1. Introduction
The identification of efficient and effective but low-cost energy storage technologies is still a challenge, preventing the full integration of renewable energy sources into the grid at present. Rechargeable batteries represent one of the best promises for such a target, at least for a number of applications. Here, electrical energy can be efficiently stored and released according to demand, and their compact size makes them highly suitable for use at small grid-scale. Among the most promising electrode materials for such applications is certainly sulfur, which can potentially enable lithium metal anode batteries with about five times higher specific energy compared to current lithium–ion batteries, while being rather inexpensive [
1]. Nonetheless, there is still concern with the foreseen resource and supply risks of lithium [
2].
Therefore, sodium–sulfur (Na–S) batteries appear to be an ideal candidate due to the rather low economic and geopolitical impact of their raw materials [
3,
4]. The theoretical capacity of a Na–S battery is ~1672 mAh g
−1, based on the sulfur with final product of Na
2S. Despite the success of high-temperature Na–S batteries (>300 °C) for grid-scale application, the problems associated with the solid electrolyte and/or cell sealing integrity failures, resulting in the risk of molten sodium explosive reactions, motivate the exploration for alternatives. A decrease in operation temperature could, in fact, offer the opportunity to improve the durability and safety of the Na–S battery [
5]. Thus, the development of room temperature (RT) Na–S batteries with conventional, organic solvent-based electrolytes is triggering tremendous research interest [
6]. In fact, considering their expected low cost, RT Na–S batteries may be the solution for the future of stationary and large-scale energy storage, enabling efficient and effective use of sustainable energy sources [
6,
7]. So far, most of the work has focused on the exploration of polymer electrolytes, either gel-polymer or solid-polymer, to suppress the polysulfides’ shuttle pathway [
8,
9,
10,
11,
12]. Moreover, attempts to fabricate sodium polysulfides’ electrodes with carbonaceous interlayers and functional separators have been demonstrated to enhance the cycling stability of Na–S batteries [
13,
14,
15]. However, RT Na–S batteries are still suffering from low reversible capacity and poor cycling stability. The low reversible capacity is mainly ascribed to the low electric conductivity of bulk sulfur and the sluggish reaction kinetics between sulfur and sodium, resulting in the poor utilization of the sulfur cathode [
9]. In addition, the high self-discharge rate and rapid capacity fading due to sodium polysulfides’ formation and shuttling across the electrolyte appear almost inevitable for RT Na–S batteries. Recently, intensive efforts have been made to identify and prepare highly efficient S-host substrates from functional carbons and inorganic metal oxides (sulfide), improving the utilization of the sulfur active material [
16,
17,
18,
19,
20,
21,
22,
23].
Although the understanding of the sulfur reduction reactions in conventional organic electrolytes is a precondition for the realization of high performance RT Na–S batteries, only very few studies have directly been devoted to unfold the sulfur conversion reaction and intermediates under battery conditions, in these carbon frameworks. A few studies have demonstrated the pronounced effects of the organic electrolytes’ solvation properties on the lithium–sulfur (Li–S) redox reactions [
24,
25,
26,
27]. Thus, understanding the solvent–dependent Na–S redox processes, i.e., how the solvent affects the Na–S redox reactions, may provide significant insight, allowing the design of improved electrolytes for high performance Na–S batteries.
In this work, we systematically investigate the electrolyte solvent on the sulfur redox reaction pathway in RT Na-S batteries, comparing its performance in ether- and carbonate-based electrolytes. The use of tetraethyleneglycol-dimethyl-ether (TEGDME) ether-based electrolyte contributes to a more complete reduction of sulfur and enables a higher electrochemical reversibility. On the other hand, an irreversible solution phase reaction is observed in carbonate solvents. Both in situ X-ray diffraction (XRD) and ex situ X-ray photoelectron spectroscopy (XPS) measurements were performed to reveal the sulfur redox reaction process. This study presents a basic understanding on the solvent-dependent sulfur redox reactions, providing a solid basis for the systematic development of room temperature Na–S batteries.
