1. Introduction
As an environmentally sustainable process for green energy production, thermochemical energy storage (TCES) supported by concentrated solar power (CSP) has been at the center of the research and industry community for over two decades. The reason for this mobilization is that, along with TCES systems, CSP constitutes a carbon free technology that alleviates global warming and exploits the renewable aspects of solar energy, incorporating mature technologies for power production (i.e., Steam Rankine Cycle). The global installed capacity of CSP has seen a great increase over the last 10 years and approximate counts of 5 GW
e, with an escalating trend that could even double in 2020 [
1]. Increasing public attention to environmental protection and the political decisions that change the preferences of manufacturers from conventional to renewable solutions are the major factors influencing these market dynamics. Nevertheless, commercial intrusion into the global power market has so far been conservative, and definitely has not aligned with the requirements of this expanding economy.
Well-established CSP systems are used commercially nowadays, producing a sufficient amount of electricity. Sensible heat storage (i.e., molten salts, oils), latent heat storage (i.e., thermochemical energy storage (TCES) and phase change materials (PCMs)) belong to the broader family of energy storage systems. Molten salts, a sensible heat storage system, performs the most mature technology at an industrial scale. Although several commercial plants trade on these technologies, significant drawbacks have restrained their widespread use. For example, molten salt technologies suffer as a result of their corrosiveness [
2], their limited power-cycle efficiency due to limitations on their operating temperatures (i.e., temperatures above ~550 °C induce salt degradation) [
3] and the significant energy consumption continuously needed to abstain from salt solidification (molten salts suffer from severe solidification at temperatures below ~200 °C) [
4]. Moreover, due to unavoidable energy losses and intermittency, molten salts can only produce and store energy for short-term periods.
Recently, TCES systems integrated with CSP have emerged as an alternative for long-term energy storage and electricity production. In such systems, the heat is stored via an endothermic chemical reaction exploiting solar energy, which is released later during the reverse exothermic reaction. The products of both reactions can be stored according to the operating conditions of the installation, and TCES has numerous advantages over the previous technologies. Energy densities of the materials used in TCES re approximately 5–10 times higher than the materials used in latent and sensible heat storage systems, and storage periods are longer, as products can be stored at ambient conditions [
2].
Among diverse chemical systems for thermochemical energy storage, calcium looping (CaL)—which relies on the calcination–carbonation reaction system (Equation (1))—stands as one of the most promising technologies [
5]. Numerous advantages of this system such as high energy density (~3.2 GJ/m
3 compared with ~0.8 GJ/m
3 in commercially installed molten salts units) and the extremely low price of natural limestone CaCO
3 (~€10/ton) have been extensively discussed, among other factors, in the literature [
5,
6,
7]. Furthermore, the commercial potential of the CSP-CaL integration for thermochemical energy storage and power production has been theoretically studied in detail by numerous authors in terms of energy analysis [
5,
8,
9], exergy analysis [
10,
11] and the optimization of power cycles used for energy production [
12,
13], proving the potential commercialization of these systems.
The feasible transition of CSP-CaL systems from a research scale towards a highly competitive industrial scale will require the development of rigorous mathematical models to be used as tools that can simulate and predict process activities in a systematic and consistent manner. However, one can see from the current literature that has been no adequate published research work in relation to the development of steady-state or dynamic mathematical reactor models. The literature has extensively explored how calcination and carbonation reactions can be accomplished in different reactor types [
14], but recent model developments of both reactors have referred mainly to applications relevant to post-combustion CO
2 capture, and reaction conditions and kinetics in these applications are different from those used in CSP-CaL processes. Typical conditions in CO
2 capture applications involve low CO
2 partial pressures that exit from the flue gases of power plants (~10% vol.) and temperatures around 650 °C during carbonation step. Under these conditions, and due to the sulphation of CaO and the multiple number of cycles, a material’s reactivity significantly drops in its residual value by 0.07–0.10 [
15]. Moreover, the reactivity of the material depends on the calcination/carbonation conditions and grain size, as well as the origin of the solids [
7].
In recent years, there have been numerous published works that have examined the simulation of heterogeneous gas–solid reaction systems in drop-tube or downflow reactors using mathematical models with different levels of complexity and accuracy [
16,
17,
18,
19,
20,
21,
22,
23,
24]. Drop-tube reactors present a number of advantages compared to other types of reactors (i.e., risers), and are well known in the chemical and petrochemical industries. In a downer reactor, both solid and gas phases enter the reactor from the top, flowing concurrently under gravity and momentum of their inlet flows, and which results in a more uniform hydrodynamic flow and a better control of the contact between gas and solid phases [
25] than the back-mixing flow and non-uniform radial distribution of solids that occurs in riser reactors [
26,
27]. The principles of these modelling techniques are used as a prototype in the current manuscript to provide a means by which to develop a detailed mathematical model of a drop-tube carbonator reactor as a provision for scale-up studies and industrial applications in CSP-CaL plants. Deng et al. [
24] set up and implemented a one-dimensional (1D) model based on mass and momentum balance equations along with a hypothetical kinetic model for a downer reactor. The results pointed out the effect of varying superficial velocities on a reactor’s performance. Chuachuensuk et al. [
21] developed a 1D model to describe the hydrodynamic characteristics and reaction kinetics of a downer regenerator reactor during the fluid catalytic cracking (FCC) process. The control of operating variables and reaction conditions was examined in terms of reactor performance. Moreover, Kasule et al. [
18] developed a detailed mathematical model for the gasification of biomass in a single-stage, downward-firing, entrained-flow gasifier whereas the dynamic terms are involved later in [
28]. Other similar studies have focused on the modelling strategies of different kinds of heterogeneous gas–solid reaction systems such as the oxidation of copper particles at high temperatures in a downer reactor [
22], or the carbonation of CaO for CO
2 capture applications in cement kilns [
20]. Finally, two recent studies have presented the construction of novel experimental facilities for the carbonation of fine CaO particles in a drop-tube reactor [
29] and an entrained-flow reactor [
30], examining the efficiency of the decarbonization of gases when exposed to different conditions and materials. To the best of our knowledge, there has not been any published work related to an experimental or model-based investigation of CaO carbonation in a drop-tube reactor that has focused on the relevant conditions for thermochemical energy storage applications.
