Characterization of Chemically and Physically Activated Carbons from Lignocellulosic Ethanol Lignin-Rich Stream via Hydrothermal Carbonization and Slow Pyrolysis Pretreatment

 ,
,  ,
,  and
and 
Abstract

1. Introduction
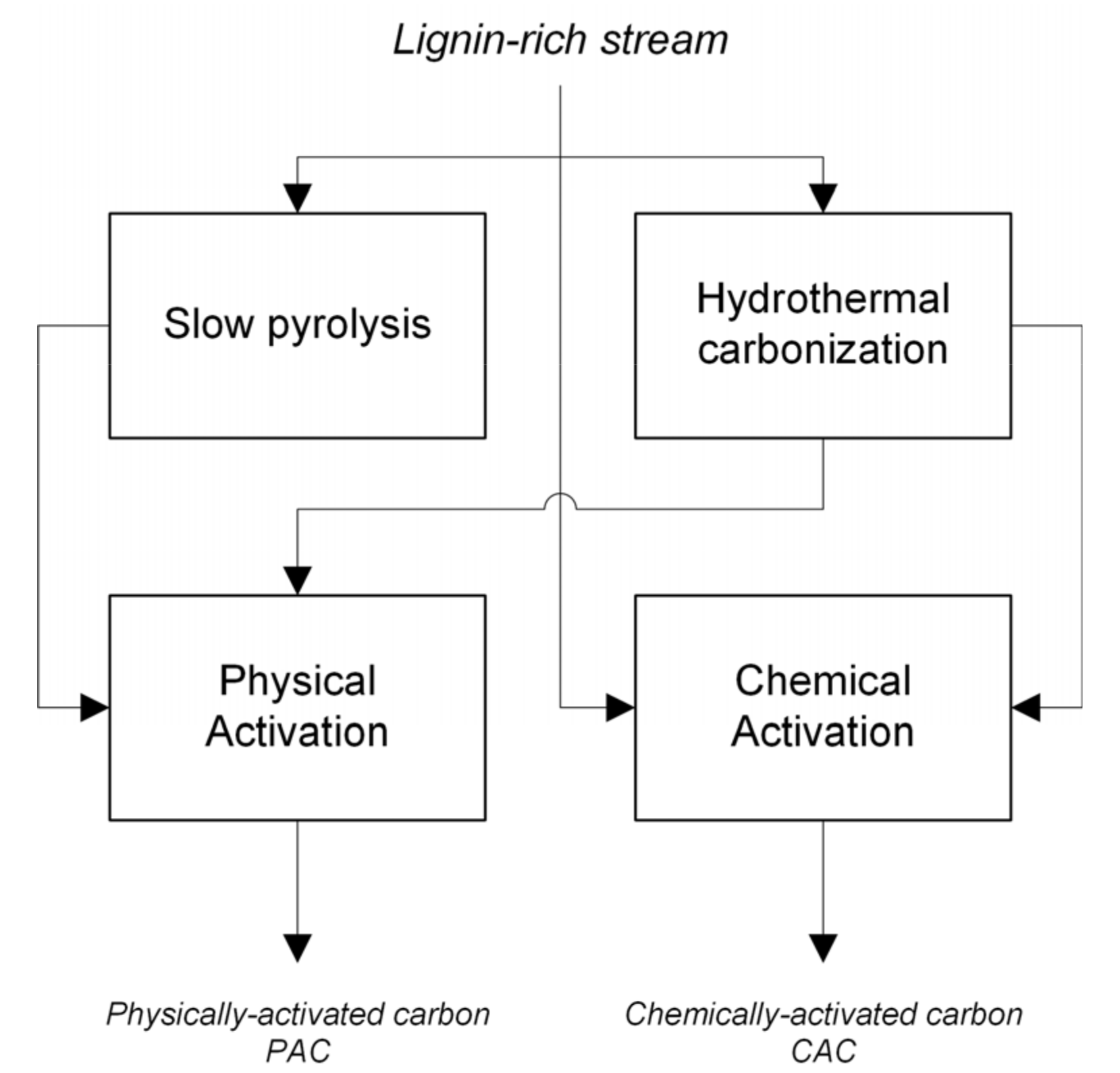
2. Materials and Methods
2.1. Feedstock
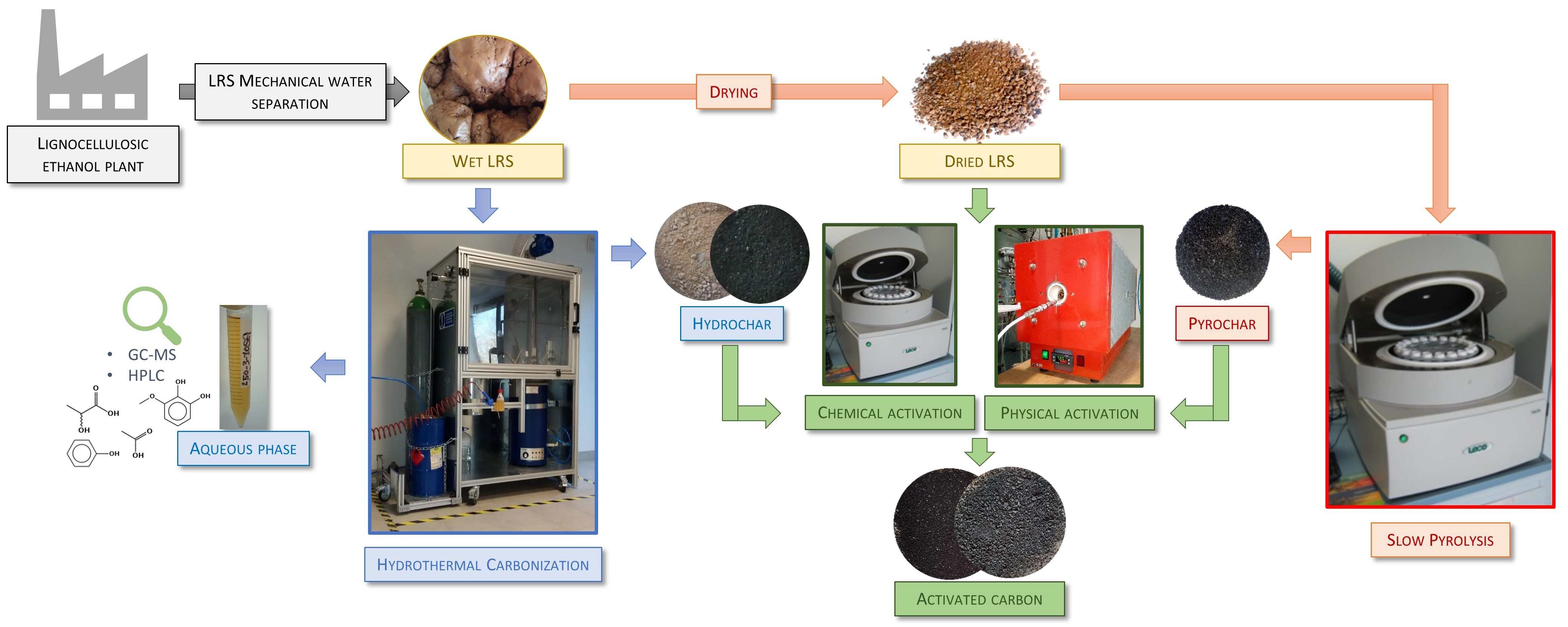
2.2. Experimental Equipment and Procedure
2.2.1. Hydrothermal Carbonization
2.2.2. Slow Pyrolysis
2.2.3. Physical Activation
2.2.4. Chemical Activation
2.3. Analytical Methods and Chemicals
2.3.1. Analysis of Solids