2. Results and Discussion
In order to address the major problem originating from the insulating nature of sulfur, the sulfur/carbon nanotube (S/CNT) composite was synthesized by a simple melt-diffusion method, which is briefly described in the Experimental section [
28]. Results on the characterization of the as-prepared S/CNT composite are presented in
Figure S1 (see Supporting Information). The scanning electron microscopy (SEM) images, revealing the morphology of the S/CNT composite, show no obvious aggregation of the sulfur particles, which indicates a homogenous distribution of the active sulfur in the carbon host. The XRD pattern confirms the orthorhombic crystal structure of cyclo-S
8 (PDF#74-1465). The broad diffraction peak located at 25° can be attributed to the graphitic CNT. The TG curve shows a single-stage sulfur loss, by evaporation between 200 and 300 °C. Quantitative evaluation yields a sulfur content of about 69 wt.%. Initially, the S/CNT electrode was investigated in the Li–S battery to confirm its activity. As shown in
Figure S2 (panels
a and
b), the composite shows a specific discharge capacity of 1020 mAh g
−1 (based on sulfur) at 100 mA g
−1 and maintained 490 mAh g
−1 at 1675 mA g
−1, without any capacity fading over 75 cycles.
The solvent-dependent redox kinetics and the reaction behavior of the RT Na–S cells were further investigated in two different electrolytes, namely, 1.5 M NaClO
4 in TEGDME with 0.2 M NaNO
3 and 1.0 M NaClO
4 in propylene carbonate/ethylene carbonate (PC/EC, 1:1 v/v) with 5 wt.% fluoroethylene carbonate (FEC), which are commonly used for Na–S batteries. The CV scans of S/CNT electrodes in these electrolytes are shown in
Figure 1. The test of the cell employing the TEGDME-based electrolyte (panel
