Green Diesel: Biomass Feedstocks, Production Technologies, Catalytic Research, Fuel Properties and Performance in Compression Ignition Internal Combustion Engines





Abstract
:1. Introduction
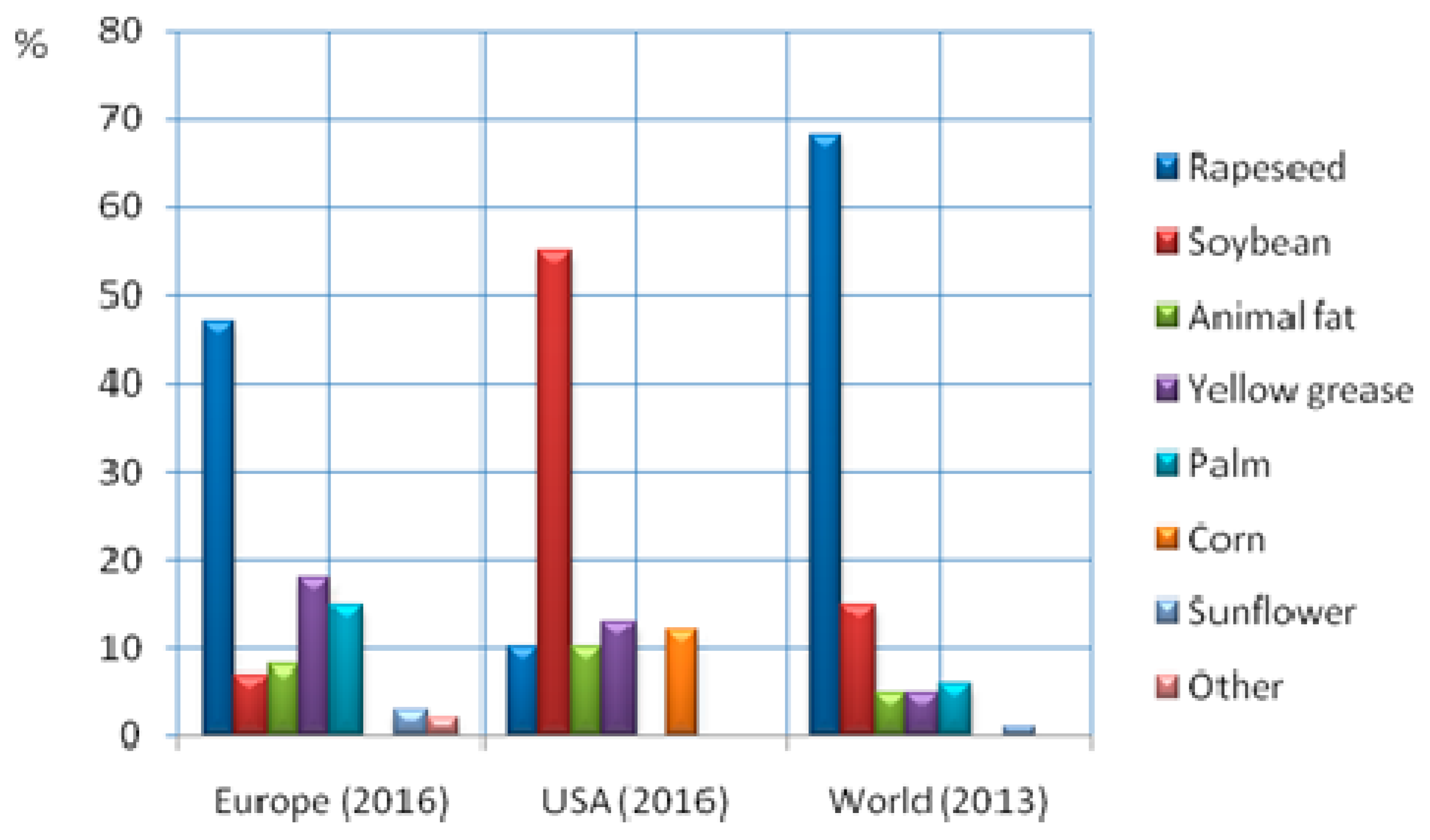
2. Biomass Feedstocks
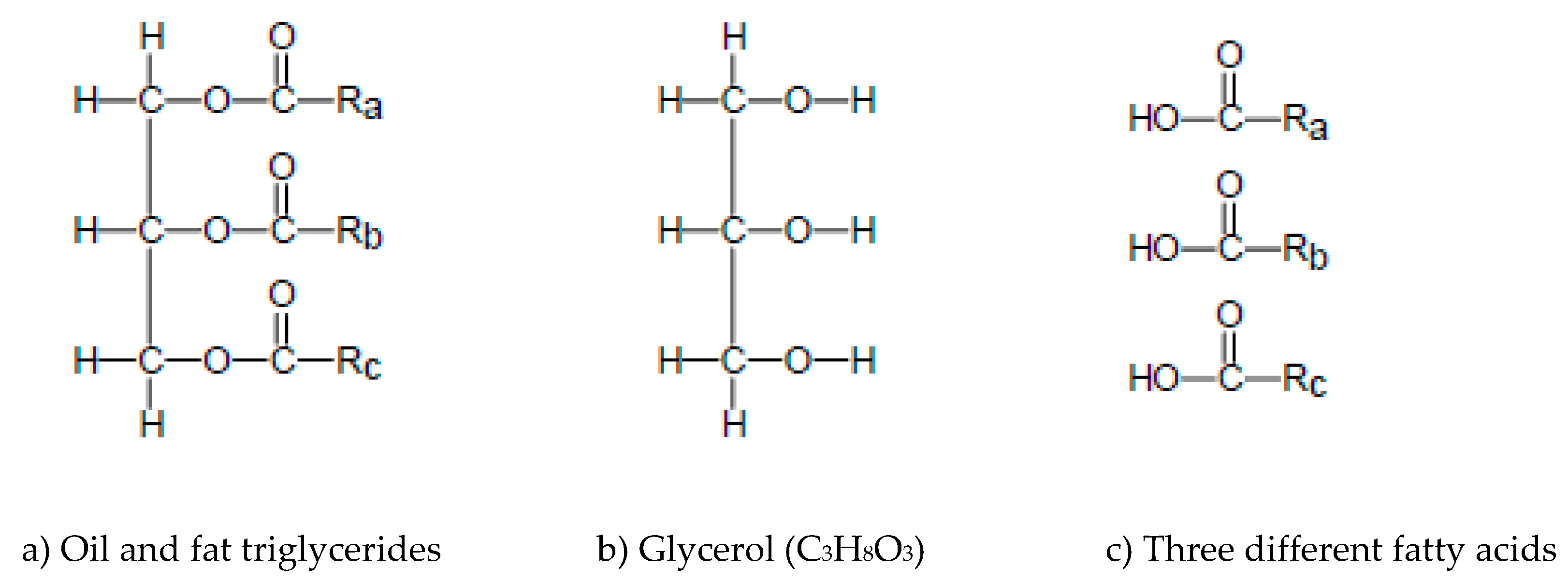
2.1. Triglycerides
2.2. Sugars and Starches
2.3. Lignocellulose
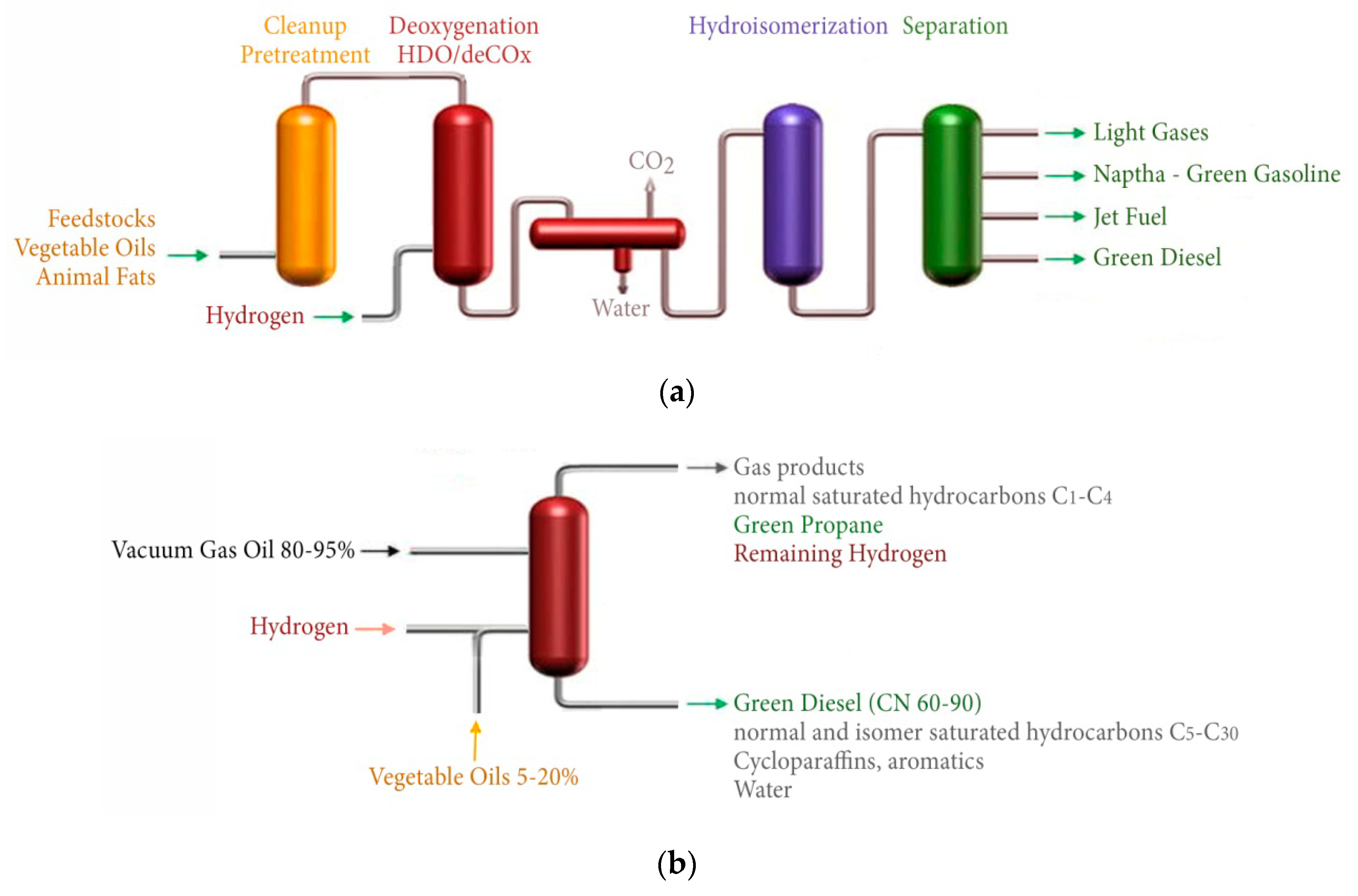
3. Hydro-Processing of Triglycerides
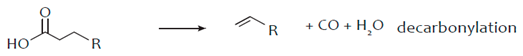
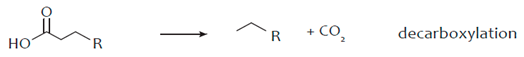
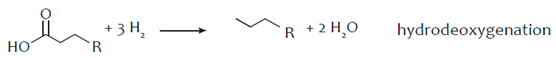
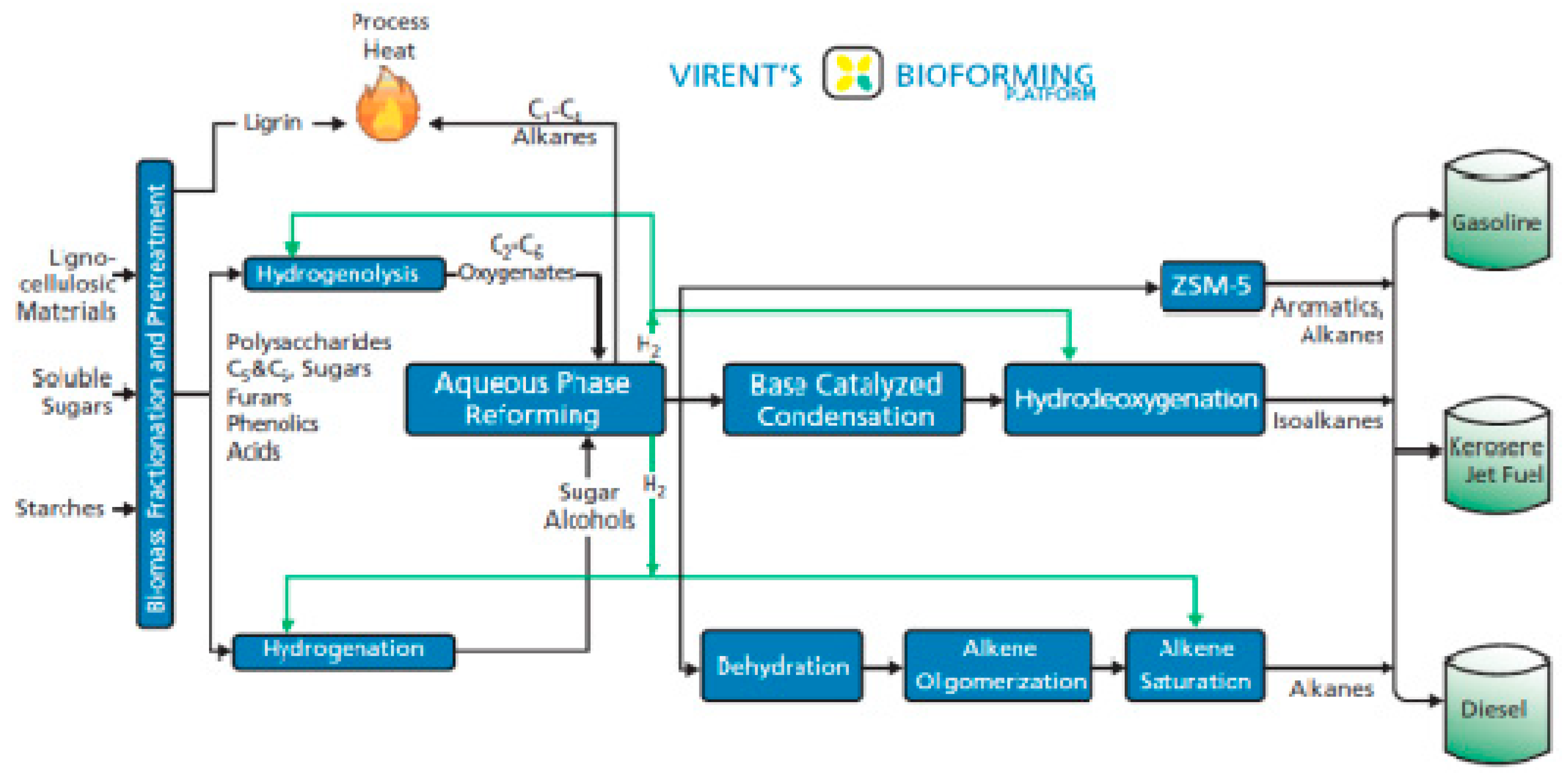
4. Catalytic Upgrading of Sugars, Starches and Alcohols
5. Thermal Conversion of Lignocellulosic Biomass
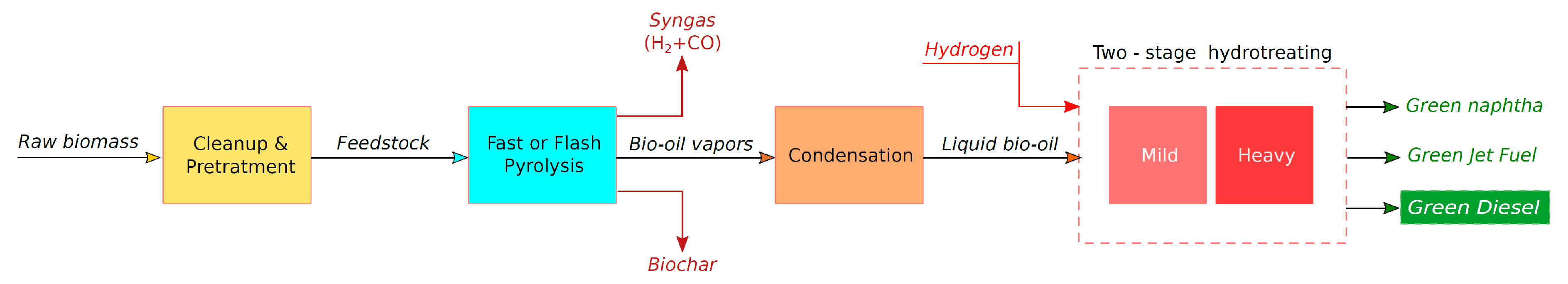
5.1. Biomass Pyrolysis and Production of Bio-Oil
5.2. Bio-Oil Upgrading through Hydrotreating
5.3. Bio-Oil Upgrading through Catalytic Vapor (Zeolite) Cracking
5.4. Hydrothermal Liquefaction of Biomass and Upgrading of Bio-Crude
6. Biomass to Liquid (BTL) Conversion of Lignocellulosic Biomass
7. Green Diesel Properties and Performance in CI Engines
7.1. Green Diesel Properties
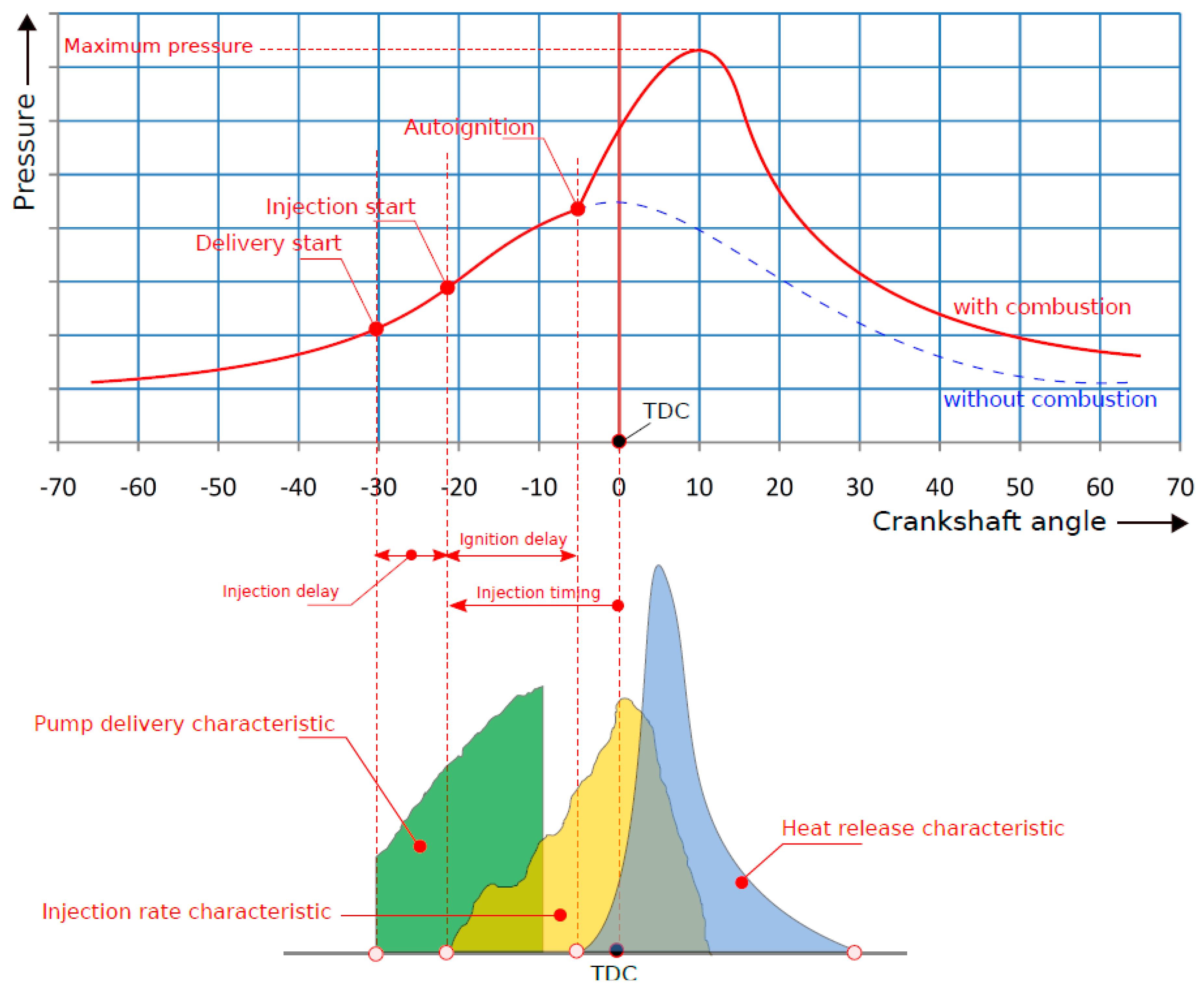
7.2. Green Diesel Performance in CI Engines
7.3. Green Diesel Emissions in Internal Combustion Engines
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Chaudhuri, U.R. Fundamentals of Petroleum and Petrochemical Engineering, 2nd ed.; Taylor and Francis Group: Boca Raton, FL, USA, 2011. [Google Scholar]
- Nejat, P.; Jomehzadeh, F.; Taheri, M.M.; Gohari, M.; Majid, M.Z. A global review of energy consumption, CO2 emissions and policy in the residential sector (with an overview of the top ten CO2 emitting countries). Renew. Sustain. Energy Rev. 2015, 43, 843–862. [Google Scholar] [CrossRef]
- Anuar, M.R.; Abdullah, A.Z. Challenges in biodiesel industry with regards to feedstock, environmental, social and sustainability issues: A critical review. Renew. Sustain. Energy Rev. 2016, 58, 208–223. [Google Scholar] [CrossRef]
- Ma, Y.; Gao, Z.; Wang, Q.; Liu, Y. Biodiesels from microbial oils: Opportunity and challenges. Bioresour. Technol. 2018, 263, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Gebremariam, S.N.; Marchetti, J.M. Economics of biodiesel production: Review. Energy Convers. Manag. 2018, 168, 74–84. [Google Scholar] [CrossRef]
- Liu, S.; Colson, G.; Wetzstein, M. Biodiesel investment in a disruptive tax-credit policy environment. Energy Policy 2018, 123, 19–30. [Google Scholar] [CrossRef]
- Rodrigues, A.; Bordado, J.C.; Dos Santos, R.G. Upgrading the glycerol from biodiesel production as a source of energy carriers and chemicals-A technological review for three chemical pathways. Energies 2017, 10, 1817. [Google Scholar] [CrossRef]
- Goula, M.A.; Charisiou, N.D.; Papageridis, K.N.; Siakavelas, G. Influence of the synthesis method parameters used to prepare nickel-based catalysts on the catalytic performance for the glycerol steam reforming reaction. Chin. J. Catal. 2016, 37, 1949–1965. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Papageridis, K.N.; Siakavelas, G.; Tzounis, L.; Kousi, K.; Baker, M.A.; Hinder, S.J.; Sebastian, V.; Polychronopoulou, K.; Goula, M.A. Glycerol steam reforming for hydrogen production over nickel supported on alumina, zirconia and silica catalysts. Top. Catal. 2017, 60, 1226–1250. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.; Papageridis, K.N.; Baklavaridis, A.; Tzounis, L.; Polychronopoulou, K.; Goula, M.A. Hydrogen production via the glycerol steam reforming reaction over nickel supported on alumina and lanthana-alumina catalysts. Int. J. Hydrogen Energy 2017, 42, 13039–13060. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Papageridis, K.N.; Siakavelas, G.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Polychronopoulou, K.; Goula, M.A. The influence of SiO2 doping on the Ni/ZrO2 supported catalyst for hydrogen production through the glycerol steam reforming reaction. Catal. Today 2018, 319, 206–219. [Google Scholar] [CrossRef]
