Femtosecond Laser-Induced Surface Modification of the Electrolyte in Solid Oxide Electrolysis Cells



Abstract
1. Introduction
2. Experimental
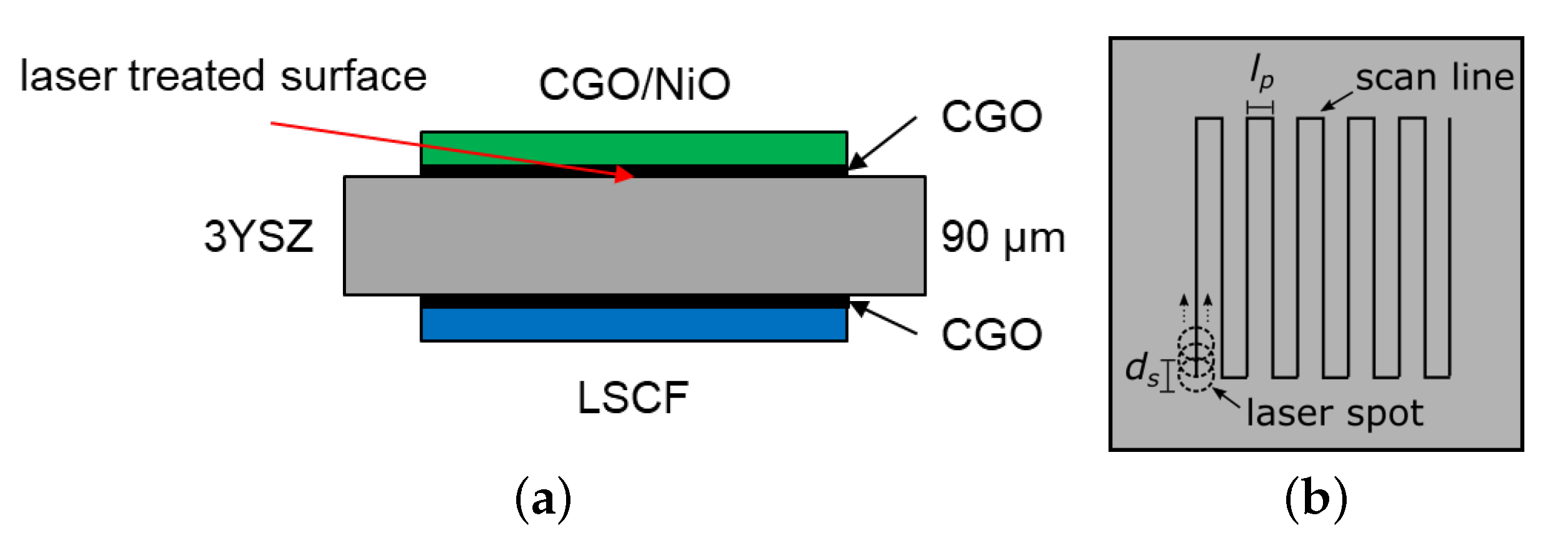
2.1. Femtosecond Laser Treatment
2.2. Physical Characterization
2.3. Electrochemical Characterization
3. Results and Discussion
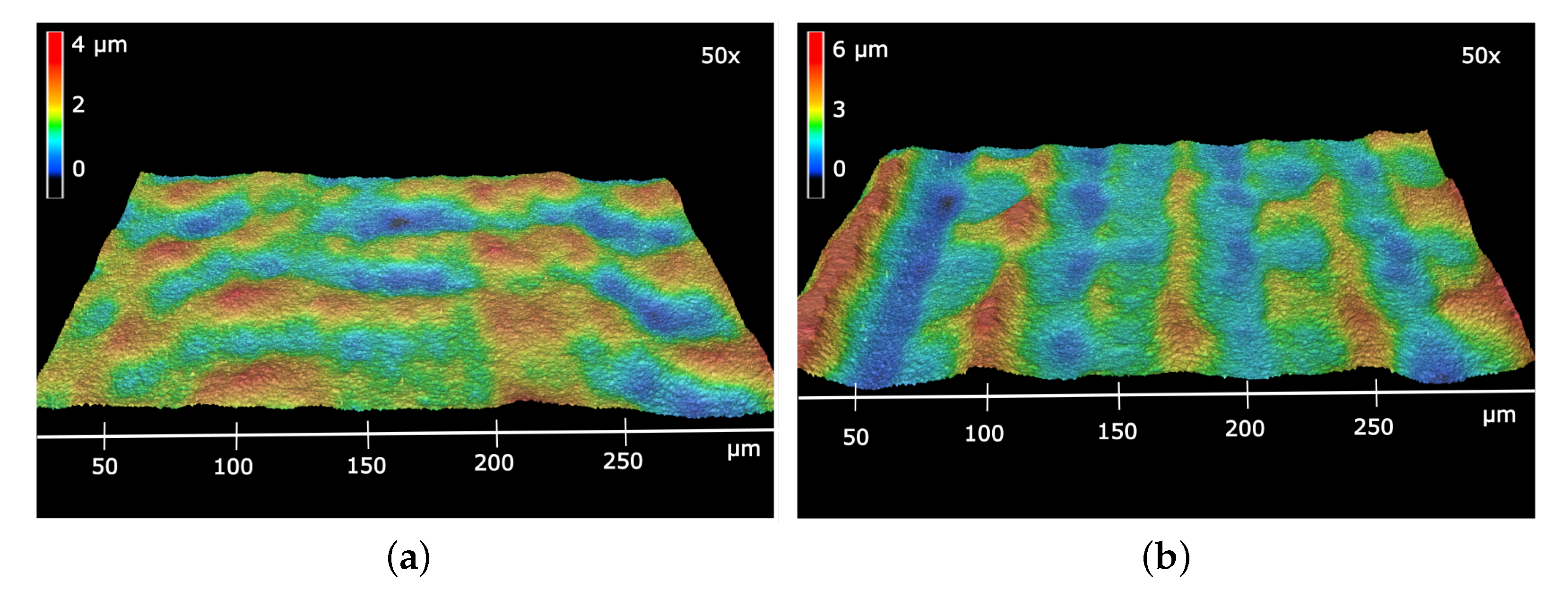
3.1. Physical Characterization
3.2. Electrochemical Characterization
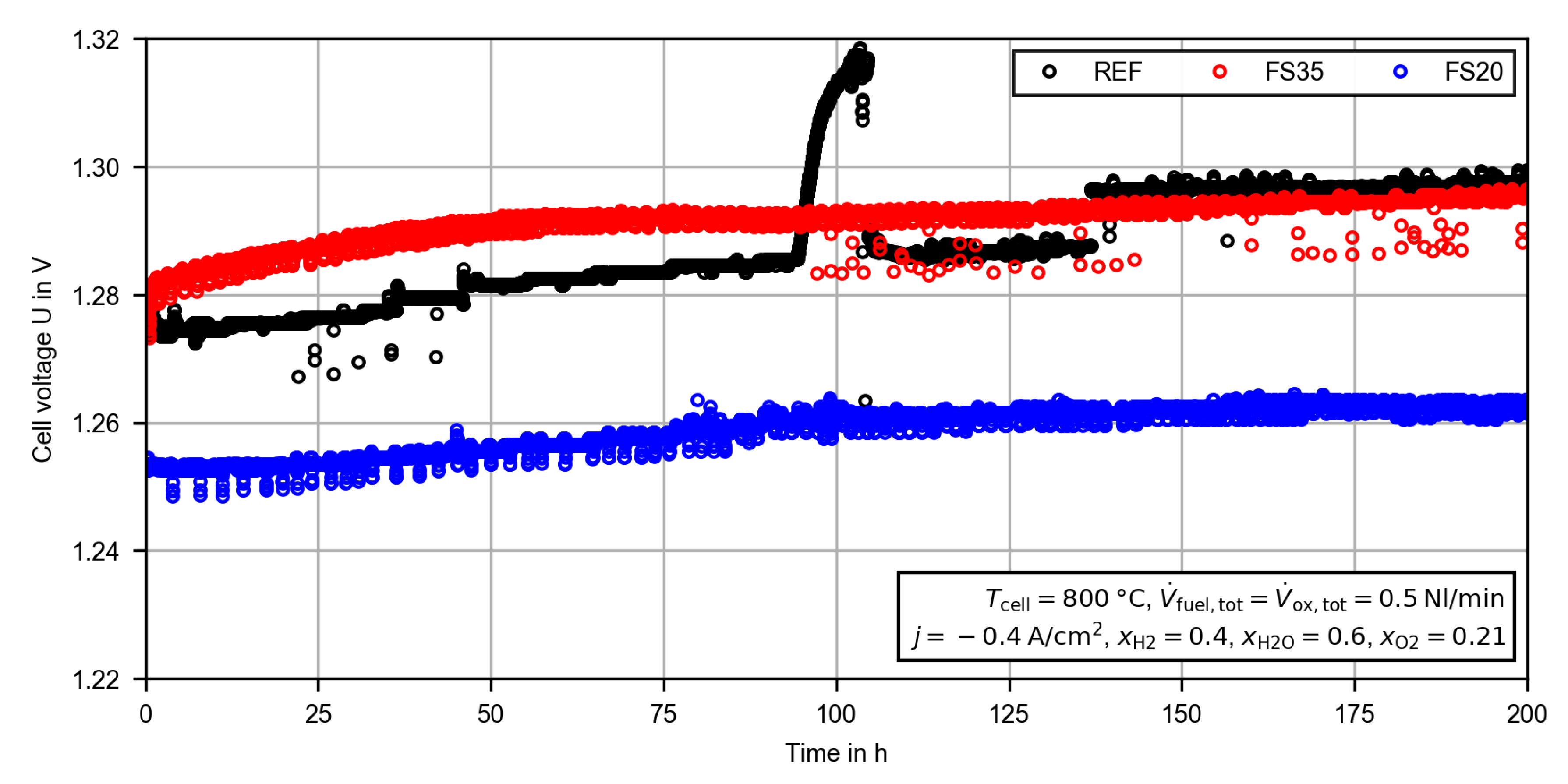
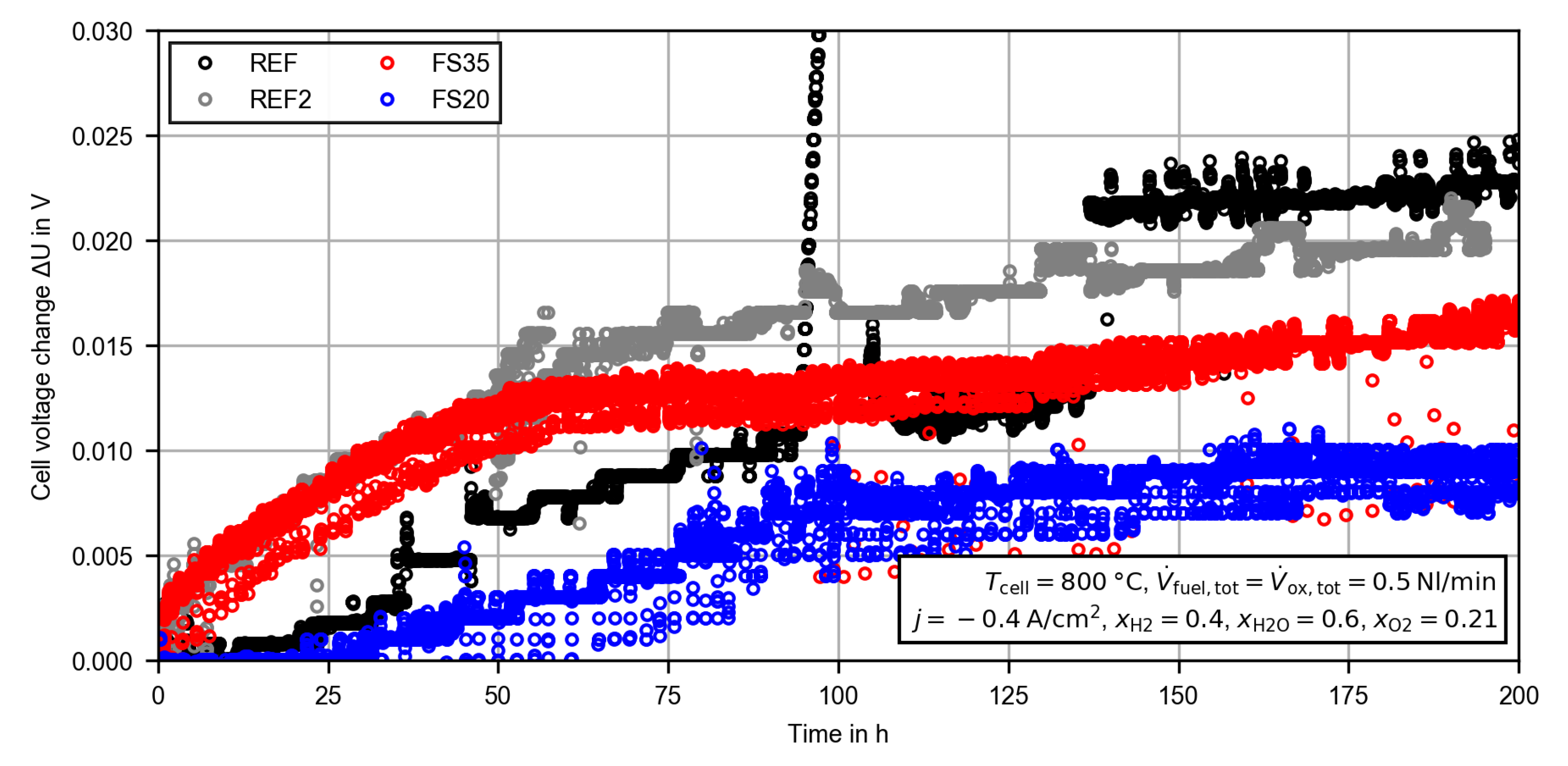
3.2.1. Constant Current Measurement
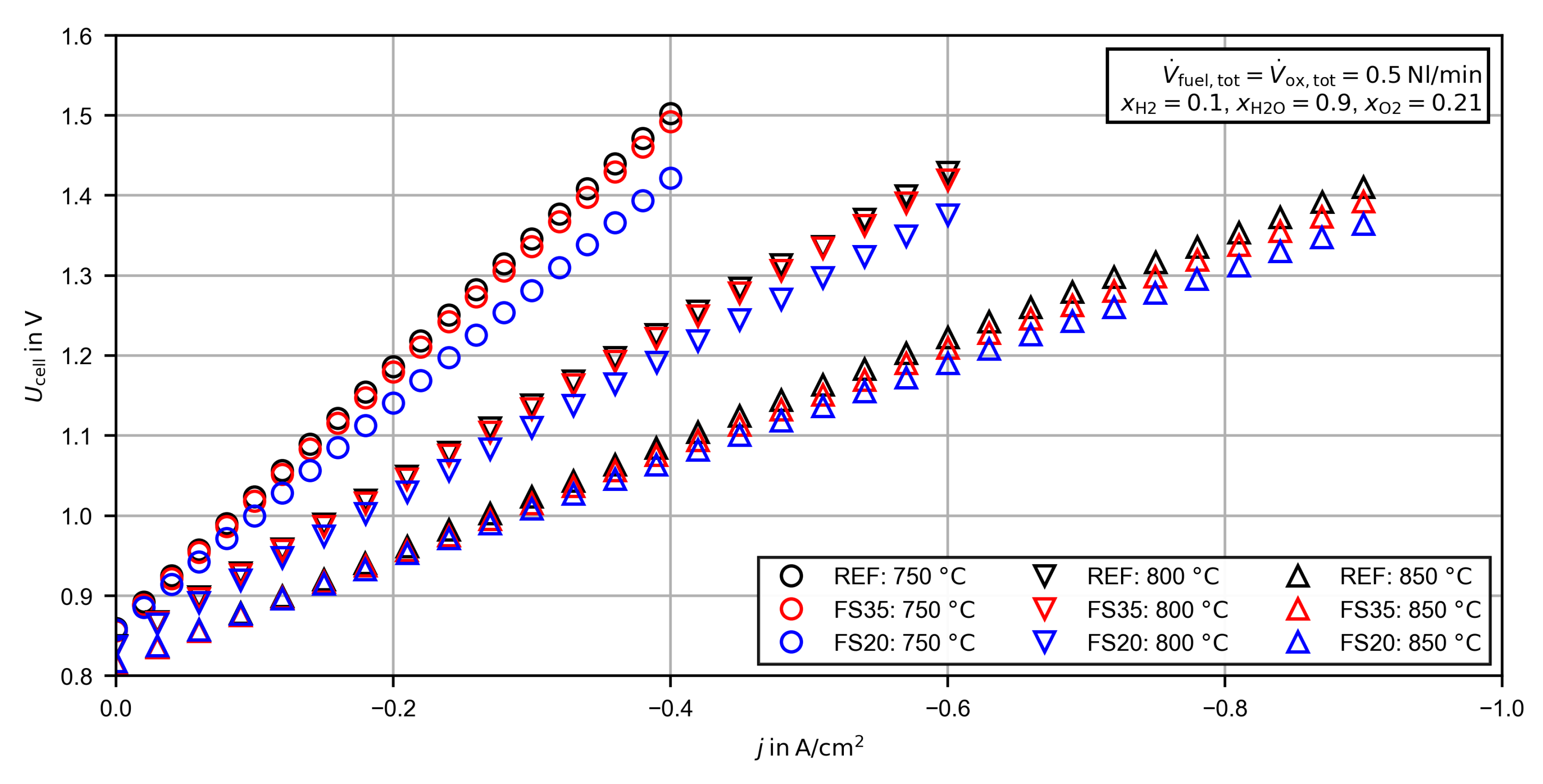
3.2.2. Voltage/Current Density Characteristic
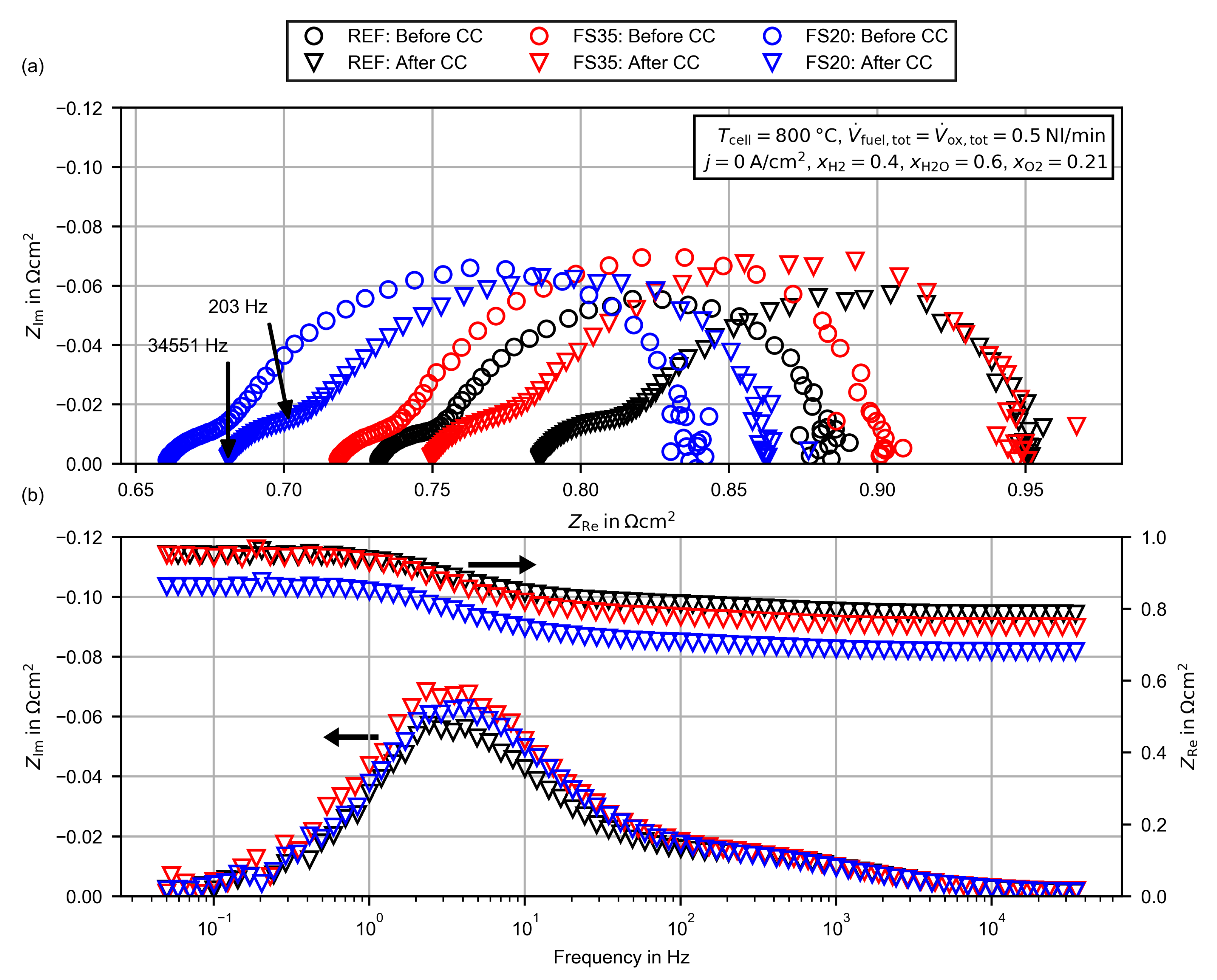
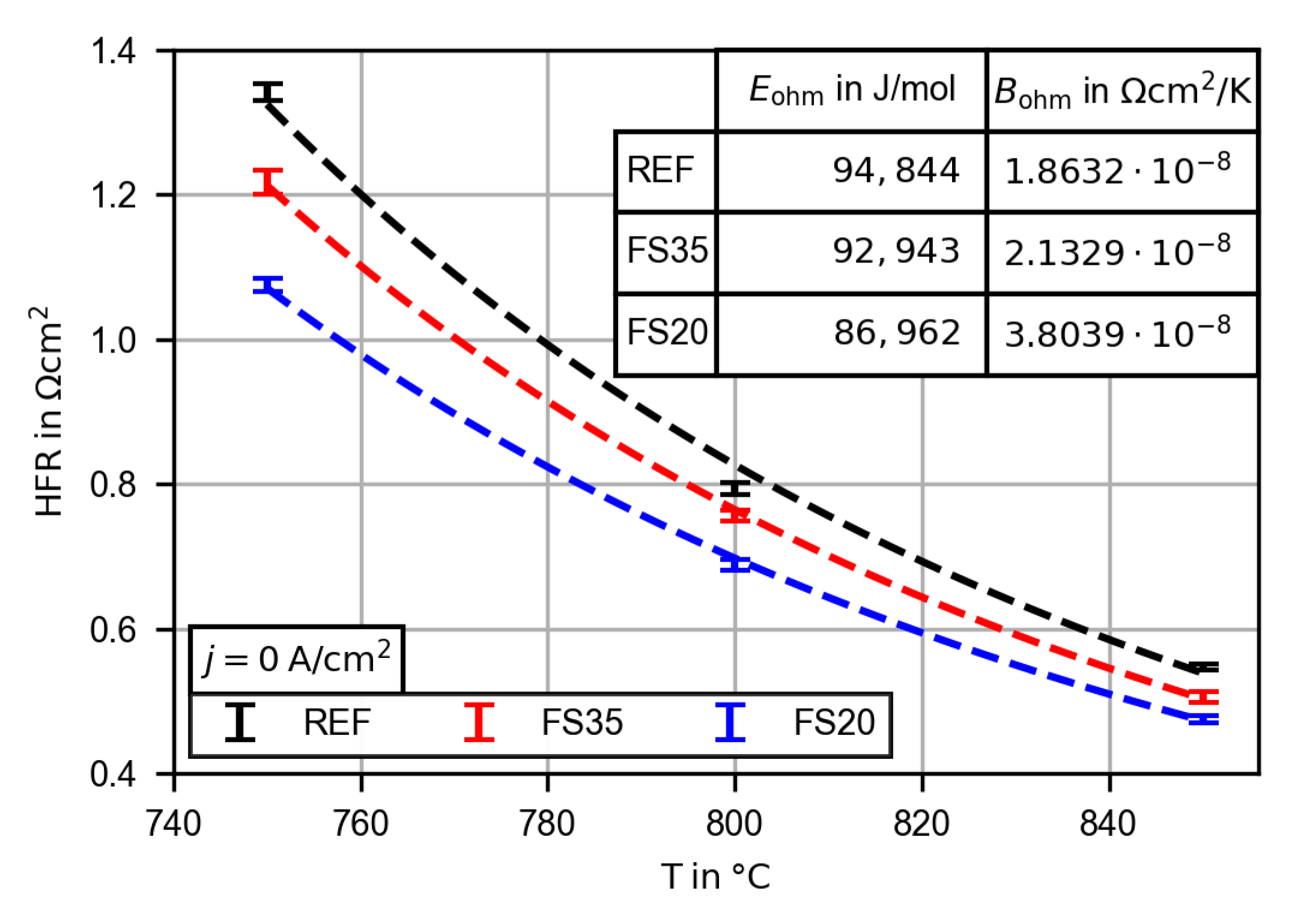
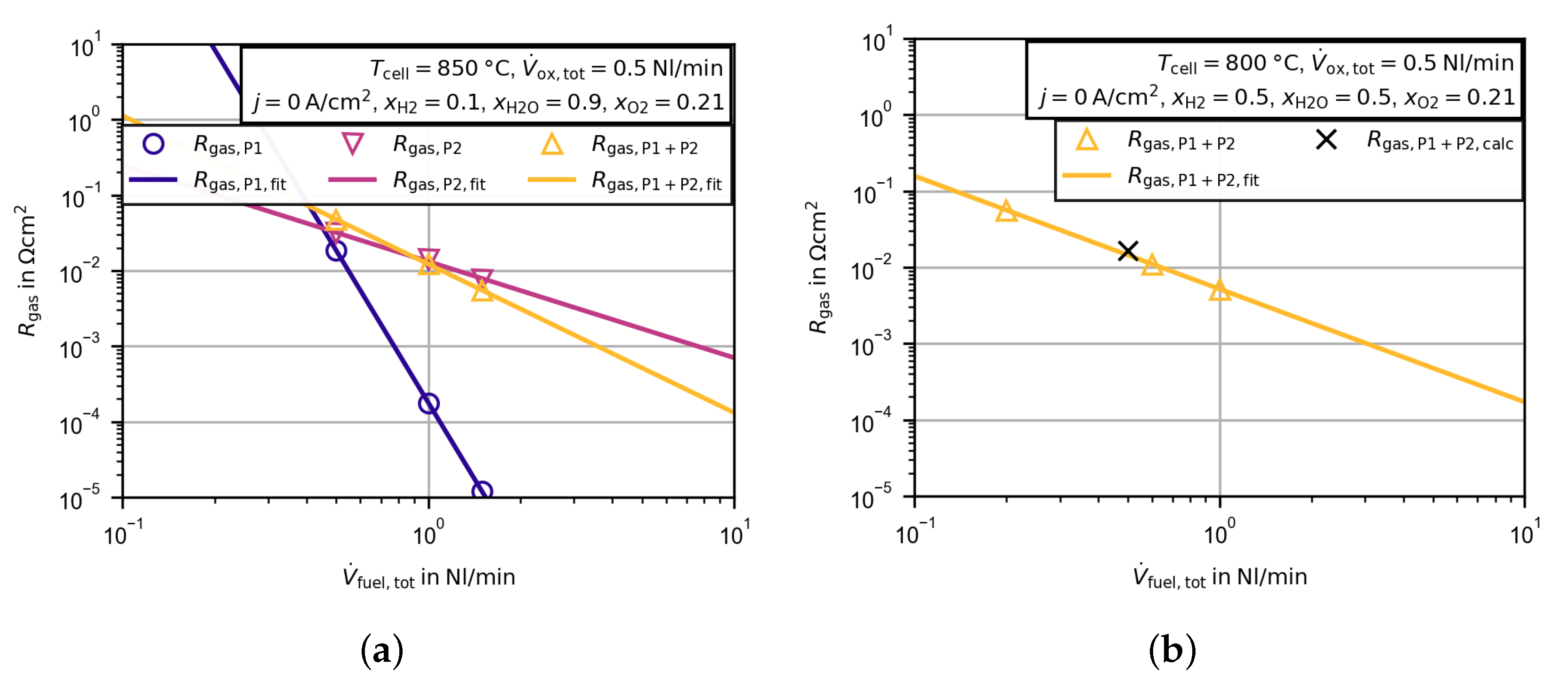
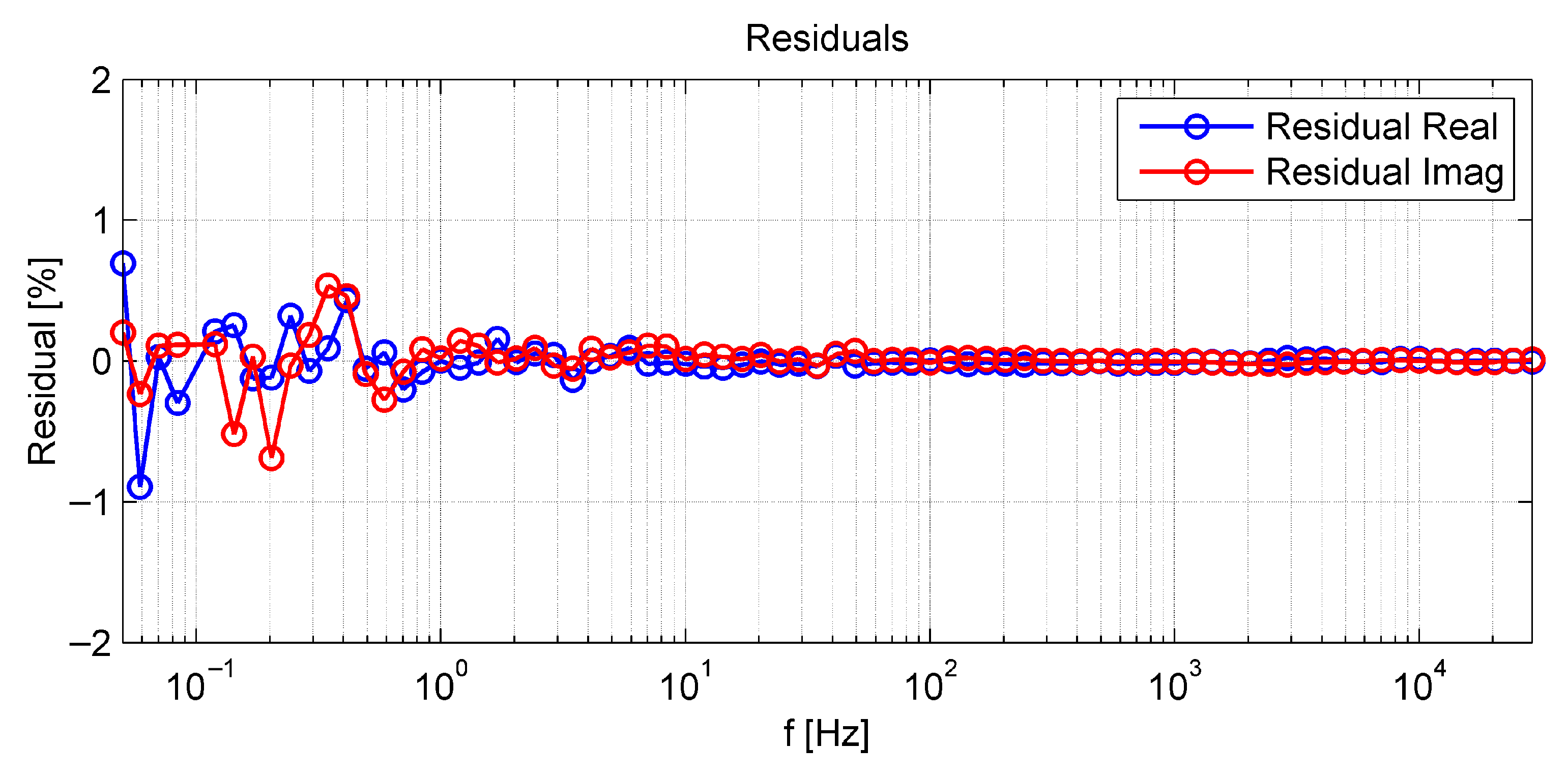
3.2.3. Evaluation of the High Frequency Resistance
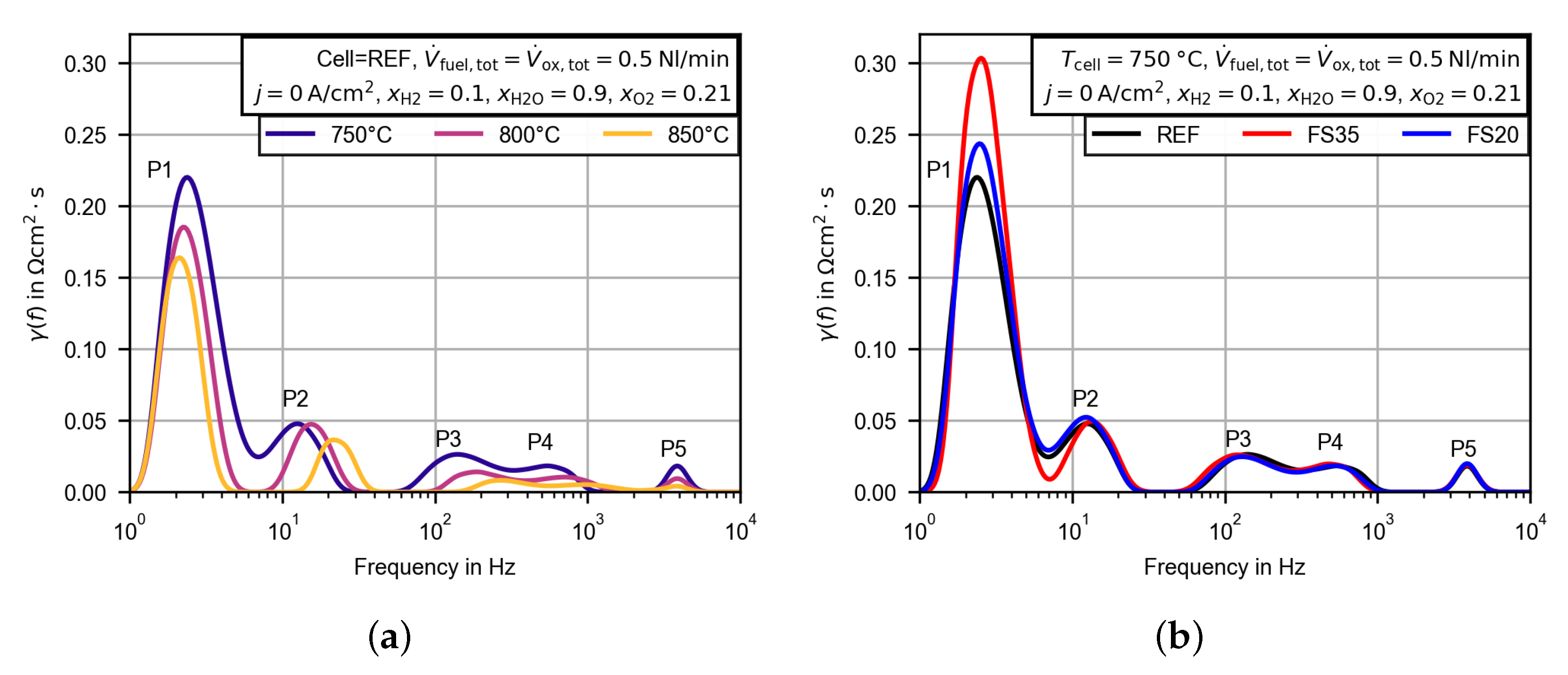
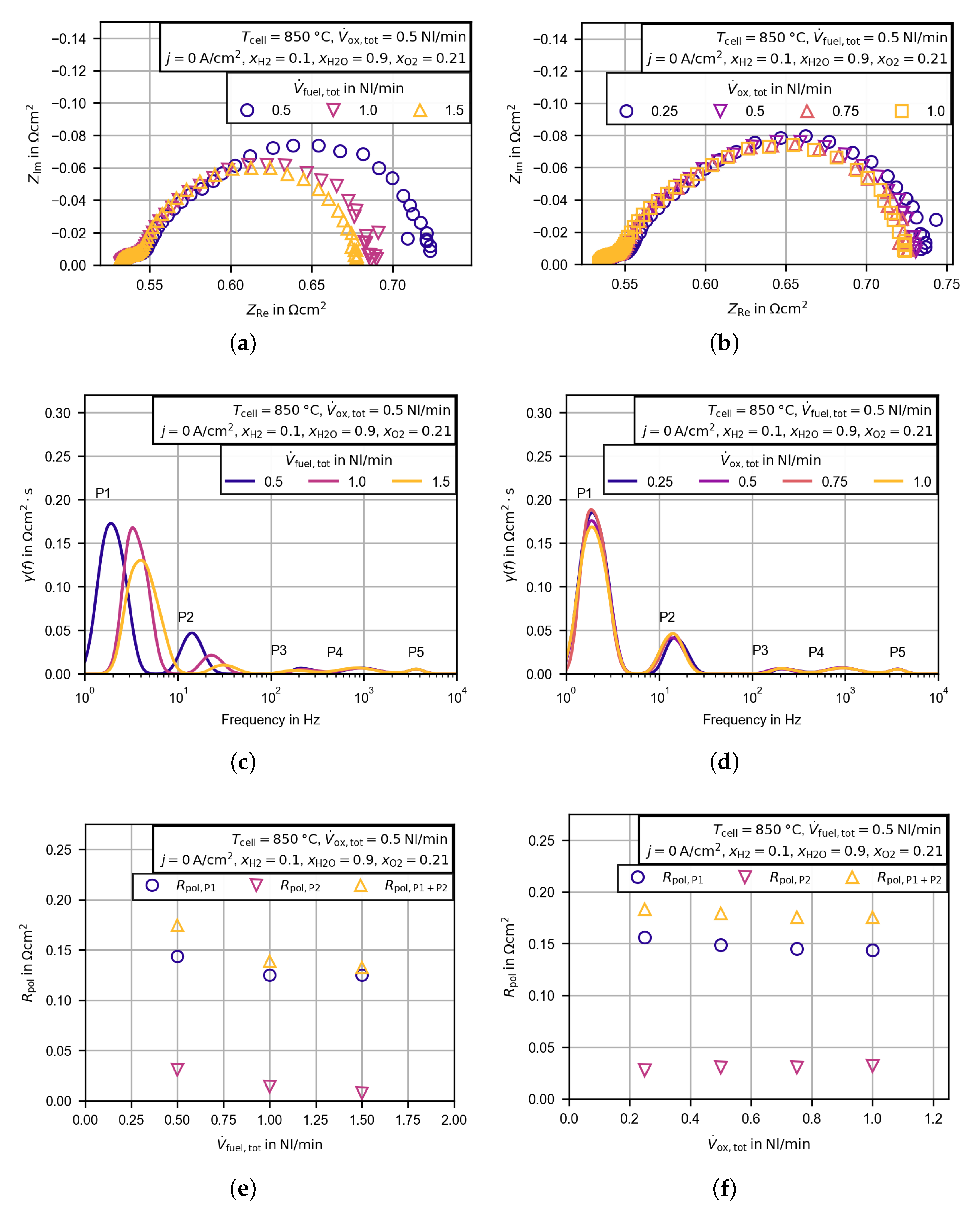
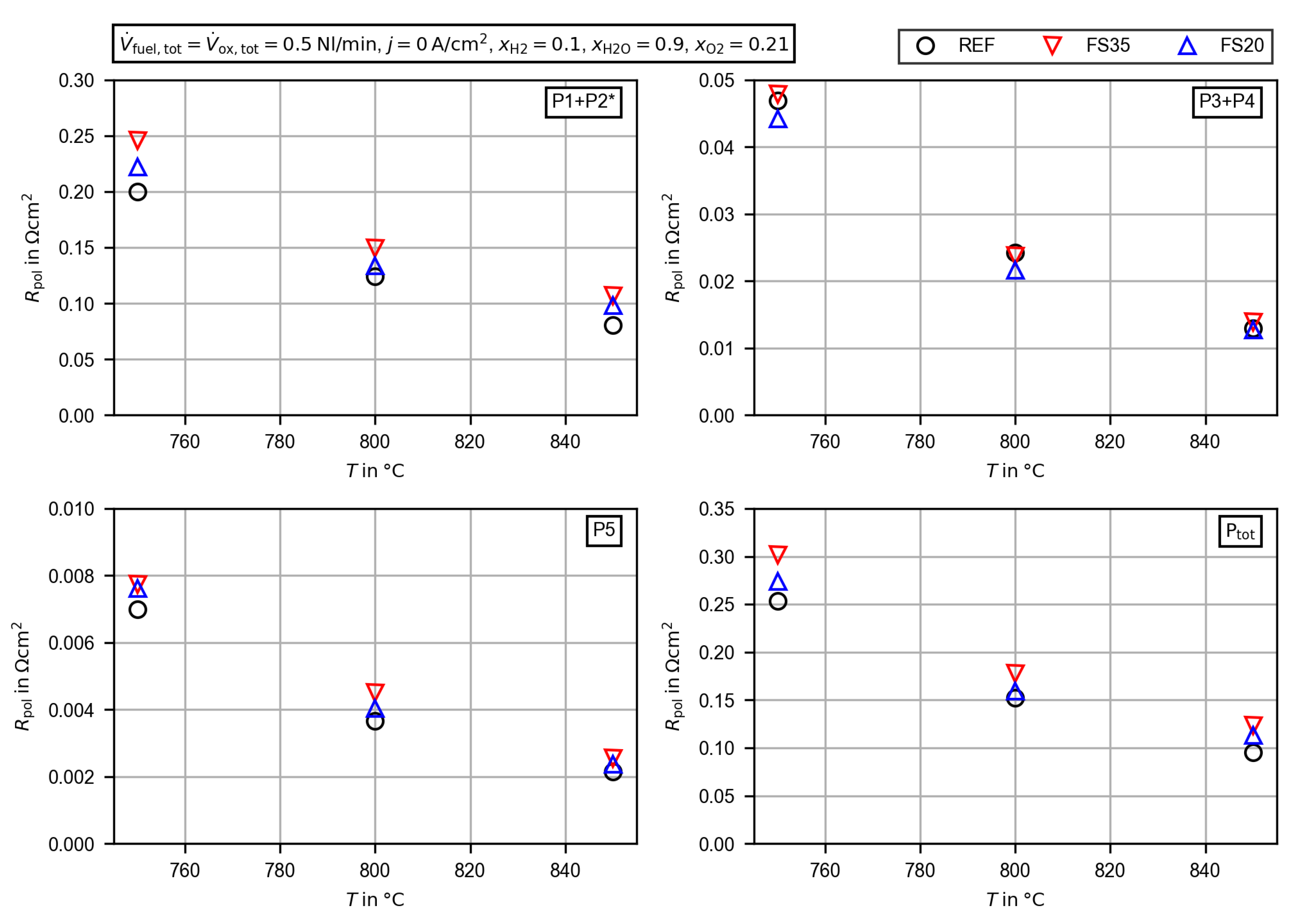
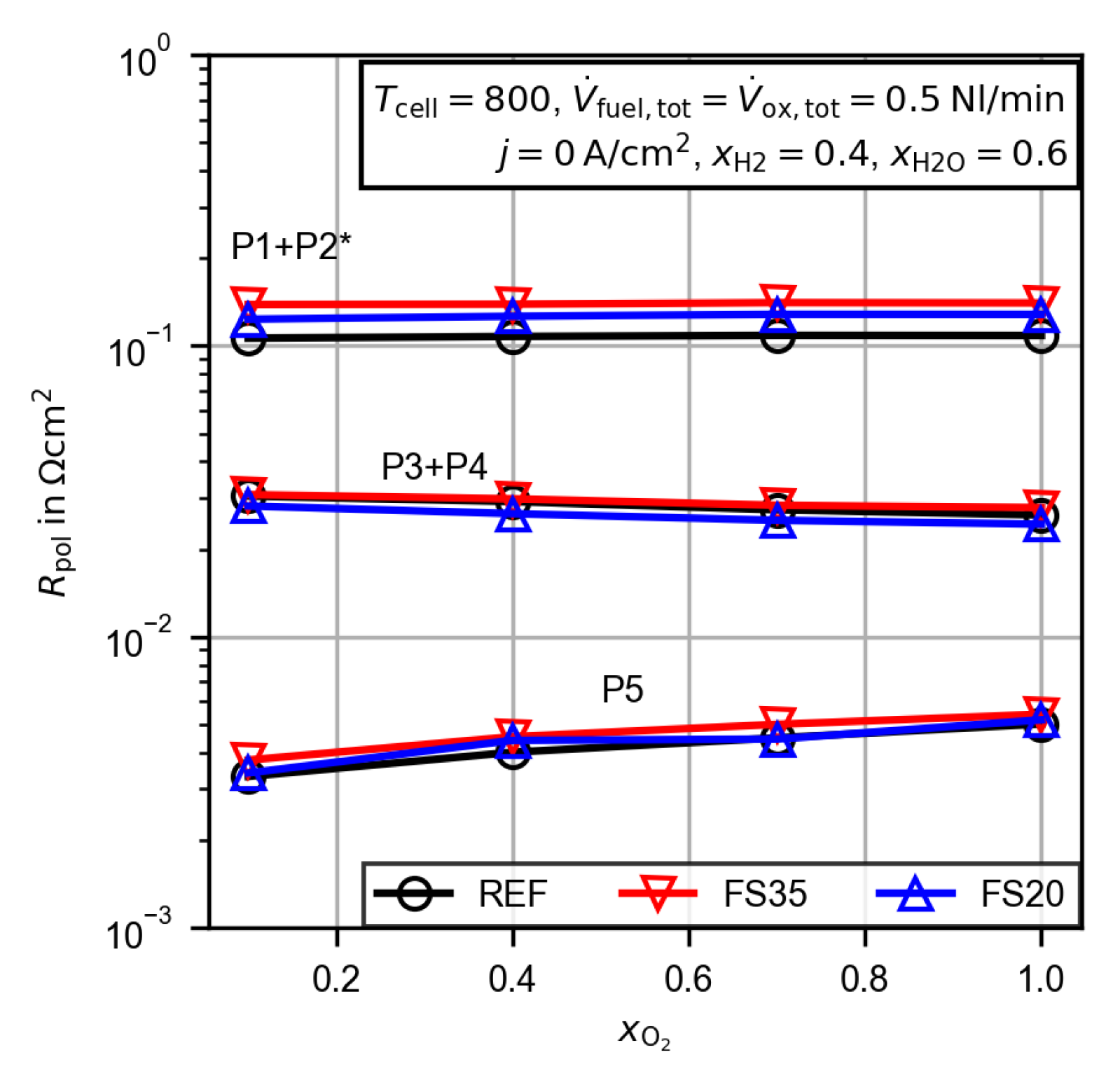
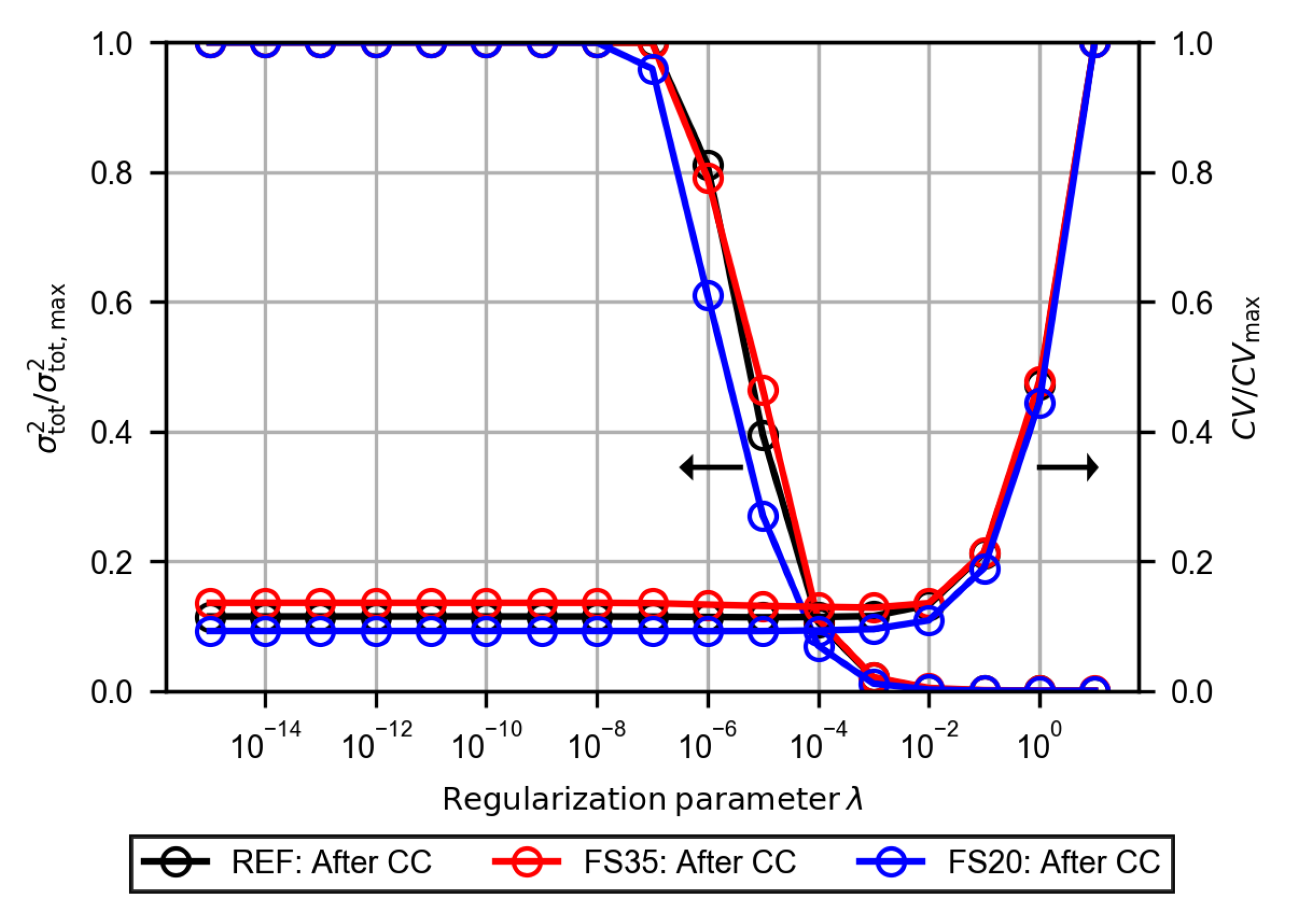
3.2.4. Evaluation of the Polarization Resistance
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASR | Area-specific resistance |
| CC | Constant current |
| DRT | Distribution of relaxation time |
| EIS | Electrochemical impedance spectroscopy |
| FS20 | Femtosecond laser-treated cell with line pitch |
| FS35 | Femtosecond laser-treated cell with line pitch |
| HFR | High frequency resistance |
| LSM | Laser scanning microscopy |
| REF | Reference cell |
| SEM | Scanning electron microscopy |