a) shows three cathodic peaks at 2.2, 1.6, and 1.0 V, supporting for a three-step reduction of sulfur in the presence of Na ions. Two broad anodic peaks centered at 2.0 and 2.4 V indicate, however, that the oxidation occurs in two steps. In contrast, for the cell employing the PC/EC-based electrolyte, we only find a single clear reduction peak at 2.1 V (panel
b), followed by two other features barely distinguishable at lower potentials. No peaks are detected during the anodic scan. Such a complete irreversibility may result from a rapid nucleophilic attack of reduced sulfur species on the carbonate solvents, as it was reported for the analogous lithium system [
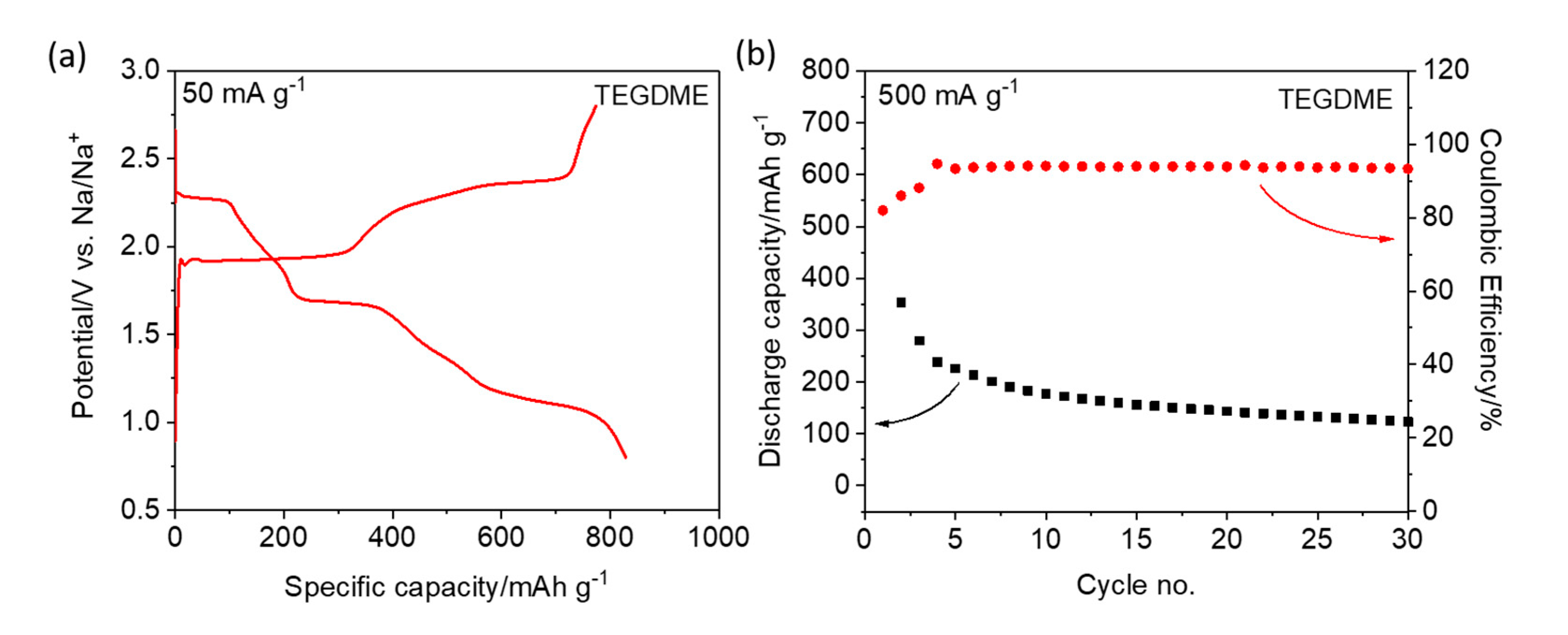
24]. The first galvanostatic charge/discharge profiles of the S/CNT electrode at 50 mA g
−1 are presented in
Figure 2a and
Figure S3 (see Supporting Information). Three distinct voltage plateaus are observed at 2.3, 1.7, and 1.1 V during the discharge of the TEGDME-based cell, which is consistent with the CV results. The electrode shows an initial specific discharge capacity of 828 mAh g
−1 and a specific charge capacity of 774 mAh g
−1, resulting in an initial Coulombic efficiency of 93.5%. The high coulombic efficiency is attributed to the presence of NaNO
3, which is proposed to reduce/suppress self-discharge. However, at higher specific currents (e.g., 500 mA g
−1), both the Coulombic efficiency and the reversible capacity are rather poor (c.f.,
Figure S4). Compared with the Li–S chemistry (c.f.,
Figure S2a,b), the Na–S cells exhibit a different voltage (kinetics) and long-term cycling behavior. In fact, a rapid capacity fade occurs, which may be due to the dissolution of sodium polysulfides and their migration through the electrolyte (see
Figure 2b). Using the PC/EC-based electrolyte, the S/CNT electrode shows a lower specific discharge capacity (i.e., 420 mAh g
−1, corresponding to an average reduction state of S