- Garcia-Olivares, A.; Sole, J.; Osychenko, O. Transportation in a 100% renewable energy system. Energy Convers. Manag. 2018, 158, 266–285. [Google Scholar] [CrossRef]
- Hosseinzadeh-Bandbafha, H.; Tabatabaei, M.; Aghbashlo, M.; Khanali, M.; Demirbas, A. A comprehensive review on the environmental impacts of diesel/biodiesel additives. Energy Convers. Manag. 2018, 174, 579–614. [Google Scholar] [CrossRef]
- Mohd Noor, C.W.; Noor, M.M.; Mamat, R. Biodiesel as alternative fuel for marine diesel engine applications: A review. Renew. Sustain. Energy Rev. 2018, 94, 127–142. [Google Scholar] [CrossRef]
- Saluja, R.K.; Kumar, V.; Sham, R. Stability of biodiesel—A review. Renew. Sustain. Energy Rev. 2016, 62, 866–881. [Google Scholar] [CrossRef]
- Taromi, A.A.; Kaliaguine, S. Green diesel production via continuous hydrotreatment of triglycerides over mesostructured γ-alumina supported NiMo/CoMo catalysts. Fuel Process. Technol. 2018, 171, 20–30. [Google Scholar] [CrossRef]
- Orozco, L.M.; Echeverri, D.A.; Sánchez, L.; Rios, L.A. Second-generation green diesel from castor oil: Development of a new and efficient continuous-production process. Chem. Eng. J. 2017, 322, 149–156. [Google Scholar] [CrossRef]
- Kordouli, E.; Pawelec, B.; Bourikas, K.; Kordulis, C.; Fierro, J.L.G.; Lycourghiotis, A. Mo promoted Ni-Al2O3 co-precipitated catalysts for green diesel production. Appl. Catal. B-Environ. 2018, 229, 139–154. [Google Scholar] [CrossRef]
- Kordouli, E.; Sygellou, L.; Kordulis, C.; Bourikas, K.; Lycourghiotis, A. Probing the synergistic ratio of the NiMo/γ-Al2O3 reduced catalysts for the transformation of natural triglycerides into green diesel. Appl. Catal. B-Environ. 2017, 209, 12–22. [Google Scholar] [CrossRef]
- Kordouli, E.; Kordulis, C.; Lycourghiotis, A.; Cole, R.; Vasudevan, P.T.; Pawelec, B.; Fierro, J.L.G. HDO activity of carbon-supported Rh, Ni and Mo-Ni catalysts. Mol. Catal. 2017, 441, 209–220. [Google Scholar] [CrossRef]
- Srifa, A.; Faungnawakij, K.; Itthibenchapong, V.; Viriya-empikul, N.; Charinpanitkul, T.; Assabumrungrat, S. Production of bio-hydrogenated diesel by catalytic hydrotreating of palm oil over NiMoS2/γ-Al2O3 catalyst. Bioresour. Technol. 2014, 158, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Gousi, M.; Andriopoulou, C.; Bourikas, K.; Ladas, S.; Sotiriou, M.; Kordulis, C.; Lycourghiotis, A. Green diesel production over nickel-alumina co-precipitated catalysts. Appl. Catal. A-Gen. 2017, 536, 45–56. [Google Scholar] [CrossRef]
- Pradhan, P.; Arora, A.; Mahajani, S.M. A semi-empirical approach towards predicting producer gas composition in biomass gasification. Bioresour. Technol. 2019, 272, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Saidur, R.; Abdelaziz, E.A.; Demirbas, A.; Hossain, M.S.; Mekhilef, S. A review on biomass as a fuel for boilers. Renew. Sustain. Energy Rev. 2011, 15, 2262–2289. [Google Scholar] [CrossRef]
- From the Sugar Platform to Biofuels and Biochemicals; Final Report for the European Commission Directorate—General Energy, No ENER/C2/423-2012/SI2.673791, V2.1; European Commission: Brussels, Belgium, 2015.