2.3.2. Analysis of HTC Aqueous Phase
2.3.3. Chemicals
3. Results
3.1. Lignin-Rich Stream Characterization
3.2. Preliminary Lignin-Rich Stream Activation
3.2.1. Physical Activation
3.2.2. Chemical Activation
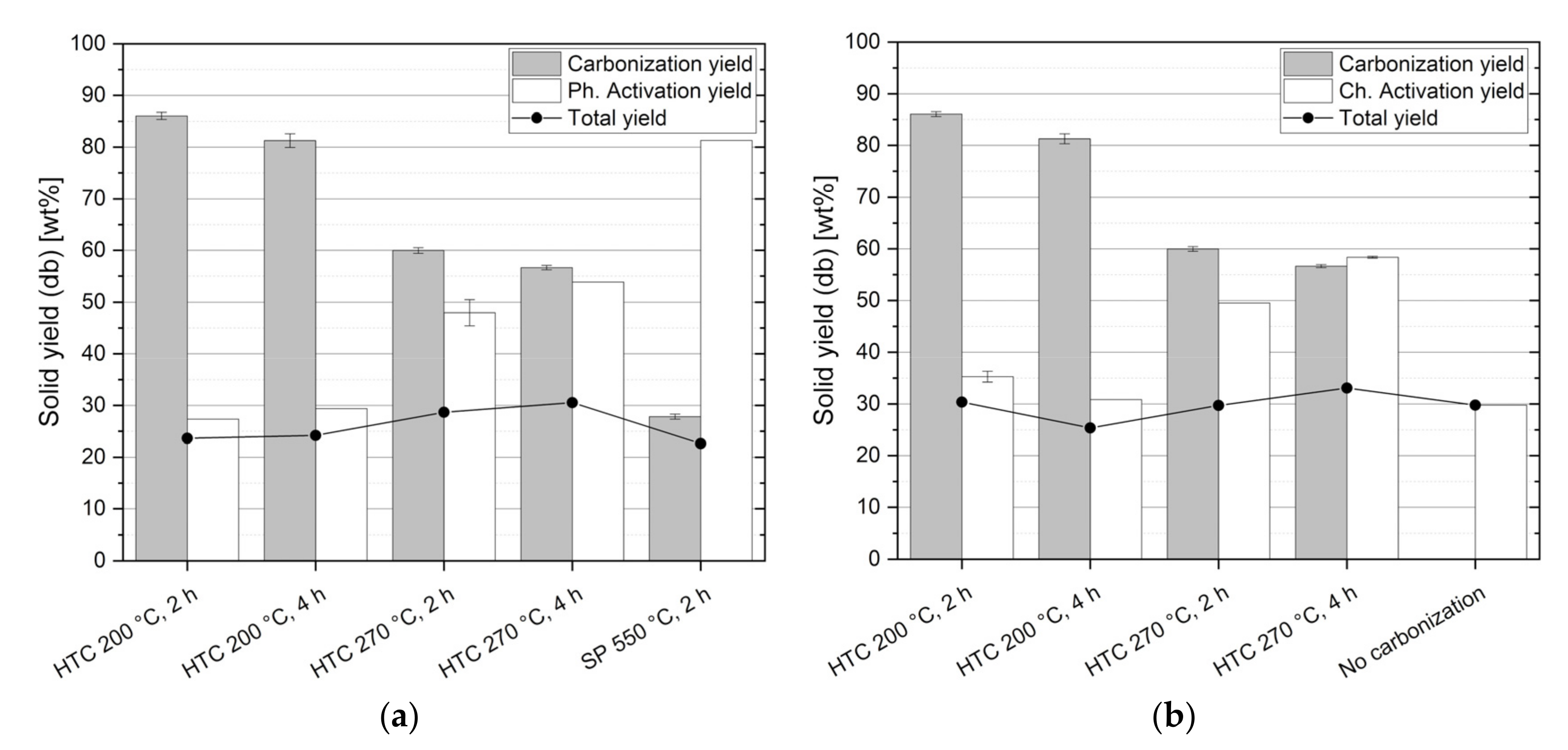
3.3. Solids Yields
3.4. FT-IR Analysis
3.5. Char Characterization
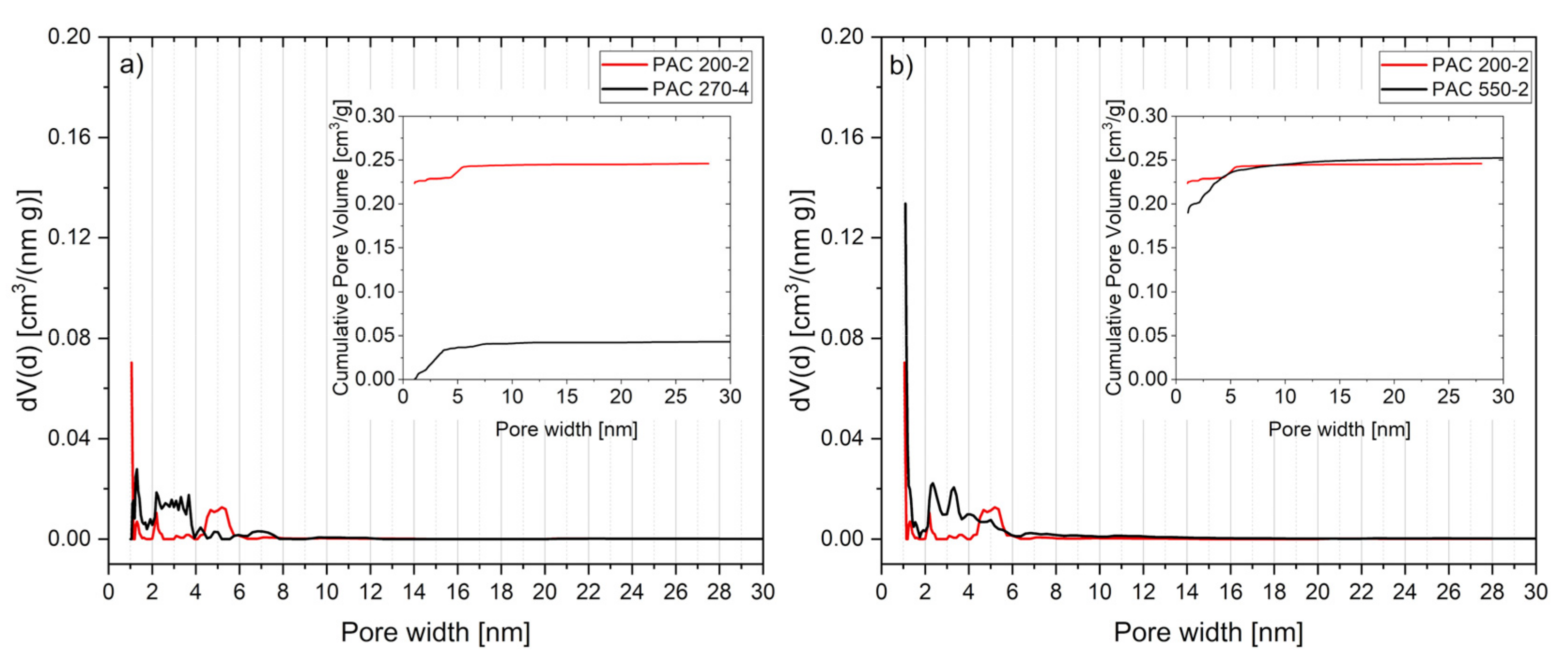
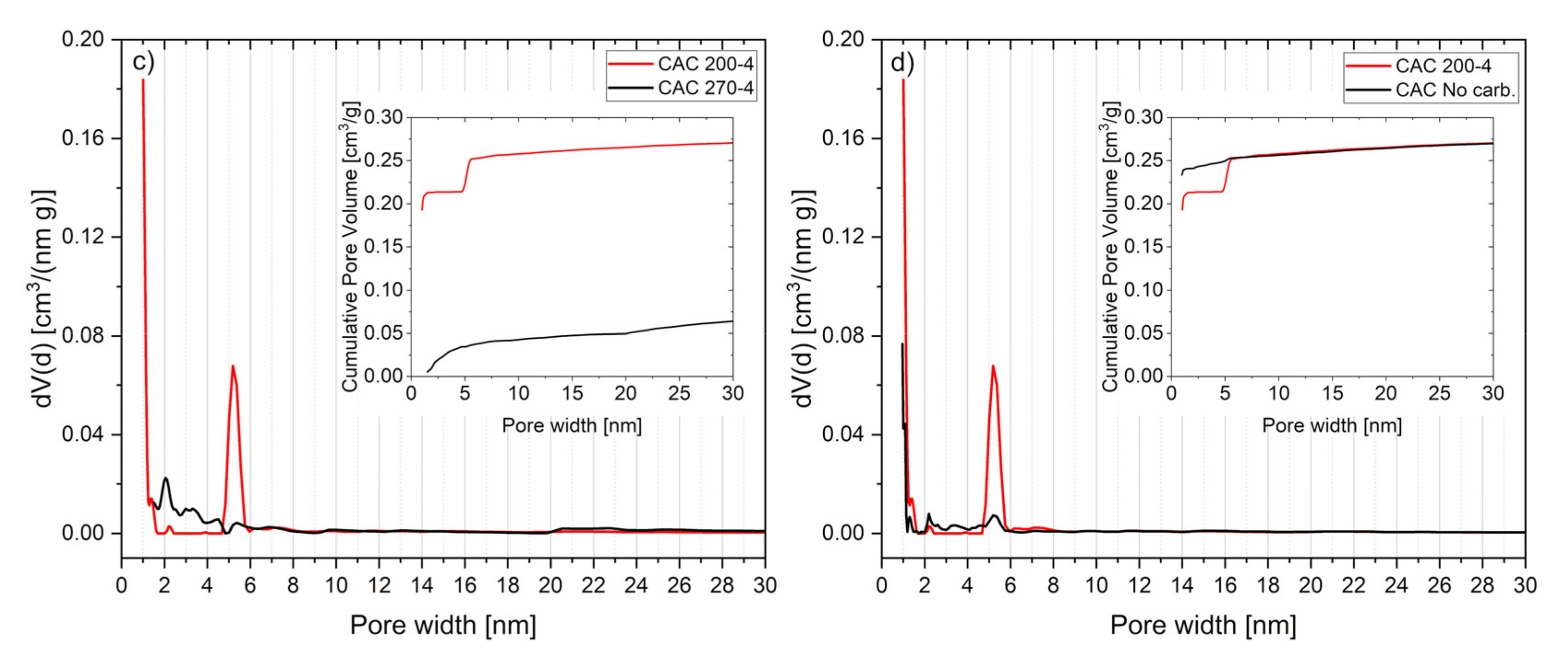
3.6. Activated Carbon Characterization
3.7. HTC Aqueous Phase Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- European Environment Agency, Sectoral