42−) and negligible charge capacity (see
Figure S3 in Supporting Information). Such an extremely poor reversibility makes the carbonate-based electrolyte unsuitable for Na/S batteries, at least under these conditions.
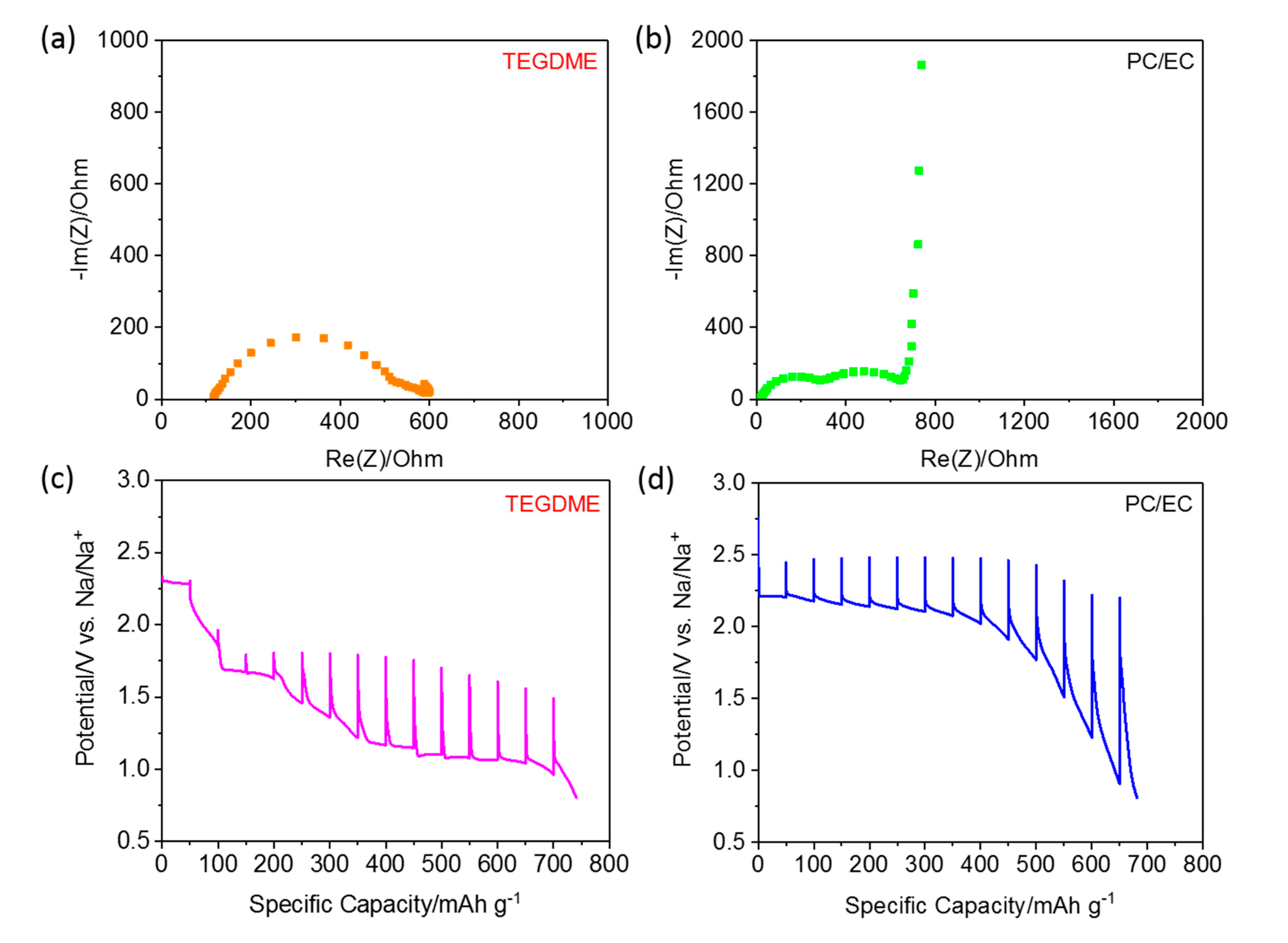
Electrochemical impedance spectroscopy (EIS) was applied on the assembled cells after resting (3 h) at the open circuit voltage (OCV) to obtain information on the self-discharge behavior. The impedance spectra exhibit two distorted semicircles in the high and medium frequency regions, followed by a steep increase in the low frequency regions. These represent the resistance of the passivation layer, the charge transfer resistance and the solid-state diffusion, respectively [
29]. The intercept with the x axis at high frequency is attributed to the electrolyte resistance. The passivation layer resistance is associated with the unstable solid-electrolyte interphase (SEI), caused by the shuttle effect in the cathode and sodium dendrites on the anode. Nyquist plots recorded using the ether and carbonate electrolytes, respectively, are presented in
Figure 3. As seen in
Figure 3a, the cell using a TEGDME electrolyte has a higher film resistance than that employing carbonate. This results from the dissolution of polysulfides and the side reaction of the sodium metal electrode with the electrolyte, leading to a thicker passivation layer in the ether-based electrolyte than in the carbonate electrolyte [
30,
31]. The cell employing the TEGDMA electrolyte shows much higher electrolyte and SEI resistances, resulting from polysulfide dissolution. This indicates a clear role of dissolved sodium polysulfides in the formation of the passivation layer. Overall, the TEGDME-based Na–S cell shows a much smaller charge transfer resistance than the carbonate-based cell, indicating faster redox kinetics in the former case.