| SOC | Solid oxide cell |
| SOEL | Solid oxide water electrolysis |
Appendix A



References
- IEA. The Future of Hydrogen. Available online: https://www.iea.org/reports/the-future-of-hydrogen (accessed on 18 August 2020).
- Da Silva Veras, T.; Mozer, T.S.; da Costa Rubim Messeder dos Santos, D.; da Silva César, A. Hydrogen: Trends, production and characterization of the main process worldwide. Int. J. Hydrogen Energy 2017, 42, 2018–2033. [Google Scholar] [CrossRef]
- Arens, M.; Worrell, E.; Eichhammer, W.; Hasanbeigi, A.; Zhang, Q. Pathways to a low-carbon iron and steel industry in the medium-term—The case of Germany. J. Clean. Prod. 2017, 163, 84–98. [Google Scholar] [CrossRef]
- Timmerberg, S.; Kaltschmitt, M.; Finkbeiner, M. Hydrogen and hydrogen-derived fuels through methane decomposition of natural gas—GHG emissions and costs. Energy Convers. Manag. X 2020, 7, 100043. [Google Scholar]
- Buttler, A.; Spliethoff, H. Current status of water electrolysis for energy storage, grid balancing and sector coupling via power-to-gas and power-to-liquids: A review. Renew. Sustain. Energy Rev. 2018, 82, 2440–2454. [Google Scholar] [CrossRef]
- Brisse, A.; Zeller, M.; Ludwig, B.; Brabandt, J. Solid Oxide Electrolyzer System Operational at the H2 Refueling Station of Karlsruhe. Fuel Cells 2019, 19, 408–416. [Google Scholar]
- Lei, L.; Zhang, J.; Yuan, Z.; Liu, J.; Ni, M.; Chen, F. Progress Report on Proton Conducting Solid Oxide Electrolysis Cells. Adv. Funct. Mater. 2019, 29, 1903805. [Google Scholar] [CrossRef]
