranzana, G.; Dubau, L.; Maillard, F.; Chatenet, M.; Lottin, O. Degradation heterogeneities induced by repetitive start/stop events in proton exchange membrane fuel cell: Inlet vs. outlet and channel vs. land. Appl. Catal. Environ. 2013, 138–139, 416–426. [Google Scholar] [CrossRef]
- Schneider, I.A.; von Dahlen, S. Start-Stop Phenomena in Channel and Land Areas of a Polymer Electrolyte Fuel Cell. Electrochem. Solid-State Lett. 2011, 14, B30–B33. [Google Scholar] [CrossRef]
- Fuller, T.F.; Gray, G. Carbon Corrosion Induced by Partial Hydrogen Coverage. ECS Trans. 2006, 8, 345–353. [Google Scholar]
- Gu, W.; Carter, R.N.; Yu, P.T.; Gasteiger, H.A. Start/Stop and Local H2 Starvation Mechanisms of Carbon Corrosion: Model vs. Experiment. ECS Trans. 2007, 11, 963–973. [Google Scholar] [CrossRef]
- Jain, K.; Gidwani, A.; Kumar, S.; Cole, J.V. CFD Study of Carbon Corrosion in PEM Fuel Cells. ECS Trans. 2008, 16, 1323–1333. [Google Scholar] [CrossRef]
- Franco, A.A.; Gerard, M.; Guinard, M.; Barthe, B.; Lemaire, O. Carbon Catalyst-Support Corrosion in Polymer Electrolyte Fuel Cells: Mechanistic Insights. ECS Trans. 2008, 13, 35. [Google Scholar]
- Maranzana, G.; Moyne, C.; Dillet, J.; Didierjean, S.; Lottin, O. About internal currents during start-up in proton exchange membrane fuel cell. J. Power Sources 2010, 195, 5990–5995. [Google Scholar] [CrossRef]
- Abbou, S.; Dillet, J.; Spernjak, D.; Mukundan, R.; Fairweather, J.D.; Borup, R.L.; Maranzana, G.; Didierjean, S.; Lottin, O. Time Evolution of Local Potentials during PEM Fuel Cell Operation with Dead-Ended Anode. ECS Trans. 2013, 58, 1631–1642. [Google Scholar] [CrossRef]
- Dillet, J.; Spernjak, D.; Lamibrac, A.; Maranzana, G.; Mukundan, R.; Fairweather, J.; Didierjean, S.; Borup, R.; Lottin, O. Impact of flow rates and electrode specifications on degradations during repeated startups and shutdowns in polymer-electrolyte membrane fuel cells. J. Power Sources 2014, 250, 68–79. [Google Scholar] [CrossRef]
- Gu, W.; Yu, P.T.; Carter, R.N.; Makharia, R.; Gasteiger, H.A. Modeling of Membrane-Electrode-Assembly Degradation in Proton-Exchange-Membrane Fuel Cells—Local H2 Starvation and Start–Stop Induced Carbon-Support Corrosion. In Modeling and Diagnostics of Polymer Electrolyte Fuel Cells; Springer: New York, NY, USA, 2010; pp. 45–87. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, P.; Chen, H.; Pei, P. A review of automotive proton exchange membrane fuel cell degradation under start-stop operating condition. Appl. Energy 2018, 223, 249–262. [Google Scholar] [CrossRef]
- Yu, Y.; Tu, Z.; Zhang, H.; Zhan, Z.; Pan, M. Comparison of degradation behaviors for open-ended and closed proton exchange membrane fuel cells during startup and shutdown cycles. J. Power Sources 2011, 196, 5077–5083. [Google Scholar] [CrossRef]
- Yano, H.; Akiyama, T.; Bele, P.; Uchida, H.; Watanabe, M. Durability of Pt/graphitized carbon catalysts for the oxygen reduction reaction prepared by the nanocapsule method. Phys. Chem. Chem. Phys. 2010, 12, 3806–3814. [Google Scholar] [CrossRef] [PubMed]
- Reiser, C.; Yang, D.; Sawyer, R. Procedure for Starting Up a Fuel Cell System Using a Fuel Purge. U.S. Patent US7410712B2, 12 August 2008. [Google Scholar]
- Ofstad, A.; Davey, J.; Sunde, S.; Borup, R.L. Carbon Corrosion of a PEMFC During Shut-down/Start-up when Using an Air Purge Procedure. ECS Trans. 2008, 16, 1301–1311. [Google Scholar] [CrossRef]
- Pei, P.; Chang, Q.; Tang, T. A quick evaluating method for automotive fuel cell lifetime. Int. J. Hydrogen Energy 2008, 33, 3829–3836. [Google Scholar] [CrossRef]
- Kim, J.H.; Cho, E.A.; Jang, J.H.; Kim, H.J.; Lim, T.H.; Oh, I.H.; Ko, J.J.; Oh, S.C. Development of a Durable PEMFC Startup Process by Applying a Dummy Load: I. Electrochemical Study. J. Electrochem. Soc. 2009, 156, B955–B961. [Google Scholar] [CrossRef]
- Jo, Y.Y.; Cho, E.A.; Kim, J.H.; Lim, T.H.; Oh, I.H.; Jang, J.H.; Kim, H.J. Effects of a hydrogen and air supply procedure on the performance degradation of PEMFCs. Int. J. Hydrogen Energy 2010, 35, 13118–13124. [Google Scholar] [CrossRef]