The sodiation overpotentials and kinetics of the S/CNT electrodes in the two electrolytes were evaluated by galvanostatic intermittent titration techniques (GITT), using specific current pulses of 50 mA g
−1 per one hour, each followed by a rest period of 6 h. The resulting voltage profiles are shown in
Figure 3c,d [
27]. The overall voltage profiles of the two cells during sodiation resemble those obtained during the galvanostatic discharge (c.f.,
Figure 2a and
Figure S3). The overpotential of the S/CNT electrode in the PC/EC-based electrolyte increases sharply with the state of discharge (SoD), reflecting the decreasing kinetics. In contrast, the electrode tested in the TEGDME-based electrolyte shows a moderate overpotential at the beginning of the sodiation, indicating that the electrode kinetics for the conversion of S
8 to long-chain polysulfides are rather fast. However, an increased polarization can be observed in the low voltage range. This phenomenon can be explained by the slow reaction kinetics for the conversion of the long-chain polysulfides into short-chain polysulfides, which is generally considered as a two-phase liquid-solid reaction. At the end, we find a small decrease on the overpotential, possibly due to the slight decrease of mass transport resistance at the end of the conversion reaction [
32]. The different overpotentials for the sodiation process, which are mainly caused by the activation overpotentials, clearly indicate different activation energies for the RT Na–S reaction in carbonate and in ether-based electrolytes. It should be noted that the discharge capacity in the GITT measurement is higher than that obtained during continuous galvanostatic discharge, indicating that the milder GITT conditions (low pulse current and long rest periods) promote the utilization of sulfur and/or polysulfides in the carbon host, which has also been noted in Li–O
2 batteries [
33].
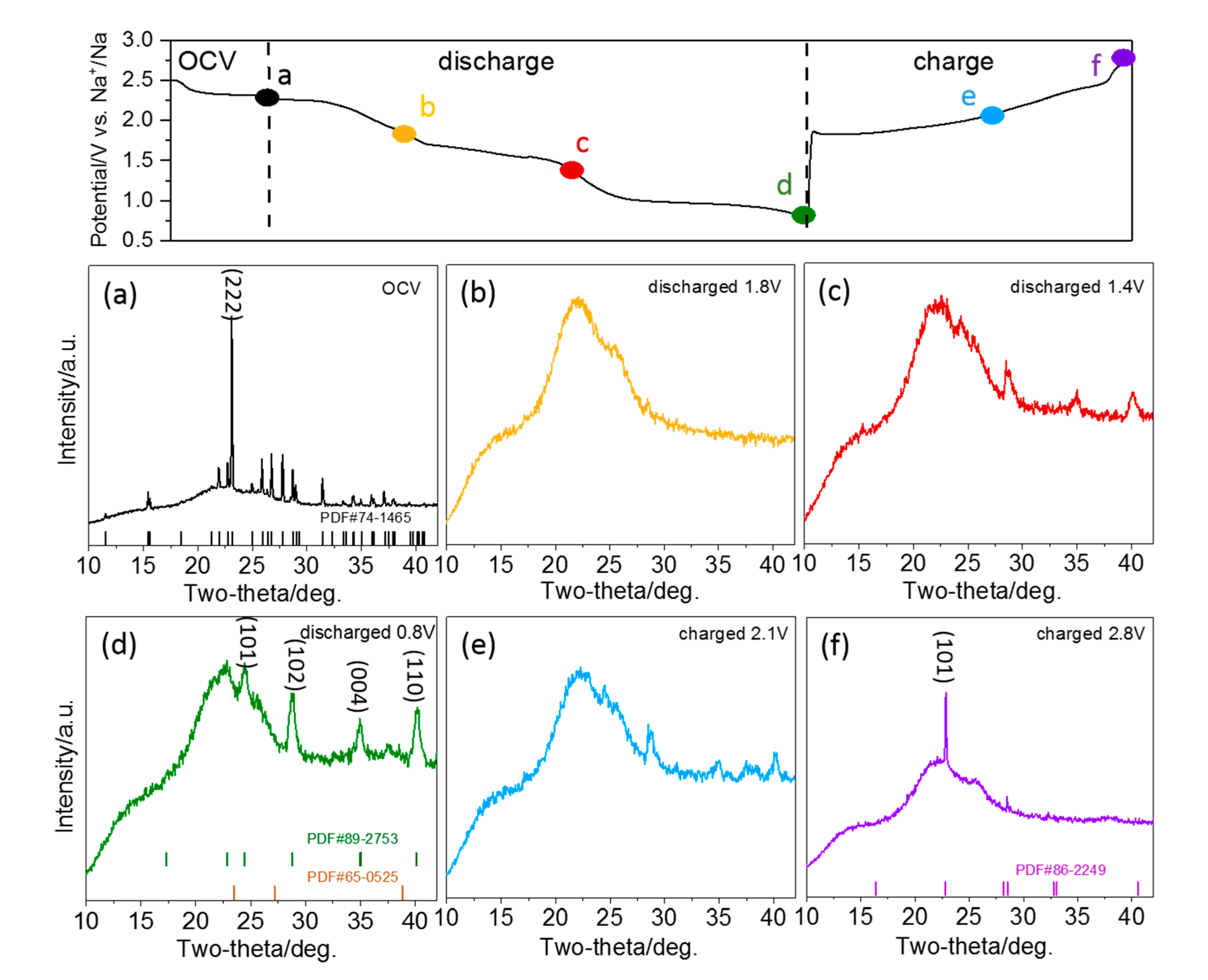
To understand the intermediates and products of the S/CNT cathode at different sodiation states, we performed an in situ X-ray diffraction (XRD) analysis [
34]. The XRD patterns of a S/CNT electrode in TEGDME collected during the first discharge-charge cycle are shown in the contour map (2θ range of 10–42°) in
Figure 4. To illustrate the phase changes more clearly, XRD diffractograms obtained at selected stages of sodiation (1.8 V, 1.4 V and 0.8 V) and desodiation (2.1 V and 2.8 V) are presented in
Figure 5 additionally. The diffractograms recorded in the course of the first discharge can be subdivided into three regions, which are consistent with the charge-discharge curves (c.f.,