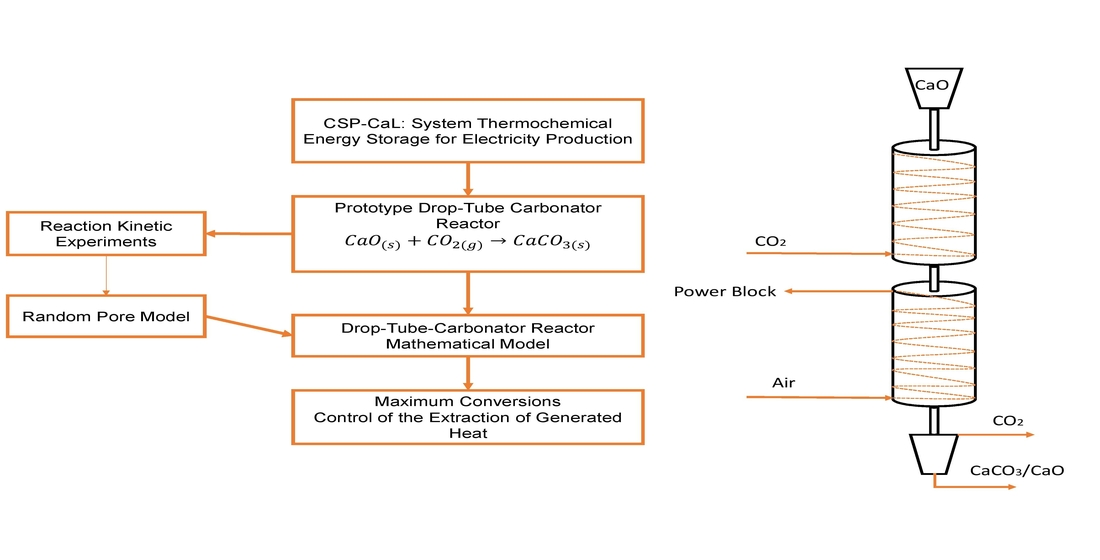
In this study, the development of a meticulous model of a tailor-made carbonator reactor for CSP-CaL integration is attempted for the first time, as part of the activities performed within the H2020 SOCRATCES project [
31]. A detailed one-dimensional (1D), steady-state, heterogeneous reaction model of a drop-tube carbonator reactor was developed, focusing on simulation studies to reveal the operating performance of a 10 kW
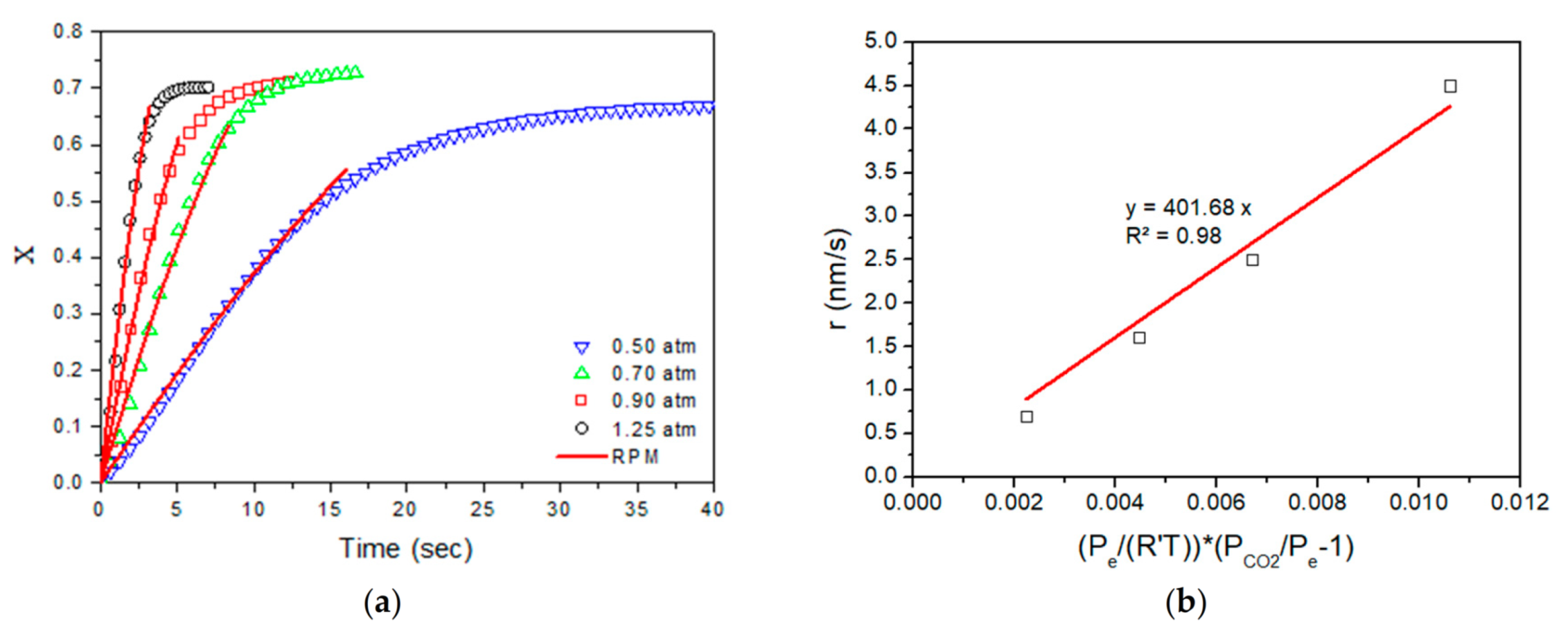
th reactor under different conditions. Mass, energy and momentum equations were considered for the two phases (gas and solid) derived by simplified Navier–Stokes equations. Moreover, the model included radiative heat transfer mechanisms and a random pore model (RPM) for carbonation kinetics that were validated through experimental work under conditions relevant to TCES. The main scope of this work is to demonstrate the model’s capabilities, and to describe the operations of a fixed-dimension prototype drop-tube carbonator reactor. The simulation studies investigate its performance, concerning the conversion and distribution of thermal power production across the length of the reactor, under different operating