- Wood, A.; He, H.; Joia, T.; Krivy, M.; Steedman, D. Communication-Electrolysis at high efficiency with remarkable hydrogen production rates. J. Electrochem. Soc. 2016, 163, F327–F329. [Google Scholar] [CrossRef]
- Sun, X.; Hendriksen, P.; Mogensen, M.; Chen, M. Degradation in Solid Oxide Electrolysis Cells During Long Term Testing. Fuel Cells 2019, 19, 740–747. [Google Scholar] [CrossRef]
- Knibbe, R.; Traulsen, M.; Hauch, A.; Ebbesen, S.; Mogensen, M. Solid oxide electrolysis cells: Degradation at high current densities. J. Electrochem. Soc. 2010, 157, B1209–B1217. [Google Scholar] [CrossRef]
- Wang, Y.; Li, W.; Ma, L.; Li, W.; Liu, X. Degradation of solid oxide electrolysis cells: Phenomena, mechanisms, and emerging mitigation strategies—A review. J. Mater. Sci. Technol. 2020, 55, 35–55. [Google Scholar] [CrossRef]
- Schefold, J.; Brisse, A.; Poepke, H. 23,000 h steam electrolysis with an electrolyte supported solid oxide cell. Int. J. Hydrogen Energy 2017, 42, 13415–13426. [Google Scholar] [CrossRef]
- Schefold, J.; Brisse, A.; Surrey, A.; Walter, C. 80,000 current on/off cycles in a one year long steam electrolysis test with a solid oxide cell. Int. J. Hydrogen Energy 2020, 45, 5143–5154. [Google Scholar] [CrossRef]
- Kusnezoff, M.; Trofimenko, N.; Müller, M.; Michaelis, A. Influence of electrode design and contacting layers on performance of electrolyte supported SOFC/SOEC single cells. Materials 2016, 9, 906. [Google Scholar] [CrossRef] [PubMed]