- O’Connell, A.; Kousoulidou, M.; Lonza, L.; Weindorf, W. Considerations on GHG emissions and energy balances of promising aviation biofuel pathways. Renew. Sustain. Energy Rev. 2019, 101, 504–515. [Google Scholar] [CrossRef]
- Searle, S.Y.; Malins, C.J. Waste and residue availability for advanced biofuel production in EU Member States. Biomass Bioenergy 2016, 89, 2–10. [Google Scholar] [CrossRef]
- Scarlat, N.; Dallemand, J.F.; Monforti-Ferrario, F.; Nita, V. The role of biomass and bioenergy in a future bioeconomy: Policies and facts. Environ. Dev. 2015, 15, 3–34. [Google Scholar] [CrossRef]
- Kim, D.S.; Hanifzadeh, M.; Kumar, A. Trend of biodiesel feedstock and its impact on biodiesel emission characteristics. Environ. Prog. Sustain. 2018, 37, 7–19. [Google Scholar] [CrossRef]
- Ambat, I.; Srivastava, V.; Sillanpää, M. Recent advancement in biodiesel production methodologies using various feedstock: A review. Renew. Sustain. Energy Rev. 2018, 90, 356–369. [Google Scholar] [CrossRef]
- Boutesteijn, C.; Drabik, D.; Venus, T.J. The interaction between EU biofuel policy and first-and second-generation biodiesel production. Ind. Crop. Prod. 2017, 106, 124–129. [Google Scholar] [CrossRef]
- Banković-Ilić, I.B.; Stojković, I.J.; Stamenković, O.S.; Veljkovic, V.B.; Hung, Y.T. Waste animal fats as feedstocks for biodiesel production. Renew. Sustain. Energy Rev. 2014, 32, 238–254. [Google Scholar] [CrossRef]
- Avinash, A.; Sasikumar, P.; Murugesan, A. Understanding the interaction among the barriers of biodiesel production from waste cooking oil in India- an interpretive structural modeling approach. Renew. Energy 2018, 127, 678–684. [Google Scholar] [CrossRef]
- Ramos, M.J.; Fernández, C.M.; Casas, A.; Rodríguez, L.; Pérez, Á. Influence of fatty acid composition of raw materials on biodiesel properties. Bioresour. Technol. 2009, 100, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Hoekman, S.K.; Broch, A.; Robbins, C.; Ceniceros, E.; Natarajan, M. Review of biodiesel composition, properties, and specifications. Renew. Sustain. Energy Rev. 2012, 16, 143–169. [Google Scholar] [CrossRef]
- Lee, D.S.; Noh, B.S.; Bae, S.Y.; Kim, K. Characterization of fatty acids composition in vegetable oils by gas chromatography and chemometrics. Anal. Chim. Acta 1998, 358, 163–175. [Google Scholar] [CrossRef]
- Ashraful, A.M.; Masjuki, H.H.; Kalam, M.A.; Rizwanul Fattah, I.M.; Imtenan, S.; Shahir, S.A.; Mobarak, H.M. Production and comparison of fuel properties, engine performance, and emission characteristics of biodiesel from various non-edible vegetable oils: A review. Energy Convers. Manag. 2014, 80, 202–228. [Google Scholar] [CrossRef]
- Sivaramakrishnan, R.; Incharoensakdi, A. Microalgae as feedstock for biodiesel production under ultrasound treatment—A review. Bioresour. Technol. 2018, 250, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Raja, R.; Shanmugam, H.; Ganesan, V.; Carvalho, I.S. Biomass from microalgae: An overview. J. Oceanogr. Mar. Res. 2014, 2, 1–7. [Google Scholar]
- Rosenberg, J.N.; Oyler, G.A.; Wilkinson, L.; Betenbaugh, M.J. A green light for engineered algae: Redirecting metabolism to fuel a biotechnology revolution. Curr. Opin. Biotech. 2008, 19, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Magnusson, M.; Brown, R.J.; Ayoko, G.A.; Nabi, M.N.; Heimann, K. Microalgal species selection for biodiesel production based on fuel properties derived from fatty acid profiles. Energies 2013, 6, 5676–5702. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Pace, R.; Marques, S.; Morgan, T.; McKelphin, C.; Mobley, J.; Crocker, M. Extraction, characterization, purification and catalytic upgrading of algae lipids to fuel-like hydrocarbons. Fuel 2016, 180, 668–678. [Google Scholar] [CrossRef]
- Hattori, T.; Morita, S. Energy crops for sustainable bioethanol production: Which, were and how? Plant Prod. Sci. 2010, 13, 221–234. [Google Scholar] [CrossRef]
- Walker, G.M. Bioethanol: Science and Technology of Fuel Alcohol, 1st ed.; Ventus Publishing ApS: Copenhagen, Denmark, 2010. [Google Scholar]
- Tamalampudi, S.; Fukuda, H.; Kondo, A. Bioethanol from starchy biomass. Part II: Hydrolysis and fermentation. In Handbook of Plant-Based Biofuels, 1st ed.; Pandey, A., Ed.; Taylor and Francis Group: Boca Raton, FL, USA, 2009. [Google Scholar]
- Chang, V.S.; Holtzapple, M.T. Fundametal factors affecting biomass enzymatic reactivity. Appl. Biochem. Biotechnol. 2000, 84, 5–37. [Google Scholar] [CrossRef]
- Laureano-Perez, L.; Teymouri, F.; Alizadeh, H.; Dale, B.E. Understanding factors that limit enzymatic hydrolysis of biomass. Appl. Biochem. Biotechnol. 2005, 124, 1081–1099. [Google Scholar] [CrossRef]
- Harmsen, P.F.H.; Huijgen, W.J.J.; López, L.M.B.; Bakker, R.R.C. Literature Review of Physical and Chemical Pretreatment Processes for Lignocellulosic Biomass; ECN-E-10-013; Energy Research Centre of the Netherlands: Petten, The Netherlands, 2010. [Google Scholar]
- Agbor, V.B.; Cicek, N.; Sparling, R.; Berlin, A.; Levin, D.B. Biomass pretreatment: Fundamentals toward application. Biotechnol. Adv. 2011, 29, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.K.; Sharma, S. Recent updates on different methods of pretreatment of lignocellulosic feedstocks: A review. Bioresour. Bioprocess. 2017, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Parakash, S. Refining Processing Handbook, 1st ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Fahim, M.A.; Al-Sahhaf, T.A.; Elkilani, A.S. Fundamentals of Petroleum Refining, 1st ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Scherzer, J.; Gruia, A.J. Hydrocracking Science and Technology, 1st ed.; Marcel Dekker: New York, NY, USA, 2003. [Google Scholar]
- Speight, J.G. The Desulfurization of Heavy Oils and Residues, 2nd ed.; Marcel Dekker: New York, NY, USA, 2000. [Google Scholar]
- Sotelo-Boyás, R.; Trejo-Zárraga, F.; Hernández-Loyo, F.J. Hydroconversion of triglycerides into green liquid fuels. In Hydrogenation, Karamé, 1st ed.; IntechOpen: London, UK, 2012. [Google Scholar]
- Murzin, D.Y. Hydrotreating of triglyceride-based feedstocks in refineries. In Advances in Chemical Engineering; Murzin, D.Y. Academic Press: London, UK, 2013; Volume 42, pp. 141–194. [Google Scholar]
- Kim, S.K.; Brand, S.; Lee, H.S.; Kim, Y.; Kim, J. Production of renewable diesel by hydrotreatment of soybean oil: Effect of reaction parameters. Chem. Eng. J. 2013, 228, 114–123. [Google Scholar] [CrossRef]
- Engineering ToolBox, Hydrocarbons—Physical Data. 2017. Available online: www.engineeringtoolbox.com/hydrocarbon-boiling-melting-flash-autoignition-point-density-gravity-molweight-d_1966.html (accessed on 24 December 2018).
- Klerk, A. Fischer-Tropsch Refining; Wiley-VCH Verlag & Co., KGaA: Weinheim, Germany, 2011. [Google Scholar]
- Deldari, H. Suitable catalysts for hydroisomerization of long-chain normal paraffins. Appl. Catal. A-Gen. 2005, 293, 1–10. [Google Scholar] [CrossRef]
- Akhmedov, V.; Al-Khowaiter, S. Recent advances and future aspects in the selective isomerization of high n-alkanes. Catal. Rev. 2007, 49, 33–139. [Google Scholar] [CrossRef]
- Drop in Fuels 2020: A Global Market Survey, Emerging Markets Online. 2018. Available online: www.emerging-markets.com (accessed on 14 December 2018).
- Hydrotreated Vegetable Oils (HVO) Biorefineries–The Rise of Renewable Diesel. Available online: biorrefineria.blogspot.com/2015/09/hydrotreated-vegetable-oils-hvo.biorefineries.html (accessed on 14 December 2018).
- Nunes, P.P.; Brodzki, D.; Bugli, G.; Djega-Mariadassou, G. Soybean oil hydrocracking under pressure: Process and general aspects of the transformation. Oil Gas Sci. Technol. 1986, 41, 421–431. [Google Scholar]
- Gusmao, J.; Brodzki, D.; Djega-Mariadassou, G.; Frety, R. Utilization of vegetable oils as an alternative source for diesel-type fuel: Hydrocracking on reduced Ni/SO2 and sulphided Ni-Mo/γ-Al2O3. Catal. Today 1989, 5, 533–544. [Google Scholar] [CrossRef]
- Huber, G.W.; O’Connor, P.; Corma, A. Processing biomass in conventional oil refineries: Production of high quality diesel by hydrotreating vegetable oils in heavy vacuum oil mixtures. Appl. Catal. A-Gen. 2007, 329, 120–129. [Google Scholar] [CrossRef]
- Donnis, B.; Egeberg, R.G.; Blom, P.; Knudsen, K.G. Hydroprocessing of bio-oils and oxygenates to hydrocarbons. Understanding the reaction routes. Top. Catal. 2009, 52, 229–240. [Google Scholar] [CrossRef]
- Nava, R.; Pawelec, B.; Castaño, P.; Álvarez-Galván, M.C.; Loricera, C.V.; Fierro, J.L.G. Upgrading of bio-liquids on different mesoporous silica-supported CoMo catalysts. Appl. Catal. B-Environ. 2009, 92, 154–167. [Google Scholar] [CrossRef]
- Šimáček, P.; Kubička, D.; Šebor, G.; Pospíšil, M. Fuel properties of hydroprocessed rapeseed oil. Fuel 2010, 89, 611–615. [Google Scholar] [CrossRef]
- Šimáček, P.; Kubička, D. Hydrocracking of petroleum vacuum distillate containing rapeseed oil: Evaluation of diesel fuel. Fuel 2010, 89, 1508–1513. [Google Scholar] [CrossRef]
- Kumar, R.; Rana, B.S.; Tiwari, R.; Verma, D.; Kumar, R.; Joshi, R.K.; Garg, M.O.; Sinha, A.K. Hydroprocessing of jatropha oil and its mixtures with gas oil. Green Chem. 2010, 12, 2232–2239. [Google Scholar] [CrossRef]
- Kubička, D.; Kaluža, L. Deoxygenation of vegetable oils over sulfided Ni, Mo and NiMo catalysts. Appl. Catal. A-Gen. 2010, 372, 199–208. [Google Scholar] [CrossRef]
- Mikulec, J.; Cvengroš, J.; Joríková, Ľ.; Banič, M.; Kleinová, A. Second generation diesel fuel from renewable sources. J. Clean. Prod. 2010, 18, 917–926. [Google Scholar] [CrossRef]
- Sotelo-Boyas, R.; Liu, Y.; Minowa, T. Renewable diesel production from the hydrotreating of rapeseed oil with Pt/zeolite and NiMo/Al2O3 catalysts. Ind. Eng. Chem. Res. 2011, 50, 2791–2799. [Google Scholar] [CrossRef]
- Kubička, D.; Šimáček, P.; Žilkova, N. Transformation of vegetable oils into hydrocarbons over mesoporous-alumina-supported CoMo catalysts. Top. Catal. 2009, 52, 161–168. [Google Scholar] [CrossRef]
- Guzman, A.; Torres, J.E.; Prada, L.P.; Nuñez, M.L. Hydroprocessing of crude palm oil at pilot plant scale. Catal. Today 2010, 156, 38–43. [Google Scholar] [CrossRef]
- Hancsók, J.; Kasza, T.; Kovács, S.; Solymosi, P.; Holló, A. Production of bioparaffins by the catalytic hydrogenation of natural triglycerides. J. Clean. Prod. 2012, 34, 76–81. [Google Scholar] [CrossRef]
- Kovacs, S.; Kasza, T.; Thernesz, A.; Horvath, I.W.; Hancsak, J. Fuel production by hydrotreating of triglycerides on NiMo/Al2O3/F catalyst. Chem. Eng. J. 2011, 176–177, 237–243. [Google Scholar] [CrossRef]