conditions. Subsequently, this will lead to future optimization, design and further scale-up studies. For this reason, critical variables were parametrically studied (i.e., the reactor’s wall temperatures, flows, carbonation pressure, etc.) with respect to process performance.
2. Concentrated Solar Power (CSP) and Calcium Looping (CaL) Concept Description
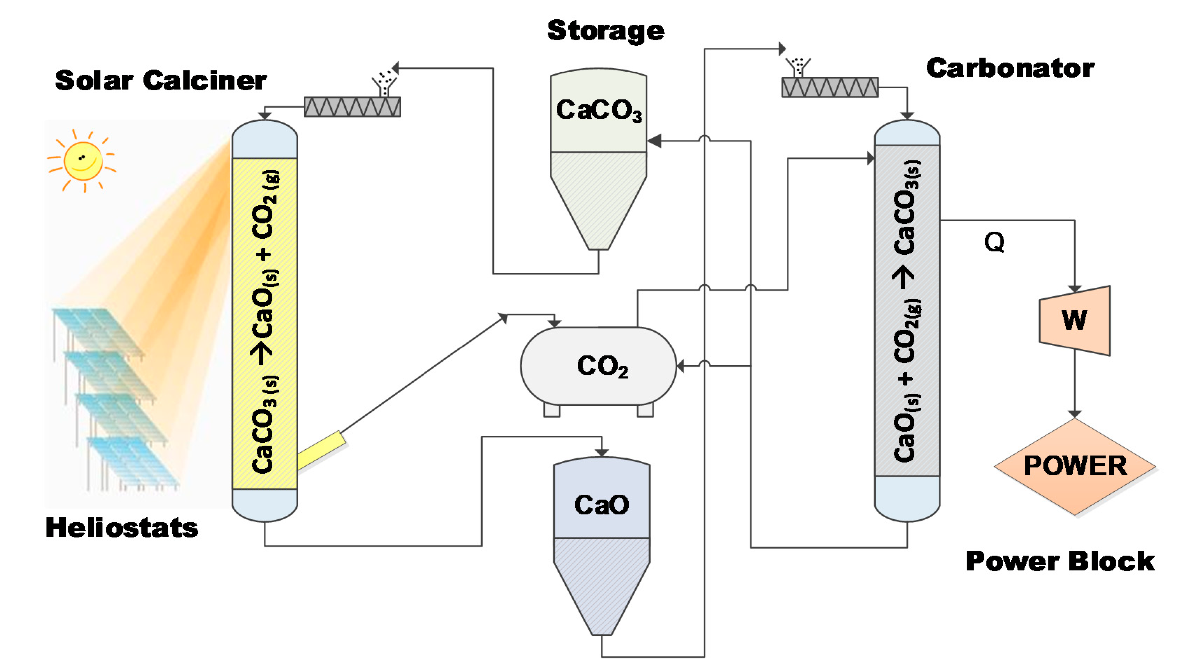
For the first time within a European Union (EU) research project [
31], a CSP-CaL integrated system was tested to prove that stored thermochemical energy can be effectively used for electricity production. The prototype pilot plant (comprising a calciner, carbonator and power block) as well as storage modules are depicted in
Figure 1. In this plant, solar energy is exploited to accomplish an endothermic calcination reaction where limestone (CaCO
3) is decomposed towards CaO and CO
2 (Equation (1)) in the calciner. The products are stored in vessels through the storage section, then react in the carbonator reactor to recover a large part of thermal energy through the exothermic carbonation reaction. The concept is completed via an engine (
Figure 1) in the power block where this energy is explicitly used to produce electricity.