- Laguna-Bercero, M. Recent advances in high temperature electrolysis using solid oxide fuel cells: A review. J. Power Sources 2012, 203, 4–16. [Google Scholar] [CrossRef]
- Larrea, A.; Sola, D.; Laguna-Bercero, M.; Pea, J.; Merino, R.; Orera, V. Self-supporting thin yttria-stabilised zirconia electrolytes for solid oxide fuel cells prepared by laser machining. J. Electrochem. Soc. 2011, 158, B1193–B1197. [Google Scholar] [CrossRef]
- Cebollero, J.; Laguna-Bercero, M.; Lahoz, R.; Silva, J.; Moreno, R.; Larrea, A. Optimization of laser-patterned YSZ-LSM composite cathode-electrolyte interfaces for solid oxide fuel cells. J. Eur. Ceram. Soc. 2019, 39, 3466–3474. [Google Scholar] [CrossRef]
- Heiroth, S.; Koch, J.; Lippert, T.; Wokaun, A.; Günther, D.; Garrelie, F.; Guillermin, M. Laser ablation characteristics of yttria-doped zirconia in the nanosecond and femtosecond regimes. J. Appl. Phys. 2010, 107, 014908. [Google Scholar] [CrossRef]
- Gabler, A.; Müller, C.; Rauscher, T.; Gimpel, T.; Hahn, R.; Köhring, M.; Kieback, B.; Röntzsch, L.; Schade, W. Ultrashort-pulse laser structured titanium surfaces with sputter-coated platinum catalyst as hydrogen evolution electrodes for alkaline water electrolysis. Int. J. Hydrogen Energy 2018, 43, 7216–7226. [Google Scholar] [CrossRef]
- Koj, M.; Gimpel, T.; Schade, W.; Turek, T. Laser structured nickel-iron electrodes for oxygen evolution in alkaline water electrolysis. Int. J. Hydrogen Energy 2019, 44, 12671–12684. [Google Scholar] [CrossRef]