- Liu, Y.; Sotelo-Boyás, R.; Murata, K.; Minowa, T.; Sakanishi, K. Hydrotreatment of vegetable oils to produce bio-hydrogenated diesel and liquefied petroleum gas fuel over catalysts containing sulfided Ni-Mo and solid acids. Energy Fuel. 2011, 25, 4675–4685. [Google Scholar] [CrossRef]
- Toba, M.; Abe, Y.; Kuramochi, H.; Osako, M.; Mochizuki, T.; Yoshimura, Y. Hydrodeoxygenation of waste vegetable oil over sulfide catalysts. Catal. Today 2011, 164, 533–537. [Google Scholar] [CrossRef]
- Sebos, I.; Matsoukas, A.; Apostolopoulos, V.; Papayannakos, N. Catalytic hydroprocessing of cottonseed oil in petroleum diesel mixtures for production of renewable diesel. Fuel 2009, 88, 145–149. [Google Scholar] [CrossRef]
- Kubička, D.; Horáček, J. Deactivation of HDS catalysts in deoxygenation of vegetable oils. Appl. Catal. A-Gen. 2011, 394, 9–17. [Google Scholar] [CrossRef]
- Templis, C.; Vonortas, A.; Sebos, I.; Papayannakos, N. Vegetable oil effect on gasoil HDS in their catalytic co-hydroprocessing. Appl. Catal. B-Environ. 2011, 104, 324–329. [Google Scholar] [CrossRef]
- Veriansyah, B.; Han, J.Y.; Kim, S.K.; Hong, S.A.; Kim, Y.J.; Lim, J.S.; Shu, Y.W.; Oh, S.G.; Kim, J. Production of renewable diesel by hydroprocessing of soybean oil: Effect of catalysts. Fuel 2012, 94, 578–585. [Google Scholar] [CrossRef]
- Krár, M.; Kovács, S.; Kalló, D.; Hancsók, J. Fuel purpose hydrotreating of sunflower oil on CoMo/Al2O3 catalyst. Bioresource Technol. 2010, 101, 9287–9293. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Liu, Y.; Inaba, M.; Takahara, I. Production of synthetic diesel by hydrotreatment of jatropha oils using Pt-Re/H-ZSM-5 catalyst. Energy Fuel. 2010, 24, 2404–2409. [Google Scholar] [CrossRef]
- Srifa, A.; Faungnawakij, K.; Itthibenchapong, V.; Assabumrungrat, S. Roles of monometallic catalysts in hydrodeoxygenation of palm oil to green diesel. Chem. Eng. J. 2015, 278, 249–258. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Sato, T.; Shimada, H.; Matsubayashi, N.; Nishijima, A. Influences of oxygen-containing substances on deactivation of sulfided molybdate catalysts. Appl. Catal. 1991, 73, 55–63. [Google Scholar] [CrossRef]
- Laurent, E.; Delmon, B. Study of the hydrodeoxygenation of carbonyl, carboxylic and guaiacyl groups over sulfided CoMo/gamma-Al2O3 and NiMo/gamma-Al2O3 catalyst. 2. Influence of water, ammonia and hydrogen-sulfide. Appl. Catal. A-Gen. 1994, 109, 97–115. [Google Scholar] [CrossRef]
- Yang, Y.; Gilbert, A.; Xu, C. Hydrodeoxygenation of bio-crude in supercritical hexane with sulfided CoMo and CoMoP catalysts supported on MgO: A model compound study using phenol. Appl. Catal. A-Gen. 2009, 360, 242–249. [Google Scholar] [CrossRef]
- Kordulis, C.; Bourikas, K.; Gousi, M.; Kordouli, E.; Lycourghiotis, A. Development of nickel based catalysts for the transformation of natural triglycerides and related compounds into green diesel: A critical review. Appl. Catal. B-Environ. 2016, 181, 156–196. [Google Scholar] [CrossRef]
- Laurent, E.; Delmon, B. Influence of water in the deactivation of a sulfide NiMo gamma-Al2O3 catalyst during hydrodeoxygenation. J. Catal. 1994, 146, 281–291. [Google Scholar] [CrossRef]
- Laurent, E.; Delmon, B. Influence of oxygen-containing, nitrogen-containing, and sulfur-containing-compounds on the hydrodeoxygenation of phenols over sulfided CoMo/gamma-Al2O3 and NiMo/gamma-Al2O3 Catalysts. Ind. Eng. Chem. Res. 1993, 32, 2516–2524. [Google Scholar] [CrossRef]
- Wildschut, J.; Mahfud, F.H.; Venderbosch, R.H.; Heeres, H.J. Hydrotreatment of fast pyrolysis oil using heterogeneous noble-metal catalysts. Ind. Eng. Chem. Res. 2009, 48, 10324–10334. [Google Scholar] [CrossRef]
- Sepúlveda, C.; Leiva, K.; García, R.; Radovic, L.R.; Ghampson, I.T.; Desisto, W.J.; Fierro, J.L.G.; Escalona, N. Hydrodeoxygenation of 2-methoxyphenol over Mo2N catalysts supported on activated carbons. Catal. Today 2011, 172, 232–239. [Google Scholar] [CrossRef]
- Echeandia, S.; Arias, P.L.; Barrio, V.L.; Pawelec, B.; Fierro, J.L.G. Synergy effect in the HDO of phenol over Ni–W catalysts supported on active carbon: Effect of tungsten precursors. Appl. Catal. B-Environ. 2010, 101, 1–12. [Google Scholar] [CrossRef]
- Popov, A.; Kondratieva, E.; Goupil, J.M.; Mariey, L.; Bazin, P.; Gilson, J.P.; Travert, A.; Mauge, F. Bio-oils hydrodeoxygenation: Adsorption of phenolic molecules on oxidic catalyst supports. J. Phys. Chem. C 2010, 114, 15661–15670. [Google Scholar] [CrossRef]
- Kikhtyanin, O.V.; Rubanov, A.E.; Ayupov, A.B.; Echevsky, G.V. Hydroconversion of sunflower oil on Pd/SAPO-31 catalyst. Fuel 2010, 89, 3085–3092. [Google Scholar] [CrossRef]
- Morgan, T.; Santillan-Jimenez, E.; Harman-Ware, A.E.; Ji, Y.; Grubb, D.; Crocker, M. Catalytic deoxygenation of triglycerides to hydrocarbons over supported nickel catalysts. Chem. Eng. J. 2012, 189–190, 346–355. [Google Scholar] [CrossRef]
- Fan, K.; Liu, J.; Yang, X.; Rong, L. Hydrocracking of Jatropha oil over Ni-H3PW12O40/nano-hydroxyapatite catalyst. Int. J. Hydrogen Energy 2014, 39, 3690–3697. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Loe, R.; Garrett, M.; Morgan, T.; Crocker, M. Effect of Cu promotion on cracking and methanation during the Ni-catalyzed deoxygenation of waste lipids and hemp seed oil to fuel-like hydrocarbons. Catal. Today 2018, 302, 261–271. [Google Scholar] [CrossRef]
- Peng, B.; Yao, Y.; Zhao, C.; Lercher, J.A. Towards quantitative conversion of microalgae oil to diesel-range alkanes with bifunctional catalysts. Angew. Chem. Int. Ed. 2012, 51, 2072–2075. [Google Scholar] [CrossRef] [PubMed]
- Thuan Minh, H.; Armbruster, U.; Pohl, M.M.; Schneider, M.; Radnik, J.; Dang-Lanh, H.; Binh Minh Quoc, P.; Duc Anh, N.; Martin, A. Hydrodeoxygenation of phenol as a model compound for bio-oil on non-noble bimetallic nickel-based catalysts. ChemCatChem 2014, 6, 1940–1951. [Google Scholar]
- Wu, S.K.; Lai, P.C.; Lin, Y.C.; Wan, H.P.; Lee, H.T.; Chang, Y.H. Atmospheric hydro- deoxygenation of guaiacol over alumina-, zirconia-, and silica-supported nickel phosphide catalysts. ACS Sustain. Chem. Eng. 2013, 1, 349–358. [Google Scholar] [CrossRef]
- Peng, B.; Zhao, C.; Kasakov, S.; Foraita, S.; Lercher, J.A. Manipulating catalytic pathways: Deoxygenation of palmitic acid on multifunctional catalysts. Chem. Eur. J. 2013, 19, 4732–4741. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Yenumala, S.R.; Maity, S.K.; Shee, D. Kinetics of hydrodeoxygenation of stearic acid using supported nickel catalysts: Effects of supports. Appl. Catal. A-Gen. 2014, 471, 28–38. [Google Scholar] [CrossRef]
- Zuo, H.; Liu, Q.; Wang, T.; Ma, L.; Zhang, Q.; Zhang, Q. Hydrodeoxygenation of methyl palmitate over supported Ni catalysts for diesel-like fuel production. Energy Fuels 2012, 26, 3747–3755. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Delichere, P.; Geantet, C. Hydrodeoxygenation of guaiacol Part II: Support effect for CoMoS catalysts on HDO activity and selectivity. Appl. Catal. B-Environ. 2011, 101, 246–255. [Google Scholar] [CrossRef]
- Ameen, M.; Azizan, M.T.; Yusup, S.; Ramli, A.; Yasir, M. Catalytic hydrodeoxygenation of triglycerides: An approach to clean diesel fuel production. Renew. Sustain. Energy Rev. 2017, 80, 1072–1088. [Google Scholar] [CrossRef]
- Sang, O.Y.; Twaiq, F.; Zakaria, R.; Mohamed, A.R.; Bhatia, S. Biofuel production from catalytic cracking of palm oil. Energy Source 2003, 25, 859–869. [Google Scholar] [CrossRef]
- Twaiq, F.A.A.; Mohamad, A.R.; Bhatia, S. Performance of composite catalysts in palm oil cracking for the production of liquid fuels and chemicals. Fuel Process. Technol. 2004, 85, 1283–1300. [Google Scholar] [CrossRef]
- Liu, Y.; Sotelo-Boyás, R.; Murata, K.; Minowa, T.; Sakanishi, K. Production of Bio-hydrogenated diesel by hydrotreatment of high-acid-value waste cooking oil over ruthenium catalyst supported on Al-polyoxocation-pillared montmorillonite. Catalysts 2012, 2, 171–190. [Google Scholar] [CrossRef]
- Lovás, P.; Hudec, P.; Hadvinová, M.; Ház, A. Conversion of rapeseed oil via catalytic cracking: Effect of the ZSM-5 catalyst on the deoxygenation process. Fuel Process. Technol. 2015, 134, 223–230. [Google Scholar] [CrossRef]
- Nasikin, M.; Susanto, B.H.; Hirsaman, M.A.; Wijanarko, A. Biogasoline from palm oil by simultaneous cracking and hydrogenation reaction over Nimo/zeolite catalyst. World Appl. Sci. J. 2009, 5, 74–79. [Google Scholar]
- Yigezu, Z.D.; Muthukumar, K. Biofuel production by catalytic cracking of sunflower oil using vanadium pentoxide. J. Anal. Appl. Pyrol. 2015, 112, 341–347. [Google Scholar] [CrossRef]
- Yigezu, Z.D.; Muthukumar, K. Catalytic cracking of vegetable oil with metal oxides for biofuel production. Energy Convers. Manag. 2014, 84, 326–333. [Google Scholar] [CrossRef]
- Zhao, X.; Wei, L.; Cheng, S.; Huang, Y.; Yu, Y.; Julson, J. Catalytic cracking of camelina oil for hydrocarbon biofuel over ZSM-5-Zn catalyst. Fuel Process. Technol. 2015, 139, 117–126. [Google Scholar] [CrossRef]
- Zhao, X.; Wei, L.; Cheng, S.; Kadis, E.; Cao, Y.; Boakye, E.; Gu, Z.; Julson, J. Hydroprocessing of carinata oil for hydrocarbon biofuel over Mo-Zn/Al2O3. Appl. Catal. B-Environ. 2016, 196, 41–49. [Google Scholar] [CrossRef]
- Charusiri, W.; Yongchareon, W.; Vitidsant, T. Conversion of used vegetable oils to liquid fuels and chemicals over HZSM-5, sulfated zirconia and hybrid catalysts. Korean J. Chem. Eng. 2006, 23, 349–355. [Google Scholar] [CrossRef]
- Blommel, P.G.; Cortright, R.D. Production of Conventional Liquid Fuels from Sugar, White Paper. 2008. Available online: http://www.biofuelstp.eu/downloads/Virent_Technology_Whitepaper.pdf (accessed on 14 December 2018).
- Cottright, R.D.; Blommel, P.G. Synthesis of Liquid Fuel and Chemicals from Oxygenated Hydrocarbons. U.S. Patent No. US 2008/0216391, 11 September 2008. [Google Scholar]
- Cottright, R.D.; Blommel, P.G. Synthesis of Liquid Fuel and Chemicals from Oxygenated Hydrocarbons. Patent No. WO 2008/109877, 12 September 2008. [Google Scholar]
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew. Chem. Int. Edit. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- Benson, T.J.; Daggolu, P.R.; Hernandez, R.A.; Liu, S.; White, M.G. Catalytic deoxygenation chemistry: Upgrading of liquids derived from biomass processing. Adv. Catal. 2013, 56, 187–353. [Google Scholar]
- Biddy, M.; Jones, S. Catalytic Upgrading of Sugars to Hydrocarbons Technology Pathway; Technical Report NREL/TP-5100-58055, PNNL-22319; National Renewable Energy Laboratory (NREL): Golden, CO, USA, 2013.