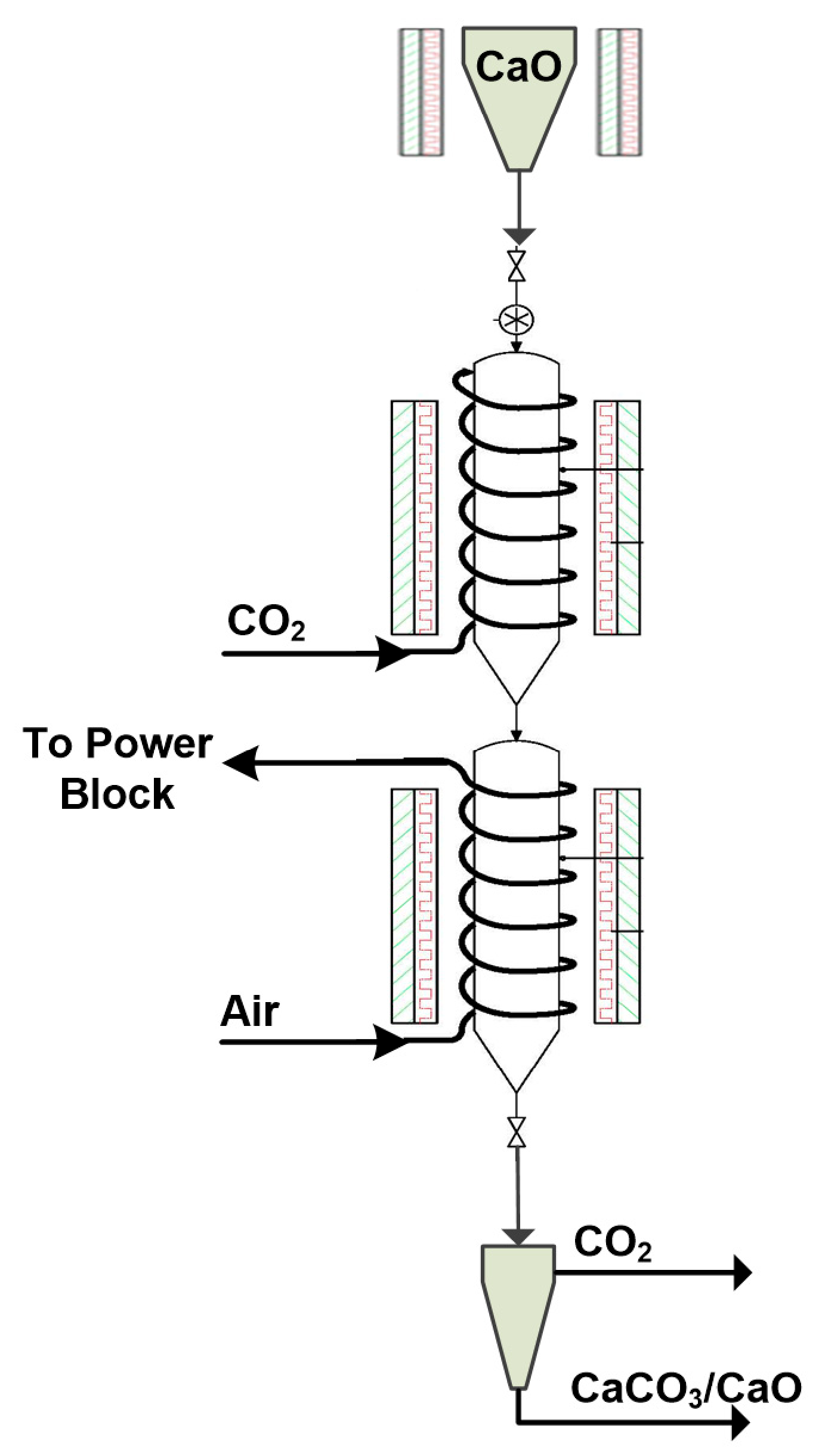
The carbonator reactor resembles a drop-tube (
Figure 2) and is suitable for a wide range of fast gas–solid reaction systems [
18,
20,
21,
22]. It is divided into two sections and the dimensions selected (~0.154 m diameter and ~4 m length) are appropriate to achieve a long solid phase residence time. The initially calcined solids (CaO) are stored in a heated tank (~200 °C) on top of the reactor and enter into it mechanically via a screw feeder device. Pure CO
2 is provided by pressurized bottles, entering from the top. A heat exchanger composed of a set of helical coils encircling the reactor is used for the removal of the heat released by the exothermic reaction. In the first reaction section, the CO
2 reactant passes through the coils to preheat before entering the reactor, while in the second section, fresh air is used as the heat transfer fluid to act as a cheap and effective heat sink (
Figure 2) [
17]. The produced hot air is later used in the power block to provide heat for electricity generation. The two reactor segments are surrounded by electric furnaces that preheat the carbonator system in order to trigger the carbonation reaction (~400–500 °C).
7. Conclusions
In the present study, a one-dimensional, steady-state mathematical model was developed for the first time for a heterogenous gas–solid carbonation reaction, in a drop-tube reactor, focusing on thermochemical energy storage conditions and based on the calcium looping process. The reactor model related mass, energy and momentum balances for both gas and solid phases under the Euler–Euler approach, while a random pore model (RPM) was developed for the kinetics and verified experimentally in a laboratory fix-bed reactor using fine particles (i.e., ~60 μm average diameter). In this manuscript, the reactor model was implemented to simulate a fix-dimensioned, drop-tube carbonator reactor prototype (i.e., 0.154 m diameter and 4 m length) with 10 kW
th thermal energy that was designed and constructed as part of the SOCRATCES project [
31].