Figure 4, left panel). The first region starts at the beginning of sodiation and ends with the first plateaus region in the charge-discharge curve at ~1.7 V. At the end of this region (see also diffractogram at 1.8 V in
Figure 5b), the features of S
8 have disappeared completely. Instead, a broad peak located at 21.5° appears, which can be attributed to the presence of soluble long-chain polysulfides. Hence, all elemental sulfur is transformed to soluble long-chain polysulfides in this region. Further reduction of the polysulfides occurs in the second region, and at the end of it, the (101), (102), (004) and (110) signals of crystalline Na
2S
2 (PDF#89-2753) appear in the diffractogram recorded at 1.4 V (c.f.,
Figure 5c). In the final region, the reduction of polysulfides to Na
2S
2 proceeds, until the features of crystalline Na
2S
2 reach the highest intensity at the end of the discharge process at 0.8 V (c.f.,
Figure 5d). Upon charging, the intensity of the features related to crystalline Na
2S
2 decrease again, indicating the gradual re-oxidation to polysulfides. After recharging to 2.1 V (c.f.,
Figure 5e), the peaks of Na
2S
2 are weakened, but are still detectable. Close to the end of the charging process (at 2.8 V in
Figure 5f), the Na
2S
2 peaks disappear, while new diffraction peaks appear at 22.8° and 28.5°, which correspond to the (101) and (300) features of cyclo-S
6 (PDF#86-2249). They possibly indicate a direct oxidation from S
62- to elemental S
6.
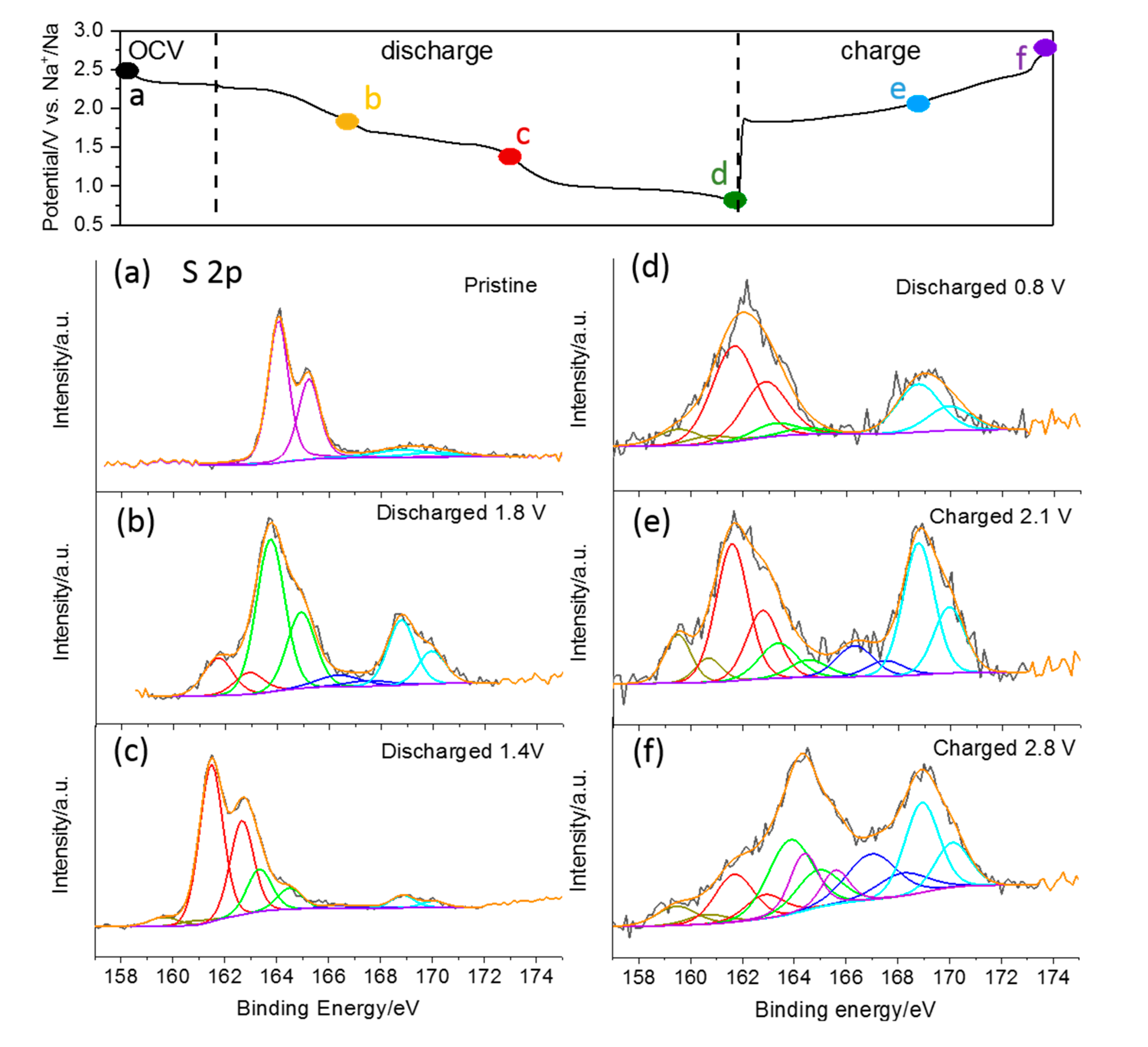
To reveal the changes of the chemical state of sulfur in the S/CNT electrode upon discharge and charge in TEGDME, ex situ XPS analysis was performed on the cycled electrodes at various discharge and charge states. Representative spectra in the S 2p range are shown in
Figure 6. The spectrum of the pristine electrode exhibits a spin–orbit doublet centered at 164.0 and 165.2 eV, which is in good agreement with the binding energy reported for the S 2p
1/2 and 2p
3/2 peaks of S
8 molecules [
35]. It may be noted that peaks at higher binding energy (beyond 166 eV) are observed in this and all following spectra originating from oxidized species. The next XPS measurement was performed on an electrode sodiated to the end of the first discharge plateau at 1.8 V (c.f., charge-discharge curve in
Figure 6). Here the main peak doublet is detected at 163.7/164.9 eV, i.e., 0.3 eV lower than in the first spectrum. It most probably contains contributions from bridging S atoms (S
B0) in polysulfide species (S
x2−) and from elemental sulfur. Additionally, another peak doublet with lower intensity appears at a lower binding energy (161.6/162.8 eV), which we assign to the terminal S atoms (S