- Shabaker, J.W.; Huber, G.W.; Davda, R.R.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of ethylene glycol over supported platinum catalysts. Catal. Lett. 2003, 88, 1–8. [Google Scholar] [CrossRef]
- Huber, G.W.; Shabaker, J.W.; Dumesic, J.A. Raney Ni-Sn catalyst for H2 production from biomass-derived hydrocarbons. Science 2003, 300, 2075–2077. [Google Scholar] [CrossRef] [PubMed]
- Chaminand, J.; Djakovitch, L.; Gallezot, P.; Marion, P.; Pinel, C.; Rosier, C. Glycerol hydrogenolysis on heterogeneous catalysts. Green Chem. 2004, 6, 359–361. [Google Scholar] [CrossRef]
- Lahr, D.G.; Shanks, B.H. Kinetic analysis of the hydrogenolysis of lower polyhydric alcohols: Glycerol to glycols. Ind. Eng. Chem. Res. 2003, 42, 5467–5472. [Google Scholar] [CrossRef]
- Lahr, D.G.; Shanks, B.H. Effect of sulfur and temperature on ruthenium-catalyzed glycerol hydrogenolysis to glycols. J. Catal. 2005, 232, 386–394. [Google Scholar] [CrossRef]
- Dasari, M.A.; Kiatsimkul, P.P.; Sutterlin, W.R.; Suppes, G.J. Low-pressure hydrogenolysis of glycerol to propylene glycol. Appl. Catal. A-Gen. 2005, 281, 225–231. [Google Scholar] [CrossRef]
- Chen, D.; Moljord, K.; Fuglerud, T.; Holmen, A. The effect of crystal size of SAPO-34 on the selectivity and deactivation of the MTO reaction. Microporous Mesoporous Mater. 1999, 29, 191–203. [Google Scholar] [CrossRef]
- Chen, J.Q.; Bozzano, A.; Glover, B.; Fuglerud, T.; Kvisle, S. Recent advancements in ethylene and propylene production using the UOP/Hydro MTO process. Catal. Today 2005, 106, 103–107. [Google Scholar] [CrossRef]
- Wang, P.; Yang, D.; Hu, J.; Xu, J.; Lu, G. Synthesis of SAPO-34 with small and tunable crystallite size by two-step hydrothermal crystallization and its catalytic performance for MTO reaction. Catal. Today 2013, 212, 62.e1–62.e8. [Google Scholar] [CrossRef]
- Jun, J.W.; Jeon, J.; Kim, C.U.; Jeong, K.E.; Jeong, S.Y.; Jhung, S.H. Synthesis of mesoporous SAPO-34 molecular sieves and their applications in dehydration of butanols and ethanol. J. Nanosci. Nanotechnol. 2013, 13, 2782–2788. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, Y.; Tao, L.; Dai, B.; Yang, M.; Chen, Z.; Zhu, X. Dehydration reaction of bio-ethanol to ethylene over modified SAPO catalysts. J. Ind. Eng. Chem. 2010, 16, 717–722. [Google Scholar] [CrossRef]
- Ermakov, R.V.; Plakhotnik, V.A. Conversion of lower alcohols into C2-C4 olefins over acid-base catalysts. Petrol. Chem. 2008, 48, 1–5. [Google Scholar] [CrossRef]
- Takahara, I.; Saito, M.; Inaba, M.; Murata, K. Dehydration of ethanol into ethylene over solid acid catalysts. Catal. Lett. 2005, 105, 249–252. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, R.; Yang, X.; Zhang, F. Comparison of four catalysts in the catalytic dehydration of ethanol to ethylene. Microporous Mesoporous Mater. 2008, 116, 210–215. [Google Scholar] [CrossRef]
- Ouyang, J.; Kong, F.; Su, G.; Hu, Y.; Song, Q. Catalytic conversion of bio-ethanol to ethylene over La-modified HZSM-5 catalysts in a bioreactor. Catal. Lett. 2009, 132, 64–74. [Google Scholar] [CrossRef]
- Ramasamy, K.K.; Wang, Y. Catalyst activity comparison of alcohols over zeolites. J. Energy Chem. 2013, 22, 65–71. [Google Scholar] [CrossRef]
- Zhang, M.; Yu, Y. Dehydration of ethanol to ethylene. Ind. Eng. Chem. Res. 2013, 52, 9505–9514. [Google Scholar] [CrossRef]
- Gucbilmez, Y.; Dogu, T.; Balci, S. Ethylene and acetaldehyde production by selective oxidation of ethanol using mesoporous V-MCM-41 catalysts. Ind. Eng. Chem. Res. 2006, 45, 3496–3502. [Google Scholar] [CrossRef]
- Iwamoto, M.; Kasai, K.; Haishi, T. Conversion of ethanol into polyolefin building blocks: Reaction pathways on nickel ion-loaded mesoporous silica. ChemSusChem 2011, 4, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Haishi, T.; Kasai, K.; Iwamoto, M. Fast and quantitative dehydration of lower alcohols to corresponding olefins on mesoporous silica catalyst. Chem. Lett. 2011, 40, 614–616. [Google Scholar] [CrossRef]
- Pan, Q.; Ramanathan, A.; Snavely, W.K.; Chaudhari, R.V.; Subramaniam, B. Synthesis and dehydration activity of novel lewis acidic ordered mesoporous silicate: Zr-KIT-6. Ind. Eng. Chem. Res. 2013, 52, 15481–15487. [Google Scholar] [CrossRef]
- Pan, Q.; Ramanathan, A.; Snavely, W.K.; Chaudhari, R.V.; Subramaniam, B. Intrinsic kinetics of ethanol dehydration over Lewis acidic ordered mesoporous silicate, Zr-KIT-6. Top. Catal. 2014, 57, 1407–1411. [Google Scholar] [CrossRef]
- Müller, J.M.; Mesquita, G.C.; Franco, S.M.; Borges, L.D.; De Macedo, J.L.; Dias, J.A.; Dias, S.C.L. Solid-state dealumination of zeolites for use as catalysts in alcohol dehydration. Microporous Mesoporous Mater. 2015, 204, 50–57. [Google Scholar] [CrossRef]
- Nash, C.P.; Ramanathan, A.; Ruddy, D.A.; Behl, M.; Gjersing, E.; Griffin, M.; Zhu, H.; Subramaniam, B.; Schaidle, J.A.; Hensley, J.E. Mixed alcohol dehydration over Brønsted and Lewis acidic catalysts. Appl. Catal. A-Gen. 2016, 510, 110–124. [Google Scholar] [CrossRef]
- O’Connor, C.T.; Kojima, M. Alkene oligomerization. Catal. Today 1990, 6, 329–349. [Google Scholar] [CrossRef]
- Quann, R.J.; Green, L.A.; Tabak, S.A.; Krambeck, F.J. Chemistry of olefin oligomerization over ZSM-5 catalyst. Ind. Eng. Chem. Res. 1988, 27, 565–570. [Google Scholar] [CrossRef]
- Schwarz, S.; Kojima, M.; O’Connor, C.T. Effect of silicon-to-aluminium ratio and synthesis time on high-pressure olefin oligomerization over ZSM-5. Appl. Catal. 1989, 56, 263–280. [Google Scholar] [CrossRef]
- Petrik, L.F.; O’Connor, C.T.; Schwarz, S. The influence of various synthesis parameters on the morphology and crystalsize of ZSM-5 and the relationship between morphology and crystal size and propene oligomerization activity. Stud. Surf. Sci. Catal. 1995, 94, 517–524. [Google Scholar]
- Tabak, S.A.; Krambeck, F.J.; Garwood, W.E. Conversion of propylene and butylene over ZSM-5 catalyst. AIChE J. 1986, 32, 1526–1531. [Google Scholar] [CrossRef]
- Chen, N.Y.; Garwood, W.E.; Dwyer, F.G. Shape Selective Catalysis in Industrial Applications, 1st ed.; Marcel Dekker: New York, NY, USA, 1989. [Google Scholar]
- De Pontes, M. Process for diesel production from low molecular weight Fischer-Tropsch products. Proceedings of Worldwide Catalyst Industry Conference CatCon’96, New Orleans, LA, USA, 29–30 October 1996. [Google Scholar]
- Düker, A.; Lichtscheidl, J.; Altwicker, C. Dieselization of the world—How to increase Diesel yield in a refinery. In Proceedings of the ERTC 14th Annual Meeting, Berlin, Germany, 9–11 November 2009. [Google Scholar]
- Kholer, E.; Scmidt, F.; Wernicke, H.J.; de Pontes, M.; Roberts, H.L. Converting olefins to diesel—The COD process. Hydrocarb. Technol. Int. 1995, 37–40. [Google Scholar]
- Vamvuka, D. Bio-oil, solid and gaseous biofuels from biomass pyrolysis processes-An overview. Int. J. Energy Res. 2011, 35, 853–862. [Google Scholar] [CrossRef]
- Guo, M.; Song, W.; Buhain, J. Bioenergy and biofuels: History, status, and perspective. Renew. Sustain. Energy Rev. 2015, 42, 712–725. [Google Scholar] [CrossRef]
- Silber, A.; Levkovitch, I.; Graber, E.R. pH-dependent mineral release and surface properties of cornstraw biochar: Agronomic implications. Environ. Sci. Technol. 2010, 44, 9318–9323. [Google Scholar] [CrossRef] [PubMed]
- Day, D.; Evans, R.J.; Lee, J.W.; Reicosky, D. Economical CO2, SOx, and NOx capture from fossil-fuel utilization with combined renewable hydrogen production and large-scale carbon sequestration. Energy 2005, 30, 2558–2579. [Google Scholar] [CrossRef]
- Winsley, P. Biochar and bioenergy production for climate change mitigation. N. Z. Sci. Rev. 2007, 64, 5–10. [Google Scholar]
- Lehmann, J. A handful of carbon. Nature 2007, 447, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J. Bio-energy in the black. Front. Ecol. Environ. 2007, 5, 381–387. [Google Scholar] [CrossRef]
- Schlogl, R. Chemical Energy Storage; Walter de Gruyter GmbH: Boston, MA, USA, 2013. [Google Scholar]
- Czernik, S.; Bridgwater, A.V. Overview of applications of biomass fast pyrolysis oil. Energy Fuel. 2004, 18, 590–598. [Google Scholar] [CrossRef]
- Balat, M.; Balat, M.; Kirtay, E.; Balat, H. Main routes for the thermo-conversion of biomass into fuels and chemicals. Part 1: Pyrolysis systems. Energy Convers. Manag. 2009, 50, 3147–3157. [Google Scholar] [CrossRef]
- Jahirul, M.I.; Rasul, M.G.; Chowdhury, A.A.; Ashwath, N. Biofuels production through biomass pyrolysis- A technological review. Energies 2012, 5, 4952–5001. [Google Scholar] [CrossRef]
- Laird, D.A.; Brown, R.C.; Amonette, J.E.; Lehmann, J. Review of the pyrolysis platform for coproducing bio-oil and biochar. Biofuel Bioprod. Biorefin. 2009, 3, 547–562. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Fast Pyrolysis and Hydroprocessing; DOE/EE-0808; U.S. Department of Energy: Washington, DC, USA, 2012.
- Wang, H.; Elliott, D.C.; French, R.J.; Deutch, S.; Iisa, K. Biomass conversion to produce hydrocarbon liquid fuel via hot-vapor filtered fast pyrolysis and catalytic hydrotreating. J. Vis. Exp. 2016, 118, 54088. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Kumar, A. Production of renewable diesel through the hydroprocessing of lignocellulosic biomass-derived bio-oil: A review. Renew. Sustain. Energy Rev. 2016, 58, 1293–1307. [Google Scholar] [CrossRef]
- Boscagli, C.; Morgano, M.T.; Raffelt, K.; Leibold, H.; Grunwaldt, J.D. Influence of feedstock, catalyst, pyrolysis and hydrotreatment temperature on the composition of upgraded oils from intermediate pyrolysis. Biomass Bioenergy 2018, 116, 236–248. [Google Scholar] [CrossRef]
- Baldauf, W.; Balfanz, U.; Rupp, M. Upgrading of flash pyrolysis oil and utilization in refineries. Biomass Bioenergy 1994, 7, 237–244. [Google Scholar] [CrossRef]
- Sheu, Y.H.E.; Anthony, R.G.; Soltes, E.J. Kinetic studies of upgrading pine pyrolytic oil by hydrotreatment. Fuel Process. Technol. 1988, 19, 31–50. [Google Scholar] [CrossRef]
- Elliott, D.C.; Hart, T.R.; Neuenschwander, G.G.; Rotness, L.J.; Zacher, A.H. Catalytic hydroprocessing of biomass fast pyrolysis bio-oil to produce hydrocarbon products. Environ. Prog. Sustain. 2009, 28, 4410–4449. [Google Scholar] [CrossRef]