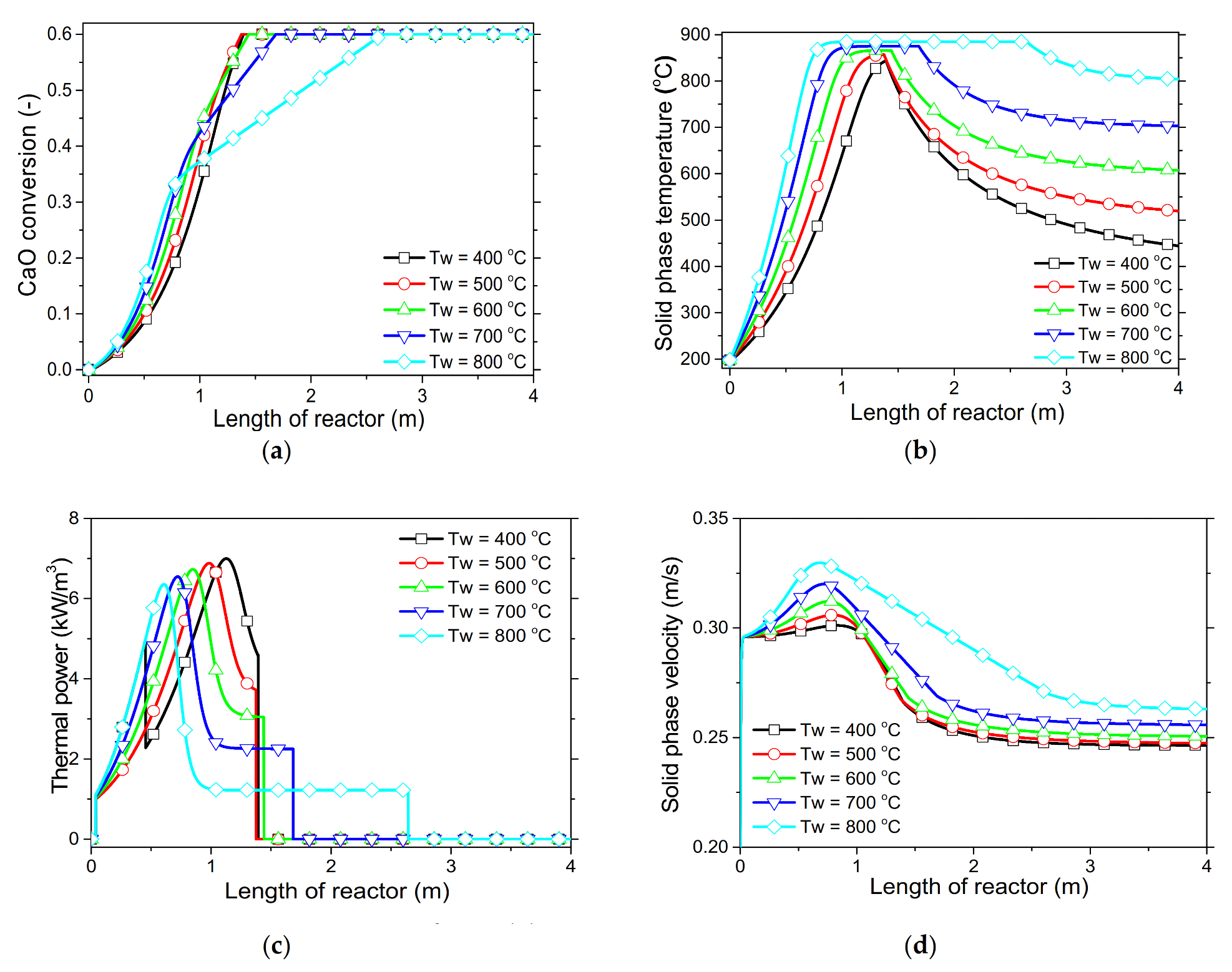
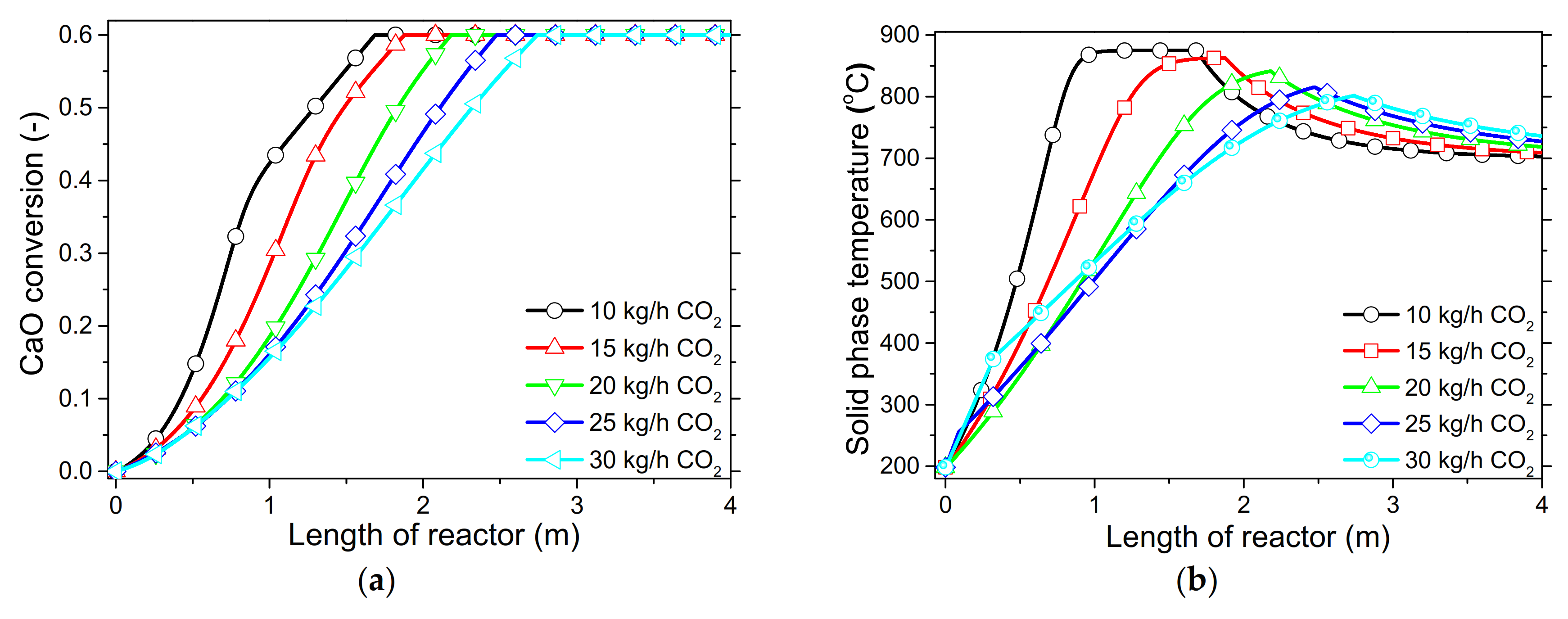
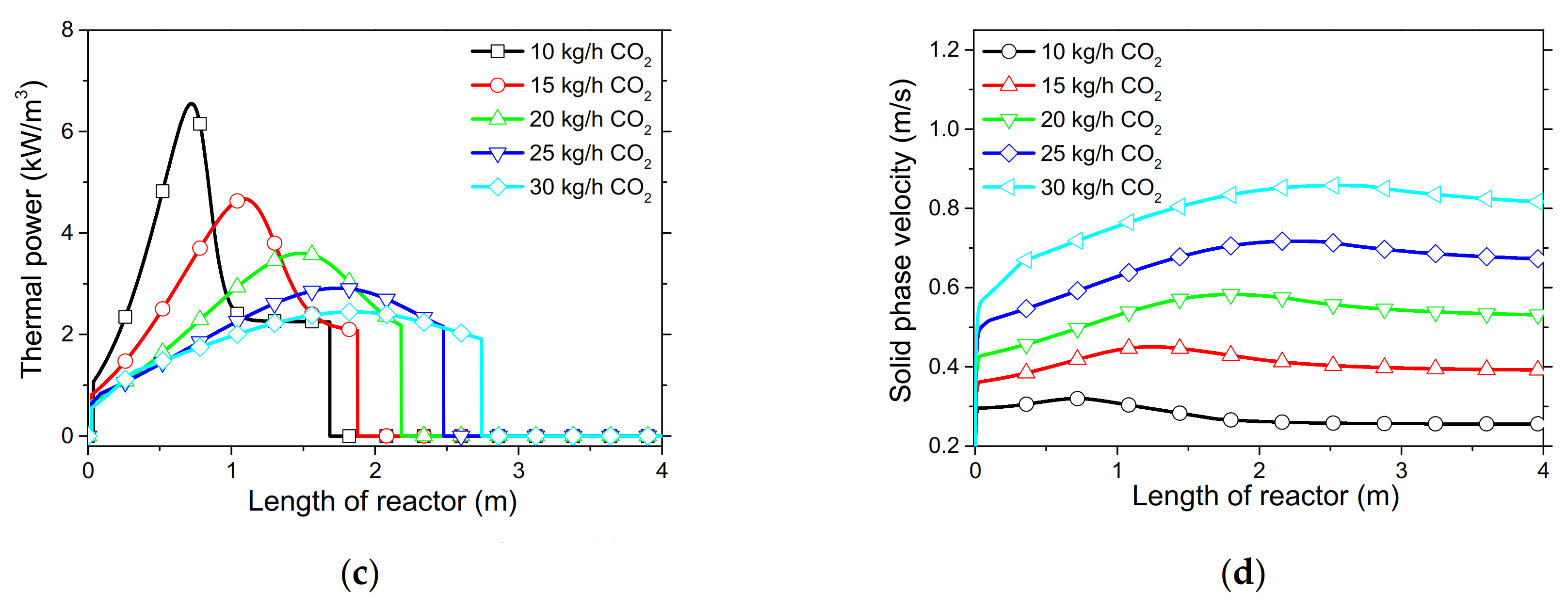
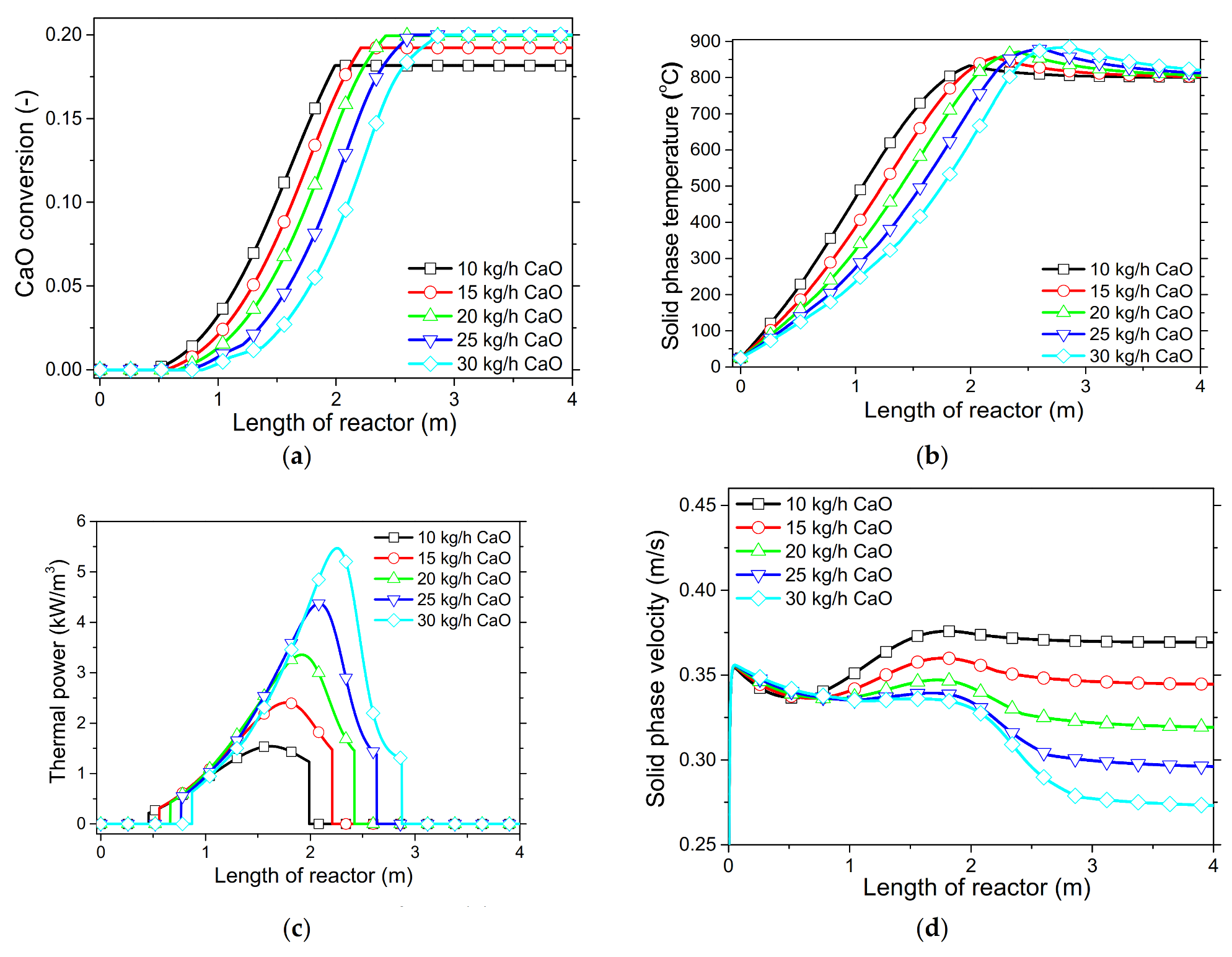
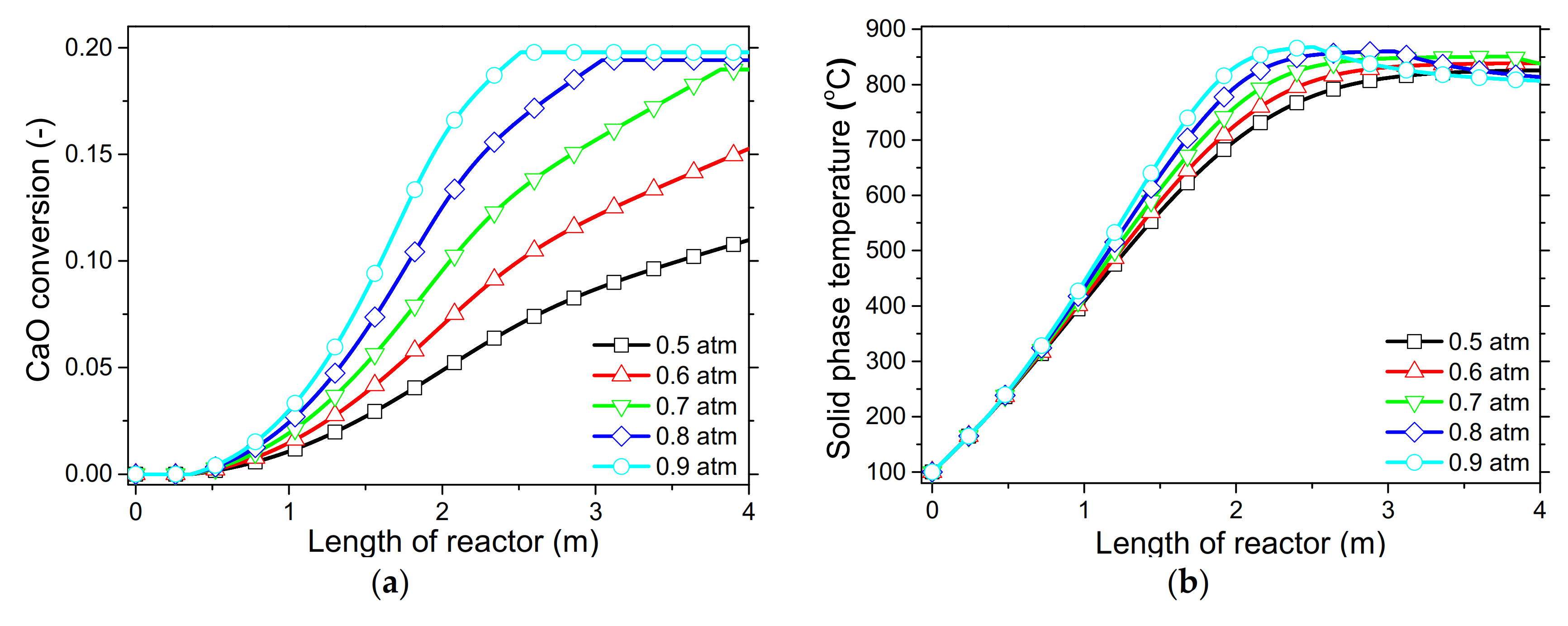
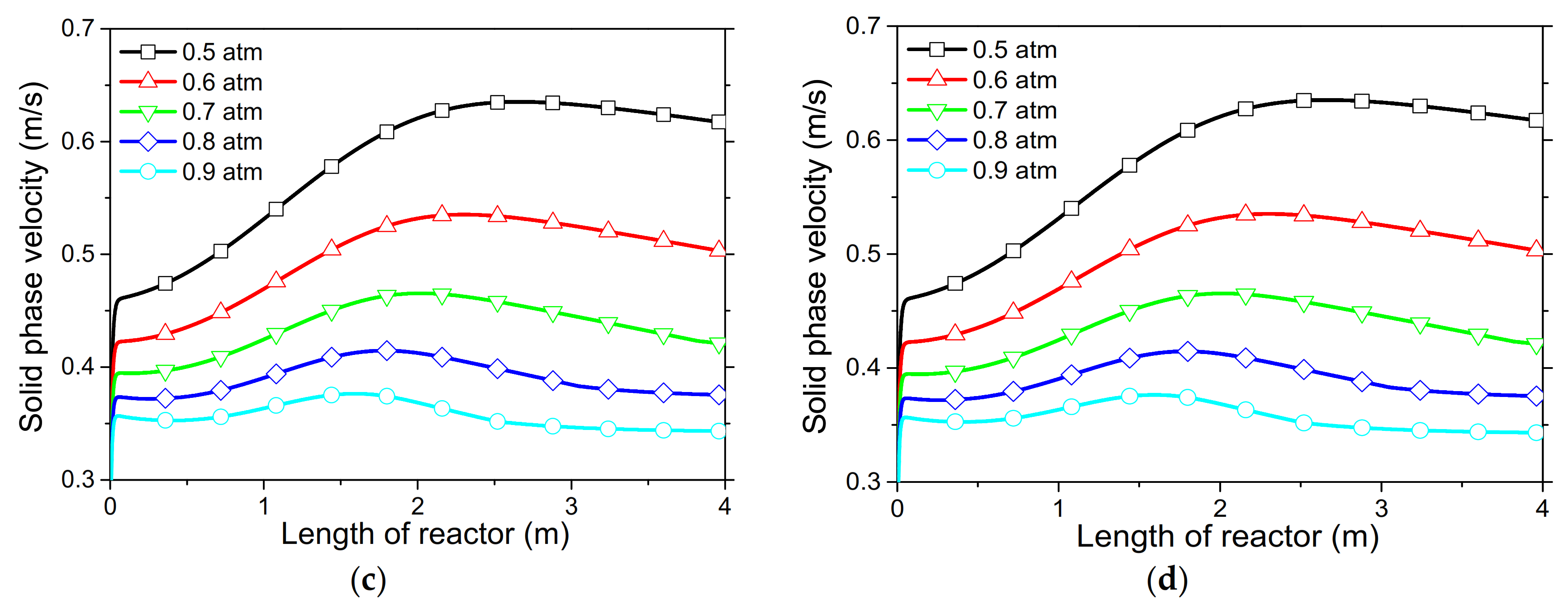
In order to demonstrate the model’s capabilities and assess the reactor’s performance concerning the conversion and distribution of thermal energy produced along the length of reactor, several simulations were performed that focused on the variations of critical process variables. Initially, the reactor’s wall temperature and CO2 flow rate were parametrically examined by