Greenhouse Gas Emissions by IPCC Sector. Available online: https://www.eea.europa.eu/data-and-maps/daviz/change-of-co2-eq-emissions-2#tab-dashboard-01 (accessed on 9 January 2019).
- Lynd, L.R. The grand challenge of cellulosic biofuels. Nat. Biotechnol. 2017, 35, 912–915. [Google Scholar] [CrossRef]
- Chiaramonti, D.; Goumas, T. Impacts on industrial-scale market deployment of advanced biofuels and recycled carbon fuels from the EU Renewable Energy Directive II. Appl. Energy 2019, 251, 113351. [Google Scholar] [CrossRef]
- Lee, S.; Speight, J.G.; Loyalka, S.K. Handbook of Alternative Fuel Technologies; CRC Press, Taylor & Francis Group LLC: Boca Raton, FL, USA, 2007. [Google Scholar] [CrossRef]
- Aden, A.; Ruth, M.; Ibsen, K.; Jechura, J.; Neeves, K.; Sheehan, J.; Wallace, B.; Montague, L.; Slayton, A.; Lukás, J. Lignocellulosic Biomass to Ethanol Process Design and Economics Utilizing Co-Current Dilute Acid Prehydrolysis and Enzymatic Hydrolysis for Corn Stover; NREL: Golden, CO, USA, 2002; Available online: https://www.nrel.gov/docs/fy02osti/32438.pdf (accessed on 8 February 2017).