- Brightman, E.; Hinds, G. In situ mapping of potential transients during start-up and shut-down of a polymer electrolyte membrane fuel cell. J. Power Sources 2014, 267, 160–170. [Google Scholar] [CrossRef]
- Randrianarizafy, B.; Schott, P.; Chandesris, M.; Gerard, M.; Bultel, Y. Design optimization of rib/channel patterns in a PEMFC through performance heterogeneities modelling. Int. J. Hydrogen Energy 2018, 43, 8907–8926. [Google Scholar] [CrossRef]
- Young, J.; Todd, B. Modelling of multi-component gas flows in capillaries and porous solids. Int. J. Heat Mass Transf. 2005, 48, 5338–5353. [Google Scholar] [CrossRef]
- Lampinen, M.J.; Fomino, M. Analysis of Free Energy and Entropy Changes for Half-Cell Reactions. J. Electrochem. Soc. 1993, 140, 3537–3546. [Google Scholar] [CrossRef]
- Willsau, J.; Heitbaum, J. The influence of Pt-activation on the corrosion of carbon in gas diffusion electrodes-A dems study. J. Electroanal. Chem. Interfacial Electrochem. 1984, 161, 93–101. [Google Scholar] [CrossRef]
- Maass, S.; Finsterwalder, F.; Frank, G.; Hartmann, R.; Merten, C. Carbon support oxidation in PEM fuel cell cathodes. J. Power Sources 2008, 176, 444–451. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Reference |
|---|---|---|
| Anode channel inlet pressure (Pa) | ||
| Cathode channel inlet pressure (Pa) | ||
| Anode rib/channel temperature (K) | 353 | |
| Cathode rib/channel temperature (K) | 353 | |
| Anode channel relative humidity (%) | 50 | |
| Cathode channel relative humidity (%) | 50 | |
| Anode inlet channel hydrogen molar fraction | 1 | |
| Cathode inlet channel oxygen molar fraction | ||
| Hydrogen stoichiometry ratio | ||
| Oxygen stoichiometry ratio | 2 | |
| (kJ/mol) | [29] | |
| (kJ/mol) | 0 | [29] |
| (J/mol/K) | [29] | |
| (J/mol/K) | [29] | |
| (kJ/mol) | [27] | |
| (J/mol/K) | [27] | |
| (kJ/mol) | [27] | |
| (J/mol/K) | [27] | |
| (V) | ||
| (V) | ||
| (V) | 0 | |
| (A/cm2) | [17] | |
| [17] | ||
| (m2/m3) | in-house data | |
| (m2/m3) | in-house data | |
| (m2/m3) | in-house data | |
| (m2/m3) | in-house data | |
| Channel depth (m) | in-house data | |
| Channel length (m) | in-house data | |
| Channel width (m) | in-house data | |
| Rib width (m) | in-house data | |
| GDL thickness (m) | in-house data | |
| GDL thickness at 1MPa (m) | in-house data | |
| MPL thickness (m) | in-house data | |
| Anode CL thickness (m) | in-house data | |
| Cathode CL thickness (m) | in-house data | |
| MB thickness (m) | in-house data |
| Operating Conditions | Values |
|---|---|
| Anode channel inlet pressure (Pa) | |
| Cathode channel inlet pressure (Pa) | |
| Anode rib/channel temperature (K) | 353 |
| Cathode rib/channel temperature (K) | 353 |
| Anode channel relative humidity (%) | 50 |
| Cathode channel relative humidity (%) | 50 |
| Anode inlet channel hydrogen molar fraction | 1 |
| Cathode inlet channel oxygen molar fraction | |
| Hydrogen stoichiometry ratio | |
| Oxygen stoichiometry ratio | 2 |
| Velocity | Elapsed Time in Corrosion (s) | Total Amount of Carbon Corroded (g/cm of MEA) | Catalyst Layer Thickness Reduction (m) |
|---|---|---|---|
| m/s | s | 0.84 | |
| m/s | s | 1.47 | |
| m/s | s | 0.52 | |
| m/s | s | 0.44 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Randrianarizafy, B.; Schott, P.; Gerard, M.; Bultel, Y. Modelling Carbon Corrosion during a PEMFC Startup: Simulation of Mitigation Strategies. Energies 2020, 13, 2338. https://doi.org/10.3390/en13092338
Randrianarizafy B, Schott P, Gerard M, Bultel Y. Modelling Carbon Corrosion during a PEMFC Startup: Simulation of Mitigation Strategies. Energies. 2020; 13(9):2338. https://doi.org/10.3390/en13092338
Chicago/Turabian StyleRandrianarizafy, Bolahaga, Pascal Schott, Mathias Gerard, and Yann Bultel. 2020. "Modelling Carbon Corrosion during a PEMFC Startup: Simulation of Mitigation Strategies" Energies 13, no. 9: 2338. https://doi.org/10.3390/en13092338
APA StyleRandrianarizafy, B., Schott, P., Gerard, M., & Bultel, Y. (2020). Modelling Carbon Corrosion during a PEMFC Startup: Simulation of Mitigation Strategies. Energies, 13(9), 2338. https://doi.org/10.3390/en13092338