- Venderbosch, R.H.; Ardiyanti, A.R.; Wildschut, J.; Oasmaa, A.; Heeres, H.J. Stabilization of biomass-derived pyrolysis oils. J. Chem. Technol. Biot. 2010, 85, 674–686. [Google Scholar] [CrossRef]
- Guo, X.Y.; Yan, Y.J.; Li, T.C. Influence of catalyst type and regeneration on upgrading of crude bio-oil through catalytical thermal cracking. Chin. J. Process Eng. 2004, 4, 53–58. [Google Scholar]
- Adjaye, J.D.; Bakhshi, N.N. Production of hydrocarbons by catalytic upgrading of a fast pyrolysis bio-oil. Part I: Conversion over various catalysts. Fuel Process. Technol. 1995, 45, 161–183. [Google Scholar] [CrossRef]
- Williams, P.T.; Horne, P.A. Characterisation of oils from the fluidised bed pyrolysis of biomass with zeolite catalyst upgrading. Biomass Bioenergy 1994, 7, 223–236. [Google Scholar] [CrossRef]
- Katikaneni, S.P.R.; Adjaye, J.D.; Bakhshi, N.N. Performance of Aluminophosphate Molecular Sieve Catalysts for the Production of Hydrocarbons from Wood-Derived and Vegetable Oils. Energy Fuel. 1995, 9, 1065–1078. [Google Scholar] [CrossRef]
- Jiang, W.; Kumar, A.; Adamopoulos, S. Liquefaction of lignocellulosic materials and its application in wood adhesives—A review. Ind. Crop. Prod. 2018, 124, 325–342. [Google Scholar] [CrossRef]
- Toor, S.S.; Rosendahl, L.; Rudolf, A. Hydrothermal liquefaction of biomass: A review of subcritical water technologies. Energy 2011, 36, 2328–2342. [Google Scholar] [CrossRef]
- Gollakota, A.R.K.; Kishore, N.; Gu, S. A review on hydrothermal liquefaction of biomass. Renew. Sustain. Energy Rev. 2018, 81, 1378–1392. [Google Scholar] [CrossRef]
- Ramirez, J.A.; Brown, R.J.; Rainey, T.J. A review of hydrothermal liquefaction bio-crude properties and prospects for upgrading to transportation fuels. Energies 2015, 8, 6765–6794. [Google Scholar] [CrossRef]
- Elliot, D.C.; Baker, E.G. Catalytic Hydrotreating of Biomass Liquefaction Products to Produce Hydrocarbon Fuels: Interim Report; Report No. PNL-5844; Pacific Northwest Laboratory: Richland, WA, USA, 1986. [Google Scholar]
- Dimitriadis, A.; Bezergianni, S. Hydrothermal liquefaction of various biomass and waste feedstocks for biocrude production: A state of the art review. Renew. Sustain. Energy Rev. 2017, 68, 113–125. [Google Scholar] [CrossRef]
- Xu, D.; Lin, G.; Guo, S.; Wang, S.; Guo, Y.; Jing, Z. Catalytic hydrothermal liquefaction of algae and upgrading of biocrude: A critical review. Renew. Sustain. Energy Rev. 2018, 97, 103–118. [Google Scholar] [CrossRef]
- Goudriaan, F.; Peferoen, D.G.R. Liquid fuels from biomass via a hydrothermal process. Chem. Eng. Sci. 1990, 45, 2729–2734. [Google Scholar] [CrossRef]
- Klass, D.L. Energy from Biomass and Wastes XI; IGT: Chicago, IL, USA, 1988. [Google Scholar]
- Grilc, M.; Likozar, B.; Levec, J. Hydrodeoxygenation and hydrocracking of solvolysed lignocellulosic biomass by oxide, reduced and sulphide form of NiMo, Ni, Mo and Pd catalysts. Appl. Catal. B-Environ. 2014, 150–151, 275–287. [Google Scholar] [CrossRef]
- Duan, P.; Savage, P.E. Upgrading of crude algal bio-oil in supercritical water. Bioresour. Technol. 2011, 102, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Savage, P.E. Catalytic treatment of crude algal bio-oil in supercritical water: Optimization studies. Energy Environ. Sci. 2011, 4, 1447–1456. [Google Scholar] [CrossRef]
- Duan, P.; Savage, P.E. Catalytic hydrotreatment of crude algal bio-oil in supercritical water. Appl. Catal. B-Environ. 2011, 104, 136–143. [Google Scholar] [CrossRef]
- Adjaye, J.D.; Bakshi, N.N. Upgrading of a wood-derived oil over various catalysts. Biomass Bioenergy 1994, 7, 201–211. [Google Scholar] [CrossRef]
- Gevert, B.S.; Otterstedt, J.E. Upgrading of directly liquefied biomass to transportation fuels: Catalytic cracking. Biomass 1987, 14, 173–183. [Google Scholar] [CrossRef]
- Higman, C. Gasification process technology. In Advances in Clean Hydrocarbon Fuel Processing: Science and Technology; Khan, M.R., Ed.; Woodhead Publishing Ltd.: Cambridge, UK, 2011; pp. 155–185. [Google Scholar]
- Storch, H.H.; Golumbic, N.; Anderson, R.B. The Fischer-Tropsch and Related Syntheses; Wiley: New York, NY, USA, 1951. [Google Scholar]
- Snehesh, S.A.; Dasappa, S. Biomass to liquid transportation fuel via Fischer Tropsch synthesis - Technology review and current scenario. Renew. Sustain. Energy Rev. 2016, 58, 267–286. [Google Scholar]
- Mahmoudi, H.; Mahmoudi, M.; Doustdar, O.; Jahangiri, H.; Tsolakis, A.; Gu, S.; LechWyszynski, M. A review of Fischer Tropsch synthesis process, mechanism, surface chemistry and catalyst formulation. Biofuels Eng. 2017, 2, 11–31. [Google Scholar] [CrossRef]
- Torres Galvis, H.M.; de Jong, K.P. Catalysts for production of lower olefins from synthesis gas: A review. ACS Catal. 2013, 3, 2130–2149. [Google Scholar] [CrossRef]
- Dry, M. Chemical concepts used for engineering purposes. Stud. Surf. Sci. Catal. 2004, 152, 196–257. [Google Scholar]
- Dancuart, L.P.; de Haan, R.; de Klerk, A. Processing of primary Fischer-Tropsch products. Stud. Surf. Sci. Catal. 2004, 152, 482–532. [Google Scholar]
- Calemma, V.; Peratello, S.; Pavoni, S.; Clerici, G.; Perego, C. Hydroconversion of a mixture of long chain n-paraffins to middle distillate: Effect of the operating parameters and products properties. Stud. Surf. Sci. Catal. 2001, 136, 307–312. [Google Scholar]
- Calemma, V.; Correra, S.; Perego, C.; Pollesel, P.; Pellegrini, L. Hydroconversion of Fischer-Tropsch waxes: Assessment of the operating conditions effect by factorial design experiments. Catal. Today 2005, 106, 282–287. [Google Scholar] [CrossRef]
- Leckel, D. Noble metal wax hydrocracking catalysts supported on high-siliceous alumina. Ind. Eng. Chem. Res. 2007, 46, 3505–3512. [Google Scholar] [CrossRef]
- Calemma, V.; Gambaro, C.; Parker, W.O.; Carbone, R.; Giardino, R.; Scorletti, P. Middle distillates from hydrocracking of FT waxes: Composition, characteristics and emission properties. Catal. Today 2010, 149, 40–46. [Google Scholar] [CrossRef]
- EN 590:2013+A1:2017. Automotive Fuels—Diesel—Requirements and Test Methods; European Committee for Standardization: Brussels, Belgium, 2017. [Google Scholar]
- ASTM D975-17a. Standard Specification for Diesel oil Fuel Oils; ASTM International: West Conshohocken, PA, USA, 2017. [Google Scholar]
- EN 14214/A2:2018-02. Liquid Petroleum Products—Fatty Acid Methyl Esters (FAME) for Use in Diesel Engines and Heating Applications—Requirements and Test Methods; European Committee for Standardization: Brussels, Belgium, 2018. [Google Scholar]
- EN 15940:2016/A1:2018. Automotive Fuels—Paraffinic DIESEL fuel from Synthesis of Hydrotreatment—Requirements and Test Methods; European Committee for Standardization: Brussels, Belgium, 2018. [Google Scholar]
- Singh, D.; Subramanian, K.A.; Garg, M.O. Comprehensive review of combustion, performance and emissions characteristics of a compression ignition engine fueled with hydroprocessed renewable diesel. Renew. Sustain. Energy Rev. 2018, 81, 2947–2954. [Google Scholar] [CrossRef]
- Mancaruso, E.; Sequino, L.; Vaglieco, B.M. First and second generation biodiesels spray characterization in a diesel engine. Fuel 2011, 90, 2870–2883. [Google Scholar] [CrossRef]
- Neste Renewable Diesel Handbook; Neste Proprietary Publication: Espoo, Finland, 2016.
- Smagala, T.G.; Christensen, E.; Christison, K.M.; Mohler, R.E.; Gjersing, E.; McCormick, R.L. Hydrocarbon renewable and synthetic diesel fuel blendstocks: Composition and properties. Energy Fuel 2013, 27, 237–246. [Google Scholar] [CrossRef]
- Matzke, M.; Litzow, U.; Jess, A.; Caprotti, R.; Balfour, G. Diesel Lubricity Requirements of Future Fuel Injection Equipment. SAE Int. J. Fuels Lubr. 2009, 2, 273–286. [Google Scholar] [CrossRef]
- Hunger, H.; Litzow, U.; Genze, S.; Dőrr, N.; Karner, D.; Eisenmenger-Sittner, C. Tribological characterization and surface analysis of diesel lubricated sliding contacts. Tribologie Schmierungstechnik 2010, 57, 6–13. [Google Scholar]
- Lois, E.; Arkoudeas, P. Lubricating Aspects of Automotive Fuels; Intechopen: London, UK, 2012. [Google Scholar]
- Taylor, C.F. The Internal Combustion Engine in Theory and Practice Vol. 1, 2nd ed.; MIT Press: Cambridge, MA, USA, 1985. [Google Scholar]
- Heywood, J.B. Internal Combustion Engine Fundamentals, 1st ed.; McGraw-Hill Education: New York, NY, USA, 1988. [Google Scholar]
- Reif, K. Diesel Engine Management—Systems and Components, 1st ed.; Springer Vieweg: Kranzberg, Germany, 2014. [Google Scholar]
- Caton, P.A.; Williams, S.A.; Kamin, R.A.; Luning-Prak, D.; Hamilton, L.J.; Cowart, J.S. Hydrotreated algae renewable fuel performance in a military diesel engine. In Proceedings of the ASME 2012 Internal Combustion Engine Division Spring Technical Conference, Torino, Italy, 6–9 May 2012; pp. 121–132. [Google Scholar]
- Petersen, J.; Seivwright, D.; Caton, P.; Millsaps, K. Combustion characterization and ignition delay modeling of low and high-cetane alternative diesel fuels in a marine diesel engine. Energy Fuel. 2014, 28, 5463–5471. [Google Scholar] [CrossRef]
- Ogunkoya, D.; Roberts, W.L.; Fang, T.; Thapaliya, N. Investigation of the effects of renewable diesel fuels on engine performance, combustion, and emissions. Fuel 2015, 140, 541–554. [Google Scholar] [CrossRef]
- Mangus, M.; Mattson, J.; Depcik, C. Performance and emissions characteristics of hydroprocessed renewable jet fuel blends in a single-cylinder compression ignition engine with electronically controlled fuel injection. Combust. Sci. Technol. 2015, 187, 857–873. [Google Scholar] [CrossRef]
- Zubel, M.; Bhardwaj, O.P.; Heuser, B.; Holderbaum, B.; Doerr, S.; Nuottimäki, J. Advanced fuel formulation approach using blends of paraffinic and oxygenated biofuels: Analysis of emission reduction potential in a high efficiency diesel combustion system. SAE Int. J. Fuels Lubr. 2016, 9, 481–492. [Google Scholar] [CrossRef]
- Mattson, J.M.S.; Depcik, C. First and second law heat release analysis in a single cylinder engine. SAE Int. J. Engines 2016, 9, 536–545. [Google Scholar] [CrossRef]