- Grand View Research Inc., Activated Carbon Market Size, Share & Trends Analysis Report by Product (Powdered Activated Carbon, Granular Activated Carbon), by Application, by End-use, and Segment Forecasts, 2018–2024. 2018. Available online: https://www.grandviewresearch.com/industry-analysis/activated-carbon-market (accessed on 11 March 2020).
- Marsh, H.; Rodríguez-Reinoso, F. Activated Carbon; Elsevier: Amsterdam, The Netherlands, 2006; pp. 27–316. [Google Scholar]
- Correa, C.R.; Stollovsky, M.; Hehr, T.; Rauscher, Y.; Rolli, B.; Kruse, A. Influence of the Carbonization Process on Activated Carbon Properties from Lignin and Lignin-Rich Biomasses. ACS Sustain. Chem. Eng. 2017, 5, 8222–8233. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, G.; Zhang, J.; Wang, Y.; Yao, M. Activated Carbon Preparation from Lignin by H3PO4 Activation and Its Application to Gas Separation. Chem. Eng. Technol. 2011, 35, 309–316. [Google Scholar] [CrossRef]
- Sebbahi, S.; Ahmido, A.; Kifani-Sahban, F.; El Hajjaji, S.; Zoulalian, A. Preoxidation and Activation of the Lignin Char: Carbonization and Oxidation Procedures. J. Eng. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Kang, S.; Li, X.; Fan, J.; Chang, J. Characterization of Hydrochars Produced by Hydrothermal Carbonization of Lignin, Cellulose, d-Xylose, and Wood Meal. Ind. Eng. Chem. Res. 2012, 51, 9023–9031. [Google Scholar] [CrossRef]
- Demir, M.; Kahveci, Z.; Aksoy, B.; Palapati, N.K.R.; Subramanian, A.; Cullinan, H.T.; El-Kaderi, H.M.; Harris, C.T.; Gupta, R.B. Graphitic Biocarbon from Metal-Catalyzed Hydrothermal Carbonization of Lignin. Ind. Eng. Chem. Res. 2015, 54, 10731–10739. [Google Scholar] [CrossRef]
- Dinjus, E.; Kruse, A.; Tröger, N. Hydrothermal Carbonization - 1. Influence of Lignin in Lignocelluloses. Chem. Eng. Technol. 2011, 34, 2037–2043. [Google Scholar] [CrossRef]
- Nowakowski, D.; Bridgwater, A.; Elliott, D.C.; Meier, D.; De Wild, P. Lignin fast pyrolysis: Results from an international collaboration. J. Anal. Appl. Pyrolysis 2010, 88, 53–72. [Google Scholar] [CrossRef]
- Dell’Orco, S.; Miliotti, E.; Lotti, G.; Rizzo, A.M.; Rosi, L.; Chiaramonti, D. Hydrothermal Depolymerization of Biorefinery Lignin-Rich Streams: Influence of Reaction Conditions and Catalytic Additives on the Organic Monomers Yields in Biocrude and Aqueous Phase. Energies 2020, 13, 1241. [Google Scholar] [CrossRef]
- Kuznetsov, B.; Shchipko, M. The conversion of wood lignin to char materials in a fluidized bed of AlCuCr oxide catalysts. Bioresour. Technol. 1995, 52, 13–19. [Google Scholar] [CrossRef]
- Baklanova, O.; Plaksin, G.; Drozdov, V.A.; Duplyakin, V.; Chesnokov, N.; Kuznetsov, B. Preparation of microporous sorbents from cedar nutshells and hydrolytic lignin. Carbon 2003, 41, 1793–1800. [Google Scholar] [CrossRef]
- Mergner, R.; Janssen, R.; Rutz, D.; de Bari, I.; Sissot, F. Lignocellulosic Ethanol Process and Demonstration—A handbook—Part I; WIP Renewable Energies: Munich, Germany, 2013; Available online: www.biolyfe.eu (accessed on 10 May 2019).