- Rauscher, T.; Müller, C.; Gabler, A.; Gimpel, T.; Köhring, M.; Kieback, B.; Schade, W.; Röntzsch, L. Femtosecond-laser structuring of Ni electrodes for highly active hydrogen evolution. Electrochim. Acta 2017, 247, 1130–1139. [Google Scholar] [CrossRef]
- Suermann, M.; Gimpel, T.; Bühre, L.; Schade, W.; Bensmann, B.; Hanke-Rauschenbach, R. Femtosecond laser-induced surface structuring of the porous transport layers in proton exchange membrane water electrolysis. J. Mater. Chem. A 2020, 8, 4898–4910. [Google Scholar] [CrossRef]
- Schönleber, M.; Klotz, D.; Ivers-Tiffée, E. A Method for Improving the Robustness of linear Kramers-Kronig Validity Tests. Electrochim. Acta 2014, 131, 20–27. [Google Scholar] [CrossRef]
- Leonide, A.; Sonn, V.; Weber, A.; Ivers-Tiffe, E. Evaluation and modeling of the cell resistance in anode-supported solid oxide fuel cells. J. Electrochem. Soc. 2008, 155, B36–B41. [Google Scholar] [CrossRef]
- Subotić, V.; Königshofer, B.; Juričić, D.; Kusnezoff, M.; Schröttner, H.; Hochenauer, C.; Boškoski, P. Detailed insight into processes of reversible solid oxide cells and stacks using DRT analysis. Energy Convers. Manag. 2020, 226, 113509. [Google Scholar] [CrossRef]
- Weiß, A.; Schindler, S.; Galbiati, S.; Danzer, M.; Zeis, R. Distribution of Relaxation Times Analysis of High-Temperature PEM Fuel Cell Impedance Spectra. Electrochim. Acta 2017, 230, 391–398. [Google Scholar] [CrossRef]
- Hahn, M.; Rosenbach, D.; Krimalowski, A.; Nazarenus, T.; Moos, R.; Thelakkat, M.; Danzer, M. Investigating solid polymer and ceramic electrolytes for lithium-ion batteries by means of an extended Distribution of Relaxation Times analysis. Electrochim. Acta 2020, 344, 136060. [Google Scholar] [CrossRef]