- Kumar, V.; Sindhu, R.K.; Kumar, S. Comparative analysis of green diesel versus petro-diesel in compression ignition engine. Biosci. Biotechnol. Res. Commun. 2018, 11, 128–135. [Google Scholar]
- Aatola, H.; Larmi, M.; Sarjovaara, T.; Mikkonen, S. Hydrotreated vegetable oil (HVO) as a renewable diesel fuel: Trade-off between NOx, particulate emission, and fuel consumption of a heavy duty engine. SAE Int. J. Engines 2008, 1, 1251–1262. [Google Scholar] [CrossRef]
- Sugiyama, K.; Goto, I.; Kitano, K.; Mogi, K.; Honkanen, M. Effects of hydrotreated vegetable oil (HVO) as renewable diesel fuel on combustion and exhaust emissions in diesel engine. SAE Int. J. Fuels Lubr. 2011, 5, 205–217. [Google Scholar] [CrossRef]
- Armas, O.; García-Contreras, R.; Ramos, A.; López, A.F. Impact of animal fat biodiesel, GTL, and HVO fuels on combustion, performance, and pollutant emissions of a light-duty diesel vehicle tested under the NEDC. J. Energy Eng. 2015, 141. [Google Scholar] [CrossRef]
- Drenth, A.C.; Olsen, D.B.; Cabot, P.E.; Johnson, J.J. Compression ignition engine performance and emission evaluation of industrial oilseed biofuel feedstocks camelina, carinata, and pennycress across three fuel pathways. Fuel 2014, 136, 143–155. [Google Scholar] [CrossRef]
- Kim, D.; Kim, S.; Oh, S.; No, S.Y. Engine performance and emission characteristics of hydrotreated vegetable oil in light duty diesel engines. Fuel 2014, 125, 36–43. [Google Scholar] [CrossRef]
- Murtonen, T.; Aakko-Saksa, P.; Kuronen, M.; Mikkonen, S.; Lehtoranta, K. Emissions with heavy-duty diesel engines and vehicles using FAME, HVO and GTL fuels with and without DOC + POC aftertreatment. SAE Int. J. Fuels Lubr. 2009, 2, 147–166. [Google Scholar] [CrossRef]
- Singh, D.; Subramanian, K.A.; Singal, S.K. Emissions and fuel consumption characteristics of a heavy duty diesel engine fueled with hydroprocessed renewable diesel and biodiesel. Appl. Energy 2015, 155, 440–446. [Google Scholar] [CrossRef]
- Karavalakis, G.; Jiang, Y.; Yang, J.; Durbin, T.; Nuottimäki, J.; Lehto, K. Emissions and fuel economy evaluation from two current technology heavy-duty trucks operated on HVO and FAME blends. SAE Int. J. Fuels Lubr. 2016, 9, 177–190. [Google Scholar] [CrossRef]
- Erkkila, K.; Nylund, N.O.; Hulkkonen, T.; Tilli, A.; Mikkonen, S.; Saikkonen, P.; Makinen, R.; Amberla, A. Emission performance of paraffinic HVO diesel fuel in heavy duty vehicles. SAE Int. 2011. [Google Scholar] [CrossRef]
- Hajbabaei, M.; Johnson, K.C.; Okamoto, R.A.; Mitchell, A.; Pullman, M.; Durbin, T.D. Evaluation of the impacts of biodiesel and second generation biofuels on NOx emissions for CARB diesel fuels. Environ. Sci. Technol. 2012, 46, 9163–9173. [Google Scholar] [CrossRef] [PubMed]
- Westphal, G.A.; Krahl, J.; Munack, A.; Rosenkranz, N.; Schröder, O.; Schaak, J.; Pabst, C.; Brüning, T.; Bünger, J. Combustion of hydrotreated vegetable oil and jatropha methyl ester in a heavy duty engine: Emissions and bacterial mutagenicity. Environ. Sci. Technol. 2013, 47, 6038–6046. [Google Scholar] [CrossRef] [PubMed]
- Happonen, M.; Heikkilä, J.; Murtonen, T.; Lehto, K.; Sarjovaara, T.; Larmi, M.; Keskinen, J.; Virtanen, A. Reductions in particulate and NOx emissions by diesel engine parameter adjustments with HVO fuel. Environ. Sci. Technol. 2012, 46, 6198–6204. [Google Scholar] [CrossRef] [PubMed]
- Jaroonjitsathian, S.; Tipdecho, C.; Sukajit, P.; Namthirach, N.; Suppatvech, S. Bio-hydrogenated diesel (BHD): Renewable fuel for advanced diesel technology. SAE Int. 2013. 2013-01-0070. [Google Scholar]
- Pflaum, H.; Hofmann, P.; Geringer, B.; Weissel, W. Potential of hydrogenated vegetable oil (HVO) in a modern diesel engine. SAE Int. 2010. 2010-32-0081. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feedstock Group | Feedstock | Structural Analysis (wt% dry) | Ultimate Analysis (wt% daf) | Ash (wt% dry) | Cell. + Semihell/Lignin | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cellulose | Hemicellulose | Lignin | Sum C5 | Sum C6 | C | H | N | S | O | ||||
| Food Crops | Sugar cane | 31 | 12 | 8 | - | - | 50 | 6.2 | 0.50 | 0.17 | 44 | 3.3 | 5.7 |
| Sugar beet | 26 | 28 | 5 | - | - | 47 | 6.2 | 1.93 | 0.14 | 45 | 4.3 | 12.0 | |
| Corn | 40 | 30 | 12 | 27 | 31 | 47 | 6.2 | 0.63 | 0.08 | 46 | 2.4 | 5.7 | |
| Wheat | 31 | 20 | 7 | - | - | 48 | 5.8 | 1.40 | 0.22 | 44 | 8.1 | 7.3 | |
| Sorghum | 39 | 24 | 9 | - | - | 49 | 5.8 | 0.91 | 0.07 | 44 | 6.9 | 6.7 | |
| Forestry | Spruce wood | 45 | 21 | 28 | 6 | 63 | 49 | 5.9 | 0.17 | 0.02 | 45 | 0.9 | 2.3 |
| Pine | 44 | 25 | 26 | 7 | 57 | 52 | 6.3 | 0.14 | 0.10 | 41 | 0.7 | 2.7 | |
| Oak | 40 | 20 | 25 | 18 | 40 | 50 | 6.3 | 0.61 | 0.09 | 43 | 1.4 | 2.4 | |
| Birch | 39 | 29 | 22 | 27 | 39 | 49 | 6.2 | 0.19 | 0.15 | 45 | 0.5 | 3.2 | |
| Forest Industry Residues | Bark | 24 | 25 | 50 | - | - | 53 | 5.9 | 0.41 | 0.05 | 40 | 4.1 | 1.0 |
| Thinnings | 37 | 18 | 34 | - | - | 51 | 5.7 | 0.59 | 0.09 | 42 | 1.5 | 1.7 | |
| Shaw dust & shavings | 47 | 21 | 25 | - | - | 52 | 5.8 | 0.12 | 0.03 | 42 | 0.6 | 2.8 | |
| Short Rotation Forestry/Coppice | Poplar | 46 | 26 | 23 | 18 | 52 | 50 | 6.1 | 0.25 | 0.03 | 44 | 1.0 | 3.2 |
| Eucalyptus | 43 | 23 | 25 | 11 | 52 | 51 | 6.1 | 0.27 | 0.04 | 42 | 1.6 | 2.6 | |
| Willow | 39 | 18 | 26 | 16 | 41 | 50 | 6.1 | 0.62 | 0.05 | 43 | 2.0 | 2.1 | |
| Grassy Crops | Switch grass | 37 | 31 | 23 | 28 | 41 | 49 | 6.1 | 0.64 | 0.12 | 44 | 6.3 | 3.0 |
| Miscanthus | 45 | 24 | 21 | 17 | 45 | 50 | 5.6 | 0.54 | 0.06 | 44 | 3.7 | 3.2 | |
| Arundo donax | 33 | 27 | 18 | 27 | 33 | 47 | 5.7 | 0.47 | 0.11 | 47 | 3.9 | 3.4 | |
| Agricultural Residues | Straw | 37 | 27 | 17 | 21 | 39 | 49 | 5.9 | 0.76 | 0.10 | 44 | 8.4 | 3.6 |
| Olive tree prunings | 30 | 18 | 21 | - | - | 48 | 6.1 | 0.88 | 0.09 | 46 | 13.3 | 2.3 | |
| Grape prunings | 26 | 39 | 32 | - | - | 49 | 6.0 | 0.83 | 0.03 | 44 | 2.6 | 2.0 | |
| Bagasse | 39 | 31 | 18 | 24 | 41 | 49 | 6.0 | 0.55 | 0.10 | 44 | 5.8 | 3.9 | |
| Agro-industrial Residues | Almond shell | 36 | 29 | 29 | 29 | 30 | 50 | 6.2 | 0.89 | 0.04 | 43 | 3.7 | 2.3 |
| Hazelnut shell | 26 | 30 | 46 | - | - | 49 | 5.9 | 0.77 | 0.46 | 45 | 1.4 | 1.2 | |
| Walnut shell | 23 | 20 | 43 | 19 | 26 | 52 | 6.2 | 0.80 | 0.08 | 41 | 1.8 | 1.0 | |
| Orange peel | 16 | 7 | 5 | - | - | 50 | 6.6 | 1.29 | 0.07 | 42 | 3.8 | 4.6 | |
| Olive husk | 23 | 25 | 47 | - | - | 51 | 6.9 | 1.22 | 0.07 | 41 | 5.5 | 1.0 | |
| Others | Macroalgae | 9 | 7 | 0 | - | - | 31 | 4.3 | 2.00 | 1.50 | 38 | 30.0 | n/a |
| Paper pulp/sludge | 59 | 17 | 14 | 12 | 68 | 42 | 5.5 | 0.79 | 0.39 | 52 | 18.9 | 5.5 | |
| Municipal solid waste | 14 | 2 | 16 | - | - | 57 | 6.7 | 1.83 | 0.64 | 33 | 25.2 | 1.0 | |
| Food industry wastes | 18 | 21 | 17 | - | - | 50 | 6.7 | 1.58 | 0.21 | 41 | 6.6 | 2.3 | |
| Fatty acid | Typical Fatty Acid Composition, wt% | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vegetable Sources | Animal and Fish Sources | |||||||||||
| Rapeseed | Palm | Soybean | Sunflower | Jatropha | Camelina | Beef Fat | Chicken Fat 1 | Yellow Grease | Lard 2 | Tallow | Salmon 3 | |
| Capric (10:0) | 0.56 | 0.48 | 0.10 | |||||||||
| Lauric (12:0) | 0.09 | 0.09 | 0.10 | 0.09 | 0.36 | 0.19 | 0.19 | |||||
| Myristic (14:0) | 1.05 | 0.09 | 0.10 | 0.28 | 2.56 | 3 | 1 | 0.78 | 2 | 2.55 | 3 | |
| Palmitic (16:0) | 4.04 | 41.59 | 11.07 | 6.31 | 14.36 | 5.79 | 27 | 22 | 16.03 | 27 | 23.85 | 11 |
| Palmitoleic (16:1) | 0.09 | 0.19 | 0.18 | 0.10 | 0.93 | 11 | 6 | 0.85 | 4 | 2.55 | 5 | |
| Stearic (18:0) | 2.1 | 3.6 | 3.72 | 3.55 | 5.88 | 2.66 | 7 | 6 | 6.90 | 11 | 17.86 | 4 |
| Oleic (18:1) | 57.26 | 40.41 | 22.61 | 21.39 | 38.94 | 15.96 | 48 | 37 | 43.34 | 44 | 41.42 | 25 |
| Linoleic (18:2) | 20.69 | 9.3 | 51.33 | 63.35 | 34.89 | 16.15 | 2 | 20 | 24.39 | 11 | 4.32 | 5 |
| Linolenic (18:3) | 8.08 | 0.29 | 5.63 | 1.46 | 0.28 | 33.81 | 1 | 1.07 | 0.88 | 5 | ||
| Arachidic (20:0) | 0.37 | 0.29 | 0.27 | 0.29 | 0.19 | 1.33 | 0.28 | 0.20 | ||||
| Eicosenoic (20:1) | 2.02 | 0.10 | 0.27 | 0.19 | 0.09 | 13.68 | 0.47 | 0.59 | ||||
| Behenic (22:0) | 0.28 | 0.10 | 0.27 | 0.58 | 0.19 | 0.81 | 0.38 | 0.10 | ||||
| Erucic (22:1) | 0.46 | 0.09 | 0.10 | 0.09 | 2.94 | 0.09 | 0.10 | |||||
| Total Saturated | 7.06 | 48.85 | 15.70 | 11.22 | 23.58 | 14.18 | 37 | 29 | 24.94 | 40 | 46.87 | 18 |
| Total Unsaturated | 88.80 | 50.28 | 80.39 | 88.69 | 75.31 | 84.87 | 61 | 65 | 75.06 | 59 | 51.21 | 80 |
| Fatty acid | Typical Fatty Acid Composition, wt% | |||||
|---|---|---|---|---|---|---|
| Nannochlopsis Oculata. | Picochlorum sp. | Phaeodactylum Tricornutum | Amphidinium sp. | Bidduphia sp. | Extubocellulus sp. | |
| Capric (10:0) | 0.07 | |||||
| Lauric (12:0) | 0.41 | |||||
| Myristic (14:0) | 5.76 | 0.55 | 3.29 | 0.79 | 21.50 | 6.61 |
| Palmitic (16:0) | 32.21 | 17.03 | 23.62 | 35.68 | 23.62 | 25.56 |
| Palmitoleic (16:1) | 29.57 | 1.22 | 48.16 | 1.08 | 33.36 | 60.52 |
| Stearic (18:0) | 0.98 | 3.45 | 0.81 | 4.10 | 0.73 | 0.95 |
| Oleic (18:1) | 20.10 | 15.73 | 3.61 | 19.35 | 1.47 | 3.21 |
| Linoleic (18:2) | 1.28 | 36.22 | ||||
| Linolenic (18:3) | 15.07 | |||||
| Arachidic (20:0) | 2.11 | 5.68 | ||||
| Eicosapentaenoic acid (20:5) | 8.26 | 12.13 | 11.94 | 9.65 | ||
| Docosahexaenoic acid (22:6) | 20.21 | |||||
| Total Saturated | 40.5 | 23.7 | 28.1 | 46.3 | 48.8 | 33.6 |
| Total Unsaturated | 59.5 | 76.3 | 71.9 | 53.7 | 51.2 | 66.4 |
| Company Name | Location | Feedstocks | Capacity | Technology |
|---|---|---|---|---|
| Neste | The Netherlands | Vegetable oil and waste animal fat | 1,000,000 tn/year | NExBTL |
| Neste | Singapore | Vegetable oil and waste animal fat | 1,000,000 tn/year | NExBTL |
| Diamond Green Diesel | USA | Non-edible vegetable oils and animal fats | 900,000 tn/year | EcofiningTM |
| UOP/Eni | Italy | Vegetable oils, animal fats and used cooking oils | 780,000 tn/year | EcofiningTM |
| Neste | Finland | Vegetable oil and waste animal fat | 380,000 tn/year | NExBTL |
| Renewable Energy Group (REG) Inc. | USA | High and low free fatty acid feedstocks | 250,000 tn/year | Dynamic Fuels LLC |
| AltAir Fuels | USA | Non-edible natural oils and agricultural waste | 130,000 tn/year | EcofiningTM |