- Miliotti, E.; Dell’Orco, S.; Lotti, G.; Rizzo, A.M.; Rosi, L.; Chiaramonti, D. Lignocellulosic Ethanol Biorefinery: Valorization of Lignin-Rich Stream through Hydrothermal Liquefaction. Energies 2019, 12, 723. [Google Scholar] [CrossRef]
- Miliotti, E.; Casini, D.; Rosi, L.; Lotti, G.; Rizzo, A.M.; Chiaramonti, D. Lab-scale pyrolysis and hydrothermal carbonization of biomass digestate: Characterization of solid products and compliance with biochar standards. Biomass Bioenergy 2020, 139, 105593. [Google Scholar] [CrossRef]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D. Determination of Sugars, Byproducts, and Degradation Products in Liquid Fraction Process Samples; NREL: Golden, CO, USA, 2008. [Google Scholar]
- Lowell, S.; Shields, J.E.; Thomas, M.A.; Thommes, M. Mesopore Analysis. In Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density, 1st ed.; Kluwer Academic Press: Dordrecht, The Netherlands, 2004; pp. 101–128. [Google Scholar] [CrossRef]
- Rouquerol, J.; Llewellyn, P.; Rouquérol, F. Is the bet equation applicable to microporous adsorbents? Stud. Surf. Sci. Catal. 2007, 160, 49–56. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Stark, N.M.; Yelle, D.J.; Agarwal, U. Techniques for Characterizing Lignin. In Lignin in Polymer Composites; Omar, F., Mohini, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 49–66. [Google Scholar] [CrossRef]
- Rodríguez-Mirasol, J.; Cordero, T.; Rodriguez, J. Preparation and characterization of activated carbons from eucalyptus kraft lignin. Carbon 1993, 31, 87–95. [Google Scholar] [CrossRef]
- Antal, M.J.; Grønli, M.G. The Art, Science, and Technology of Charcoal Production†. Ind. Eng. Chem. Res. 2003, 42, 1619–1640. [Google Scholar] [CrossRef]
- Bykov, I. Characterization of Natural and Technical Lignins using FTIR Spectroscopy. Independent Thesis, 2008. Available online: http://www.diva-portal.org/smash/get/diva2:1016107/FULLTEXT01.pdf (accessed on 20 July 2020).
- Santos, J.I.; Fillat, U.; Martin-Sampedro, R.; Ballesteros, I.; Manzanares, P.; Ballesteros, M.; Eugenio, M.E.; Ibarra, D. Lignin-enriched Fermentation Residues from Bioethanol Production of Fast-growing Poplar and Forage Sorghum. Bioresources 2015, 10, 5203–5214. [Google Scholar] [CrossRef][Green Version]
- Chen, W.; Shi, S.; Nguyen, T.; Chen, M.; Zhou, X. Effect of Temperature on the Evolution of Physical Structure and Chemical Properties of Bio-char Derived from Co-pyrolysis of Lignin with High-Density Polyethylene. Bioresources 2016, 11, 3923–3936. [Google Scholar] [CrossRef]
- Júnior, A.F.D.; Oliveira, R.N.; Deglise, X.; De Souza, N.D.; Brito, J.O. Infrared spectroscopy analysis on charcoal generated by the pyrolysis of Corymbia. citriodora wood. Rev. Matéria (Rio de Janeiro) 2019, 24. [Google Scholar] [CrossRef]
- Khanna, R.; Ikram-Ul-Haq, M.; Rawal, A.; Rajarao, R.; Sahajwalla, V.; Cayumil, R.; Mukherjee, P.S. Formation of carbyne-like materials during low temperature pyrolysis of lignocellulosic biomass: A natural resource of linear sp carbons. Sci. Rep. 2017, 7, 16832. [Google Scholar] [CrossRef] [PubMed]
- Diaz, E.; Manzano, F.J.; Martínez, J.A.V.; Rodriguez, J.J.; De La Rubia, M. Ángeles Low-Cost Activated Grape Seed-Derived Hydrochar through Hydrothermal Carbonization and Chemical Activation for Sulfamethoxazole Adsorption. Appl. Sci. 2019, 9, 5127. [Google Scholar] [CrossRef]
- Purnomo, C.W.; Castello, D.; Fiori, L. Granular Activated Carbon from Grape Seeds Hydrothermal Char. Appl. Sci. 2018, 8, 331. [Google Scholar] [CrossRef]
- Correa, C.R.; Bernardo, M.; Ribeiro, R.P.; Esteves, I.A.; Kruse, A.; Esteves, I.A. Evaluation of hydrothermal carbonization as a preliminary step for the production of functional materials from biogas digestate. J. Anal. Appl. Pyrolysis 2017, 124, 461–474. [Google Scholar] [CrossRef]