- Wan, T.; Saccoccio, M.; Chen, C.; Ciucci, F. Influence of the Discretization Methods on the Distribution of Relaxation Times Deconvolution: Implementing Radial Basis Functions with DRTtools. Electrochim. Acta 2015, 184, 483–499. [Google Scholar] [CrossRef]
- Saccoccio, M.; Wan, T.; Chen, C.; Ciucci, F. Optimal regularization in distribution of relaxation times applied to electrochemical impedance spectroscopy: Ridge and Lasso regression methods—A theoretical and experimental Study. Electrochim. Acta 2014, 147, 470–482. [Google Scholar] [CrossRef]
- Schlüter, N.; Ernst, S.; Schroeder, U. Finding the Optimal Regularization Parameter in Distribution of Relaxation Times Analysis. ChemElectroChem 2019, 6, 6027–6037. [Google Scholar] [CrossRef]
- Schlüter, N.; Ernst, S.; Schröder, U. Direct Access to the Optimal Regularization Parameter in Distribution of Relaxation Times Analysis. ChemElectroChem 2020, 7, 3445–3458. [Google Scholar] [CrossRef]
- Kerafol Keramische Folien GmbH. Manufacturer Data Sheet. Available online: https://www.kerafol.com/sofc/komponenten-fuer-brennstoffzellentechnologie/elektrolytsubstrate (accessed on 20 October 2020).
- Primdahl, S.; Mogensen, M. Gas diffusion impedance in characterization of solid oxide fuel cell anodes. J. Electrochem. Soc. 1999, 146, 2827–2833. [Google Scholar] [CrossRef]
- Bessler, W.G.; Gewies, S. Gas Concentration Impedance of Solid Oxide Fuel Cell Anodes. J. Electrochem. Soc. 2007, 154, B548. [Google Scholar] [CrossRef]