T−1) of polysulfides. The intensity ratio of the two features is 4:1 in this measurement, showing just a minor excess of the expected value of S
B to S
T atoms in the initially formed long-chain polysulfide S
82− (of 3:1). Hence, assuming again the stepwise reaction from S
8 to S
82− and then to smaller polysulfides (S
62− etc.), this result suggests that most of the sulfur was transformed to S
82− species at this point. The following XPS spectrum was recorded at the end of the second discharge plateau at 1.4 V (c.f.,
Figure 6c). It exhibits two peaks’ doublets at 163.3/164.5 and at 161.6/162.8 eV. The further peak shift of the first feature to lower binding energy indicates that the transformation of elemental sulfur to polysulfides is completed at this point. This is also corroborated by the intensity ratio of the two features, which shows a predominance of the peak doublet due to the terminal S atoms (S
T−1) of polysulfides (by 0.3:1), i.e., small polysulfide species like S
22− and S
42− are present at this point. Considering that solid crystalline Na
2S
4 has not been detected in XRD measurements at this stage of sodiation, it is plausible to assume that the formation of Na
2S
4 takes place as a liquid to liquid reaction step in the TEGDME-based electrolyte. The XPS spectrum recorded after discharging to 1.4 V shows another small feature at even lower binding energy (159.5/160.7 eV), which can be assigned to S
2−. It is interesting to note that the in situ XRD measurements do not show the presence of the solid product Na
2S (PDF#65-0525) at any stage of electrochemical sodiation. At the end of discharge (at 0.8 V, c.f.,
Figure 6d), the S2p spectrum is dominated by the peak doublet related to terminal S atoms (S
T−1), while the feature of bridging S atoms (S
B0) has almost disappeared. Furthermore, the peak doublet of S
2− is detected again with small intensity. From this result we conclude that the main product upon complete discharge is Na
2S
2, with a theoretical specific capacity of 836 mAh g
−1, which agrees with previous studies of the Na–S chemistry [
36,
37]. Upon recharging, the signal of bridging S atoms (S
B0) reappears (after charging to 2.1 V) and eventually dominates the spectrum again (after charging to 2.8 V). At the same time, its peak position shifts back to higher binding energy (to 163.8/165.0 eV after charging to 2.8 V), indicating that elemental S also contributes to this feature at the end of recharge. Surprisingly, the peak doublet due to Na
2S persisted in the spectra of the recharged samples, i.e., the reaction from Na
2S back to higher sulfides seems to be kinetically hindered, and it may be assumed that this reaction occurs only in microporous carbon with a small pore size [
18].