- Kambo, H.S.; Dutta, A. A comparative review of biochar and hydrochar in terms of production, physico-chemical properties and applications. Renew. Sustain. Energy Rev. 2015, 45, 359–378. [Google Scholar] [CrossRef]
- Reza, M.T.; Rottler, E.; Herklotz, L.; Wirth, B. Hydrothermal carbonization (HTC) of wheat straw: Influence of feedwater pH prepared by acetic acid and potassium hydroxide. Bioresour. Technol. 2015, 182, 336–344. [Google Scholar] [CrossRef]
- Funke, A.; Ziegler, F. Hydrothermal carbonization of biomass: A summary and discussion of chemical mechanisms for process engineering. Biofuels Bioprod. Biorefining 2010, 4, 160–177. [Google Scholar] [CrossRef]
- Hoekman, S.K.; Broch, A.; Robbins, C. Hydrothermal Carbonization (HTC) of Lignocellulosic Biomass. Energy Fuels 2011, 25, 1802–1810. [Google Scholar] [CrossRef]
- Sınag, A.; Gülbay, S.; Uskan, B.; Güllü, M. Comparative studies of intermediates produced from hydrothermal treatments of sawdust and cellulose. J. Supercrit. Fluids 2009, 50, 121–127. [Google Scholar] [CrossRef]
- Castello, D.; Kruse, A.; Fiori, L. Low temperature supercritical water gasification of biomass constituents: Glucose/phenol mixtures. Biomass Bioenergy 2015, 73, 84–94. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pretreatment | Pretreatment Temperature [°C] | Pretreatment Time [h] | Activation | Activation Temperature [°C] | Activating Agent Ratio (d.b.) |
|---|---|---|---|---|---|
| None | - | - | Physical, CO2, 2 h | 700 | - |
| None | - | - | Physical, CO2, 2 h | 800 | - |
| None | - | - | Chemical, KOH, 1 h | 600 | 1:1 |
| None | - | - | Chemical, KOH, 1 h | 600 | 2:1 |
| None | - | - | Chemical, KOH, 1 h | 700 | 1:1 |
| None | - | - | Chemical, KOH, 1 h | 700 | 2:1 |
| None | - | - | Chemical, KOH, 1 h | 800 | 1:1 |
| None | - | - | Chemical, KOH, 1 h | 800 | 2:1 |
| Slow pyrolysis | 550 | 2 | Physical, CO2, 2 h | 700 | - |
| Hydrothermal carbonization | 200 | 2 | Physical, CO2, 2 h | 700 | - |
| Hydrothermal carbonization | 200 | 4 | Physical, CO2, 2 h | 700 | - |
| Hydrothermal carbonization | 270 | 2 | Physical, CO2, 2 h | 700 | - |
| Hydrothermal carbonization | 270 | 4 | Physical, CO2, 2 h | 700 | - |
| Hydrothermal carbonization | 200 | 2 | Chemical, KOH, 1 h | 600 | 1:1 |
| Hydrothermal carbonization | 200 | 4 | Chemical, KOH, 1 h | 600 | 1:1 |
| Hydrothermal carbonization | 270 | 2 | Chemical, KOH, 1 h | 600 | 1:1 |
| Hydrothermal carbonization | 270 | 4 | Chemical, KOH, 1 h | 600 | 1:1 |
| Parameter | u.m. | Lignin-Rich Stream (Present Study) | ALM Lignin [14] |
|---|---|---|---|
| Moisture | % w/w (w.b.) | 69.7 | n.a. |
| Ash | % w/w (d.b.) | 3.0 | <4.0 |
| Volatile matter | % w/w (d.b.) | 69.2 | n.a. |
| Fixed Carbon | % w/w (d.b.) | 27.3 | n.a. |
| Higher Heating Value | MJ kg−1 (d.b.) | 22.8 | n.a. |
| Carbon | % w/w (d.b.) | 55.1 | 61.0 |
| Hydrogen | % w/w (d.b.) | 5.9 | 7.0 |
| Nitrogen | % w/w (d.b.) | 1.2 | 0.9 |
| Sulphur | % w/w (d.b.) | 0.2 | <0.03 |
| Oxygen | % w/w (d.b.) | 34.6 | 31.0 |
| Lignin content | % w/w (d.a.f.) | 53.0 | 94.0 * |
| Structural sugars | % w/w (d.a.f.) | 35.8 | 2.2 * |
| Parameter | LRS | Pyrochar | Physically Activated Carbon | |
|---|---|---|---|---|
| Activation temperature [°C] | - | - | 700 | 800 |
| Activation time [h] | - | - | 2 | 2 |
| BET area [m2 g−1] | 3 | 370 | 600 | 1078 |
| Micropore area [m2 g−1] | - | 309 | 525 | 949 |
| External surface area [m2 g−1] | - | 61 | 68 | 132 |
| Micropore area (%) | - | 83 | 87 | 88 |
| Micropore volume [cm3 g−1] | - | 0.14 | 0.21 | 0.39 |
| Total pore volume [cm3 g−1] | 0.01 | 0.19 | 0.28 | 0.54 |
| Micropore volume (%) | - | 71 | 77 | 72 |
| Avg. Pore diameter [nm] | 17.27 | 2.06 | 1.84 | 2.00 |
| Parameter | LRS | Chemically Activated Carbon | |||||