- Geisler, H.; Kromp, A.; Hirn, S.; Weber, A.; Ivers-Tiffée, E. Model based interpretation of coupled gas conversion and diffusion in SOFC-Anodes. ECS Trans. 2013, 57, 2691–2704. [Google Scholar] [CrossRef]
- Sonn, V.; Leonide, A.; Ivers-Tiffe, E. Combined deconvolution and CNLS fitting approach applied on the impedance response of technical Ni8YSZ cermet electrodes. J. Electrochem. Soc. 2008, 155, B675–B679. [Google Scholar] [CrossRef]
- Riegraf, M.; Costa, R.; Schiller, G.; Friedrich, K.A.; Dierickx, S.; Weber, A. Electrochemical impedance analysis of symmetrical Ni/gadolinium-doped ceria (CGO10) electrodes in electrolyte-supported solid oxide cells. J. Electrochem. Soc. 2019, 166, F865–F872. [Google Scholar] [CrossRef]
- Bessler, W.G. Gas concentration impedance of solid oxide fuel cell anodes: I. Stagnation point flow geometry. J. Electrochem. Soc. 2006, 153, A1492–A1504. [Google Scholar] [CrossRef]
- Cebollero, J.; Lahoz, R.; Laguna-Bercero, M.; Larrea, A. Tailoring the electrode-electrolyte interface of Solid Oxide Fuel Cells (SOFC) by laser micro-patterning to improve their electrochemical performance. J. Power Sources 2017, 360, 336–344. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characterization Procedure | ||||||
|---|---|---|---|---|---|---|
| in | in | Fuel Electrode | Oxygen Electrode | |||
| in | in | |||||
| CC (200 h) | 800 | |||||
| H-variation (5 steps) | 800, 750, 850 | 0.1–0.9, 0.1 | ||||