means of the reactor’s performance under the condition of “fresh CaO” (lime after first calcination). Significant effects of these variables could be seen in the final CaO conversion, reaction rate variations across the reactor and temperature profiles on the inner side of the reactor. Next, the effect of CaO flow rate and carbonation pressure were investigated for the case of “sintered CaO” (lime after 10 carbonation/calcination cycles). Carbonation pressure proved to be a critical variable, as it radically affects the reaction rate.
Simulation results indicated that carbonation of fine particles can be successfully accomplished (~60% conversion for fresh CaO) in novel drop-tube reactors with a downflow gravity direction using either with fresh or sintered materials. The cooling of the outer walls of the reactor might lead to lower, safer wall temperatures (~600 °C), as it was proven that very high temperatures in the reactor inhibited the reaction rate when equilibrium was quickly reached. Moreover, the residence time of reactants constituted a decisive parameter for the process performance in regards to the deployment of the thermal energy produced along the reactor, as 20–30 kg/h of CO2 resulted in the completion of the reaction across a wider range inside the reactor. Maximum sorbent conversions (20%) were reached in the cases of 25 and 30 kg/h of sintered CaO flow rates. The above conclusions were deduced from the reactor’s specific dimensions. Lower carbonation pressures (~0.7 atm) might be used to flatten the reaction front, making it more feasible to control the extraction of the generated heat. This could steer research towards model-based optimization strategies that will enhance future design and further scale-up studies of different carbonator reactor sizes.

 ,
, 
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}