To probe the reaction mechanism of sodiation of the S/CNT electrodes in the carbonate (PC/EC) based electrolyte, we collected similar in situ XRD diffractograms and ex situ XPS spectra. Since charging of the electrode does not work in this carbonate electrolyte, these experiments were performed only after discharge to various voltages. The in situ XRD patterns during the 1st discharge are shown in a contour map of the 2θ range of 10–42° in
Figure S5 (see Supporting Information). Initially, the intensity of the main sulfur reflection peaks gradually decreases, while the peak located at 22° broadens during further discharge. Selected in situ XRD patterns recorded during discharge are shown in
Figure S6a. As expected, the 1st XRD pattern measured at a discharged state of 2.2 V can be related to the pure S/CNT composite (c.f.,
Figure S1). An obvious peak at ~31° 2θ can be attributed to the (101) reflection of sodium metal (PDF#89-4082). With increasing sodiation, all XRD features disappear except for the characteristic peak of sodium metal, i.e., the electrode materials become completely amorphous. No additional crystalline peaks can be detected in the diffractogram recorded at the end of the discharge process (at 0.8 V), suggesting that there is no crystalline product in carbonate electrolyte upon sodiation. A high-resolution XPS detailed spectrum of the S2p region of a S/CNT electrode after discharge to 0.8 V is shown in
Figure S6b. Three S2p peak doublets are detected. The dominating doublet at 163.8/165.0 eV can be assigned to the bridging S atoms (S
B0) in polysulfide species (S
x2−) or to elemental sulfur, the smaller one at lower binding energy to the terminal S atoms (S
T−1) of polysulfides (161.6/162.8 eV), and the last one to oxidized sulfur species (168.8/170.0 eV), respectively. For S
82−, the first product which is expected upon sodiation of S
8, a ratio of S
B to S
T atoms of 3:1 is expected. Experimentally, the peak intensity ratio of the doublets at 163.8/165.0 eV and 161.6/162.8 is approximately 8:1. Thus, according to the XPS measurement, there should still be a sizable amount of elemental sulfur present on the surface of the sodiated S/CNT electrode, together with higher-order polysulfides, most probably S
82−. This can be explained by a dissolution of sulfur into the electrolyte, which occurs before the initial sodiation reaction. The results of the characterization indicate that the cyclo-S
8 is only reduced to Na
2S
8 upon sodiation, while the next reaction steps are inhibited. From the Li–S system, it is well-known that long-chain polysulfides like S
82− are dissolved in the carbonate solvent and then react with each other [
24].

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