|---|---|---|---|---|---|---|---|
| Activation temperature [°C] | - | 600 | 600 | 700 | 700 | 800 | 800 |
| KOH:LRS (d.b.) | - | 1:1 | 2:1 | 1:1 | 2:1 | 1:1 | 2:1 |
| Activation yield [% w/w] d.b. | - | 29.8 | 15.7 | 8.1 | 10.7 | 0.2 | 3.6 |
| BET area [m2 g−1] | 3 | 675 | 989 | 1437 | 1188 | 1484 | 558 |
| Micropore area [m2 g−1] | <0 | 623 | 807 | 1234 | 861 | 793 | 244 |
| External surface area [m2 g−1] | <0 | 46 | 174 | 188 | 319 | 691 | 314 |
| Micropore area (%) | Null | 92 | 82 | 86 | 72 | 53 | 44 |
| Micropore volume [cm3 g−1] | <0 | 0.24 | 0.34 | 0.55 | 0.39 | 0.35 | 0.11 |
| Total pore volume [cm3 g−1] | 0.01 | 0.31 | 0.61 | 0.73 | 0.75 | 1.39 | 1.02 |
| Micropore volume (%) | Null | 79 | 56 | 75 | 52 | 25 | 10 |
| Avg. Pore diameter [nm] | 17.27 | 1.82 | 2.47 | 2.03 | 2.52 | 3.73 | 7.29 |
| Sample | Carbon | Hydrogen | Nitrogen | Oxygen * | H/C | O/C |
|---|---|---|---|---|---|---|
| % w/w (d.b.) | % w/w (d.b.) | % w/w (d.b.) | % w/w (d.b.) | |||
| LRS | 55.1 | 5.9 | 1.2 | 37.8 | 1.28 | 0.51 |
| HTC 200 °C—2 h | 57.8 | 5.9 | 1.1 | 35.2 | 1.23 | 0.46 |
| HTC 200 °C—4 h | 58.5 | 5.9 | 1.3 | 34.4 | 1.20 | 0.44 |
| HTC 270 °C—2 h | n.m. | n.m. | n.m. | n.m. | n.m. | n.m. |
| HTC 270 °C—4 h | 71.1 | 5.6 | 1.6 | 21.7 | 0.95 | 0.23 |
| SP 550 °C—2 h | 75.0 | 2.6 | 1.9 | 20.5 | 0.42 | 0.21 |
| Parameter | Physically Activated Carbon | Chemically Activated Carbon | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pretreatment | SP | HTC | None | HTC | ||||||
| Pretr. temperature [°C] | 550 | 200 | 200 | 270 | 270 | - | 200 | 200 | 270 | 270 |
| Pretr. time [h] | 2 | 2 | 4 | 2 | 4 | - | 2 | 4 | 2 | 4 |
| BET area [m2 g−1] | 600 | 631 | 540 | 161 | 77 | 675 | 502 | 600 | 230 | 81 |
| Micropore area [m2 g−1] | 525 | 597 | 524 | 139 | 58 | 623 | 452 | 541 | 185 | 49 |
| External surface area [m2 g−1] | 68 | 36 | 29 | 22 | 19 | 46 | 50 | 59 | 45 | 32 |
| Micropore area (%) | 0.87 | 0.95 | 0.97 | 0.86 | 0.75 | 92 | 0.90 | 0.90 | 0.81 | 0.61 |
| Micropore volume [cm3 g−1] | 0.21 | 0.23 | 0.20 | 0.06 | 0.03 | 0.24 | 0.18 | 0.22 | 0.08 | 0.02 |
| Total pore volume [cm3 g−1] | 0.28 | 0.27 | 0.23 | 0.08 | 0.05 | 0.31 | 0.27 | 0.32 | 0.22 | 0.08 |
| Micropore volume (%) | 0.77 | 0.85 | 0.87 | 0.73 | 0.60 | 79 | 0.66 | 0.67 | 0.34 | 0.30 |
| Avg. Pore diameter [nm] | 1.84 | 1.72 | 1.72 | 2.11 | 2.42 | 1.82 | 2.18 | 2.15 | 3.89 | 3.83 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miliotti, E.; Rosi, L.; Bettucci, L.; Lotti, G.; Rizzo, A.M.; Chiaramonti, D. Characterization of Chemically and Physically Activated Carbons from Lignocellulosic Ethanol Lignin-Rich Stream via Hydrothermal Carbonization and Slow Pyrolysis Pretreatment. Energies 2020, 13, 4101. https://doi.org/10.3390/en13164101
Miliotti E, Rosi L, Bettucci L, Lotti G, Rizzo AM, Chiaramonti D. Characterization of Chemically and Physically Activated Carbons from Lignocellulosic Ethanol Lignin-Rich Stream via Hydrothermal Carbonization and Slow Pyrolysis Pretreatment. Energies. 2020; 13(16):4101. https://doi.org/10.3390/en13164101
Chicago/Turabian StyleMiliotti, Edoardo, Luca Rosi, Lorenzo Bettucci, Giulia Lotti, Andrea Maria Rizzo, and David Chiaramonti. 2020. "Characterization of Chemically and Physically Activated Carbons from Lignocellulosic Ethanol Lignin-Rich Stream via Hydrothermal Carbonization and Slow Pyrolysis Pretreatment" Energies 13, no. 16: 4101. https://doi.org/10.3390/en13164101
APA StyleMiliotti, E., Rosi, L., Bettucci, L., Lotti, G., Rizzo, A. M., & Chiaramonti, D. (2020). Characterization of Chemically and Physically Activated Carbons from Lignocellulosic Ethanol Lignin-Rich Stream via Hydrothermal Carbonization and Slow Pyrolysis Pretreatment. Energies, 13(16), 4101. https://doi.org/10.3390/en13164101