| HO-variation (4 steps) | 800, 750, 850 | 0.1, 0.3–0.9 | ||||
| O-variation (4 steps) | 800, 750, 850 | 0.1–1 | ||||
| j-variation ( steps) | 800, 750, 850 | 0.2–0.8 | 0.2–0.8 | |||
| -characterisitc (3 steps) | 800, 750, 850 | 0.1–0.7, 0.9–0.3 | ||||
| Overload | 850 | |||||
| Parameter | REF | FS35 | FS20 | |||
|---|---|---|---|---|---|---|
| Before CC | After CC | Before CC | After CC | Before CC | After CC | |
| HFR in | 0.732 | 0.786 | 0.718 | 0.749 | 0.660 | 0.681 |
| ASR in | 0.885 | 0.952 | 0.903 | 0.950 | 0.837 | 0.864 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marquardt, T.; Hollmann, J.; Gimpel, T.; Schade, W.; Kabelac, S. Femtosecond Laser-Induced Surface Modification of the Electrolyte in Solid Oxide Electrolysis Cells. Energies 2020, 13, 6562. https://doi.org/10.3390/en13246562
Marquardt T, Hollmann J, Gimpel T, Schade W, Kabelac S. Femtosecond Laser-Induced Surface Modification of the Electrolyte in Solid Oxide Electrolysis Cells. Energies. 2020; 13(24):6562. https://doi.org/10.3390/en13246562
Chicago/Turabian StyleMarquardt, Tobias, Jan Hollmann, Thomas Gimpel, Wolfgang Schade, and Stephan Kabelac. 2020. "Femtosecond Laser-Induced Surface Modification of the Electrolyte in Solid Oxide Electrolysis Cells" Energies 13, no. 24: 6562. https://doi.org/10.3390/en13246562
APA StyleMarquardt, T., Hollmann, J., Gimpel, T., Schade, W., & Kabelac, S. (2020). Femtosecond Laser-Induced Surface Modification of the Electrolyte in Solid Oxide Electrolysis Cells. Energies, 13(24), 6562. https://doi.org/10.3390/en13246562