| UPM Biofuels | Finland | Crude tall oil | 100,000 tn/year | UPM BioVerno |
| Catalyst | Reactor Type | Oil | H2/Oil Ratio | T (°C) | P (bar) | LHSV (h−1) | Duration (h) | Conversion % | Selectivity among Oil Product (wt%) | Main Products 1 | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ni/Al2O3 | Continuous | Rapeseed | 50 | 260–280 | 35 | 0.25–4 | n.a. | 98 | 59 (C17) | C17 (zero C18) | 72 |
| Mo/Al2O3 | Continuous | Rapeseed | 50 | 260–280 | 35 | 0.25–4 | n.a. | 100 | 82 (C18) | C18 (only 2 wt% C17) | 72 |
| Ni–Mo/Al2O3 | Continuous | Rapeseed | 50 | 260–280 | 35 | 0.25–4 | n.a. | 100 | 94 (C17 and C18) | C15–C18 (C17/C18 = 0.36) | 72 |
| Ni–Mo/Al2O3 | Continuous | Rapeseed | 250 | 340 | 40 | 1 | n.a. | n.a. | 91.64 (C15–C18) | C15–C18 (C17/C18 = 1.32) | 73 |
| Ni–Mo/Al2O3 | Batch | Rapeseed | n.a. | 350 | 80 | n.a. | 3 | n.a. | 70 (C15–C18) | C15–C18 (C17/C18 = 1.63) | 74 |
| Ni–Mo/Al2O3 | Continuous | Rapeseed | 100 | 310 | 70 | 1.5 | n.a. | 100 | >90 (C17 and C18) | C17–C18 (C17/C18 = 0.25) | 75 |
| Ni–Mo/MCM-41 | Continuous | Rapeseed | 100 | 310 | 70 | 1.5 | n.a. | 100 | >90 (C17 and C18) | C17–C18 (C17/C18 = 0.5) | 75 |
| Ni–Mo/OMA | Continuous | Rapeseed | 100 | 310 | 70 | 1.5 | n.a. | 100 | >90 (C17 and C18) | C17–C18 (C17/C18 = 0.07) | 75 |
| Pt/HZSM-5 | Batch | Rapeseed | n.a. | 380 | 110 | n.a. | 3 | n.a. | 43 (C5–C12) | Gasoline (C5–C12) | 74 |
| FCC equilibrium catalyst | Continuous 2 | Rapeseed | n.a. | 525 | 1 | n.a. | n.a. | n.a. | 15.7 (C12–C20) | Gasoline (C5–C12) 32.4 wt% | 107 |
| FCC-ZSM-5 | Continuous 2 | Rapeseed | n.a. | 525 | 1 | n.a. | n.a. | n.a. | 13.2 (C12–C20) | Gasoline (C5–C12) 32.3 wt% | 107 |
| Ni–Mo/Al2O3 | Continuous | Palm | n.a. | 350 | 40 | 2 | n.a. | n.a. | 80 (C15–C18) | C15–C18 (C17/C18 = 0.58) | 76 |
| Ni–Mo/Al2O3 | Continuous | Palm | 250 | 350 | 45 | 0.6 | n.a. | n.a. | 92.69 (C15–C18) | C15–C18 (C17/C18 = 0.78) | 73 |
| Ni–Mo/zeolite | Batch | Palm | n.a. | 300–320 | 1 | n.a. | n.a. | n.a. | 47.24 (C15–C19) | C15–C19 and also 52.76% C8–C13 | 108 |
| MCM-41 | Palm | n.a. | 450 | 1 | 2.5 | n.a. | n.a. | 26 (Diesel) | Also gasoline 23.9% and kerosene 13 wt% | 105 | |
| Al–MCM-41 | Continuous | Palm | n.a. | 450 | 1 | 2.5 | n.a. | n.a. | 11.2–20.3 (Diesel) | Gasoline 30.1–31.7% and kerosene 10.6–20.7 wt% | 104 |
| Ni–Mo/Al2O3 | Batch | Soybean | n.a. | 400 | 92 | n.a. | 2 | 92.9 | 64.45 (C15–C18) | C15–C18 (C17/C18 = 2.49) | 102 |
| Pd/Al2O3 | Batch | Soybean | n.a. | 400 | 92 | n.a. | 2 | 91.9 | 79.22 (C15–C18) | C15–C18 (C17/C18 = 11.9) | 102 |
| Co–Mo/Al2O3 | Batch | Soybean | n.a. | 400 | 92 | n.a. | 2 | 78.9 | 33.67 (C15–C18) | C15–C18 (C17/C18 = 2.16) | 102 |
| Ni/Al2O3-SiO2 | Batch | Soybean | n.a. | 400 | 92 | n.a. | 2 | 60.8 | 39.24 (C15–C18) | C15–C18 (C17/C18 = 29.3)) | 102 |
| Pt/Al2O3 | Batch | Soybean | n.a. | 400 | 92 | n.a. | 2 | 50.8 | 37.71 (C15–C18) | C15–C18 (C17/C18 = 0.92) | 102 |
| Ru/Al2O3 | Batch | Soybean | n.a. | 400 | 92 | n.a. | 2 | 39.7 | 32.00 (C15–C18) | C15–C18 (C17/C18 = 39.6) | 102 |
| Ni/Al2O3 | Batch | Soybean | n.a. | 350 | 6.9 | n.a. | 4 | 68 | 51.20 (≥C18) | ≥C18 | 99 |
| Ni–Al/LDH | Batch | Soybean | n.a. | 350 | 6.9 | n.a. | 4 | 74 | 52.90 (C8–C17) | C8–C17 | 99 |
| Ni–Mg–Al/LDH | Batch | Soybean | n.a. | 350 | 6.9 | n.a. | 4 | 49 | 54.1 (≥C18) | ≥C18 | 99 |
| Mg–Al/LDH | Batch | Soybean | n.a. | 350 | 6.9 | n.a. | 4 | 72 | 47.80 (C8–C17) | C8–C17 | 99 |
| Co–Mo/Al2O3 | Continuous | Sunflower | 450 | 340–350 | 50–80 | 1 | n.a. | 94.0–99.8 | 63.1–71.5 | 91 cetane number Diesel fuel | 77 |
| Co–Mo/Al2O3 | Continuous | Sunflower | 500–600 | 380 | 40–60 | 1 | n.a. | 100 | 83.1–89.2 | C15–C18 (C17/C18 = 0.15) | 92 |
| Ni–Mo/Al2O3 | Continuous | Sunflower | 450 | 340–350 | 50–80 | 1 | n.a. | 81.8–97.4 | 42.0–51.9 | 92 cetane number Diesel fuel | 77 |
| Ni–Mo/ Al2O3 | Continuous | Sunflower | 500 | 350 | 45 | 0.8 | n.a. | n.a. | 86.59 (C15–C18) | C15–C18 (C17/C18 = 0.86) | 73 |
| Ni–W/Al2O3 | Continuous | Sunflower | 450 | 340–350 | 50–80 | 1 | n.a. | 86.7–95.6 | 39.4–49.3 | 91 cetane number Diesel fuel | 77 |
| Ni–Mo/Al2O3-F | Continuous | Sunflower | 500 | 350–370 | 20–40 | 1 | n.a. | 91–96 | 73.2–75.6 | C15–C18 of which 37–38% isomers | 78 |
| Pd/SAPO-31 | Continuous | Sunflower | 1000 | 310–350 | 20 | 0.9–1.6 | n.a. | n.a. | 83.4–100 | nSH 1 and iSH 1 of which 46.8–90.7% C17 and C18 | 98 |
| V2O5/α-Al2O3 | Batch | Sunflower | n.a. | 355 | 1 | n.a. | 0.66 | 92.1 | 48 (Heavy Diesel) | also about 23% gasoline and 4% kerozene | 109 |
| V2O5/α-Al2O3 | Batch | Sunflower | n.a. | 390 | 1 | n.a. | 0.5 | 85 | 68 (b.p > 200 °C) | Liquid oil products with boiling point > 200 °C | 110 |
| Co3O4/α-Al2O3 | Batch | Sunflower | n.a. | 390 | 1 | n.a. | 0.5 | 68 | 60 (b.p > 200 °C) | Liquid oil products with boiling point > 200 °C | 110 |
| KOH/α-Al2O3 | Batch | Sunflower | n.a. | 390 | 1 | n.a. | 0.5 | 65 | 53 (b.p > 200 °C) | Liquid oil products with boiling point > 200 °C | 110 |
| MoO3/α-Al2O3 | Batch | Sunflower | n.a. | 390 | 1 | n.a. | 0.5 | 56 | 59 (b.p > 200 °C) | Liquid oil products with boiling point > 200 °C | 110 |
| NiO/α-Al2O3 | Batch | Sunflower | n.a. | 390 | 1 | n.a. | 0.5 | 66 | 57 (b.p > 200 °C) | Liquid oil products with boiling point > 200 °C | 110 |
| ZnO/α-Al2O3 | Batch | Sunflower | n.a. | 390 | 1 | n.a. | 0.5 | 84 | 70 (b.p > 200 °C) | Liquid oil products with boiling point > 200 °C | 110 |
| Ni–Mo/Al2O3 | Continuous | Jatropha | 800 | 350 | 40 | 7.6 | n.a. | 100 | 97.2 (C11–C20) | C11–C20 (iSH/nSH 1 = 0.08) | 79 |
| Ni–Mo/SiO2 | Continuous | Jatropha | 800 | 350 | 40 | 7.6 | n.a. | 100 | 99.1 (C11–C20) | C11–C20 (iSH/nSH 1 = 0.03) | 79 |
| Ni–Mo/Al2O3-SiO2 | Continuous | Jatropha | 800 | 350 | 40 | 7.6 | n.a. | 100 | 89.6 (C11–C20) | C11–C20 and also 9.8 wt% C5–C10 (iSH/nSH 1 = 0.26) | 79 |
| Ni–Mo/H-Y | Continuous | Jatropha | 800 | 350 | 40 | 7.6 | n.a. | 100 | 50.9 (C11–C20) | C11–C20 and also 48.9 wt% C5–C10 (iSH/nSH 1 = 0.87) | 79 |
| Ni–Mo/H-ZSM-5 | Continuous | Jatropha | 800 | 350 | 40 | 7.6 | n.a. | 100 | 22.1 (C11–C20) | Also 77.8 wt% C5–C10 (iSH/nSH 1 = 1.21) | 79 |
| Ni–HPW(30%)/nHA | Continuous | Jatropha | 600 | 360 | 30 | 2 | n.a. | 100 | 83.4 | C10–C22 [(C15 + C17)/(C16 + C18)] = 4.4, (iSH/nSH 1 = 1.64) | 100 |
| Pt/H-ZSM-5 | Batch | Jatropha | n.a. | 270 | 65 | n.a. | 12 | 100 | 78.9 (C15–C18) | C15–C18 (C17/C18 = 0.40) | 93 |
| Pt/USY | Batch | Jatropha | n.a. | 270 | 65 | n.a. | 12 | 31.2 | 68.15 (C15–C18) | C15–C18 (C17/C18 = 0.51) | 93 |
| ZSM-5-Zn-20 | Continuous | Camelina | n.a. | 500 | 1 | 0.6 | n.a. | 76.8 | 52.39 (C13–C15) | C13–C15 and also C7–C8 25.09 wt% | 111 |
| Mo-Zn/Al2O3 | Batch | Carinata | n.a. | 350 | 21–58 | n.a. | n.a. | >92 | 83 (C12–C18) | C12–C18 | 112 |
| Ni/Al2O3 | Continuous | Hemp seed 3 | 300 | 350 | 40 | 4.7 | n.a. | 100 | 90% (C10–C17) | C10–C17 (C17 equal to 49–62 wt%) | 101 |
| Ni–Cu/Al2O3 | Continuous | Hemp seed 3 | 300 | 350 | 40 | 4.7 | n.a. | 100 | 100 (C10–C20) | C10–C17 (C17 equal to 80 wt%) | 101 |
| Ni–Mo/Al2O3 | Batch | WCO 4 | 666 | 300 | 70 | 2.8 | 3 | 99.6 | 93 (C15–C18) | C15–C18 (iSH/nSH 1 = 1.4/97.5) | 80 |
| Co–Mo/Al2O3 | Batch | WCO 4 | 666 | 300 | 70 | 2.8 | 3 | 99.3 | 84.7 (C15–C18) | C15–C18 (iSH/nSH 1 = 6.4/89.7) | 80 |
| Ni–W/Al2O3 | Batch | WCO 4 | 666 | 300 | 70 | 2.8 | 3 | 100 | 92.8 (C15–C18) | C15–C18 (iSH/nSH 1 = 2.2/97.3) | 80 |
| Ni–Mo/Al2O3 | Batch | WCO 4 | 666 | 350 | 70 | 2.8 | 3 | 99.8 | 90.7 (C15–C18) | C15–C18 (iSH/nSH 1 = 3.8/94.8) | 80 |
| Ni–Mo/B2O3-Al2O3 | Batch | WCO 4 | 666 | 350 | 70 | 2.8 | 3 | 99.9 | 87.1 (C15–C18) | C15–C18 (iSH/nSH 1 = 7.5/91.4) | 80 |
| Ru/SiO2 | Continuous | WCO 4 | 400 | 350 | 20 | 15.2 | n.a. | n.a. | 98.9 (C11–C20) | C11–C20 (iSH/nSH 1 = 0.08) | 106 |
| Ru/Al13-montmorillonite | Continuous | WCO 4 | 400 | 350 | 20 | 15.2 | n.a. | 100 | 89.8 (C11–C20) | C11–C20 and 9.1 wt% C5–C10 (iSH/nSH 1 = 0.2) | 106 |
| Ru/H-Y | Continuous | WCO 4 | 400 | 350 | 20 | 15.2 | n.a. | n.a. | 56.5 (C11–C20) | C11–C20 and 42.8 wt% C5–C10 (iSH/nSH 1 = 0.43) | 106 |
| HZSM-5 | Batch | WCO 4 | n.a. | 400–420 | 10 | n.a. | 1 | n.a. | 30.55-36.3 (Gas oil) | also 15.57–16.76% gasoline and 9.99–11.83% kerozene | 113 |
| Sulfated Zr2O3 | Batch | WCO 4 | n.a. | 400–420 | 10 | n.a. | 1 | n.a. | 41.18-44.61 (Gas oil) | also 10.04–14.68% gasoline and 6.51–10.32% kerozene | 113 |
| Ni–Mo/B2O3-Al2O3 | Batch | LTG 5 | 666 | 350 | 70 | 2.8 | 3 | 99.8 | 92.1 (C15–C18) | C15–C18 (iSH/nSH 1 = 2.6/96) | 80 |
| Ni–Mo/B2O3-Al2O3 | Batch | STG 6 | 666 | 350 | 70 | 2.8 | 3 | 99.3 | 86.6 (C15–C18) | C15–C18 (iSH/nSH 1 = 3.4/94.8) | 80 |
| Ni/Al2O3 | Continuous | 75 wt% YG 7 | 2400 | 375 | 40 | 2 | 3 | inadequate | - | Gas products and unconverted liquids | 101 |
| Ni–Cu/Al2O3 | Continuous | 75 wt%YG 7 | 2400 | 375 | 40 | 2 | 3 | 100 | 92% (Diesel) | Diesel (34wt% C17) | 101 |
| Ni–Cu/Al2O3 | Continuous | 75 wt% YG 7 | 2400 | 375 | 40 | 2 | 8 | 100 | 94% (Diesel) | Diesel (74 wt% C17) | 101 |
| Ni–Mo/Al2O3 | Continuous | Lard | 250 | 350 | 45 | 0.8 | n.a. | n.a. | 86.11 (C15–C18) | C15–C18 (C17/C18 = 0.47) | 73 |
| Properties | Wood | Willow | Straw | Sweet Grass | Petroleum Distillate Fuel |
|---|---|---|---|---|---|
| Water Content, wt% | 15–30 | 17.4 | 47.4 | 24.7 | 0.1 |
| Carbon, wt% | 54–58 | 43.17 | 28.2 | 38.3 | 85 |
| Hydrogen, wt% | 5.5–7 | 7.15 | 8.78 | 7.42 | 11 |
| Oxygen, wt% | 35–40 | 49.49 | 62.83 | 54.08 | 1 |
| Nitrogen, wt% | 0–0.2 | 0.1 | 0.1 | 0.1 | 0.3 |
| Ash, wt% | 0–0.2 | 0.1 | |||
| pH | 2–3 | 2.68 | 3.45 | 2.87 | - |
| Viscosity, mm2/s | 40–100 | 53.2 | 17.2 | 34.2 | 2.39 |
| Density, kg/m3 | 1.2 | 0.94 | |||
| Higher Heating Value (HHV), MJ/kg | 16–19 | 18.4 | 13.6 | 16.4 | 40 |
| Solid Particulates, wt% | 0.2–1 | 1 | |||
| Distillation Residue, wt% | up to 50 | 1 |
| Catalyst | Reactor Type | Time, h | Temperature, °C | Pressure, bar | DOD 1, % | Yield of Oil, wt% | Ref. |
|---|---|---|---|---|---|---|---|
| Co–Mo/Al2O3 | Batch | 4 | 350 | 200 | 81 | 26 | 175 |
| Co–Mo/Al2O3 | Continuous | 4 | 370 | 300 | 100 | 33 | 176 |
| Ni–Mo/Al2O3 | Batch | 4 | 350 | 200 | 74 | 28 | 175 |
| Ni–Mo/Al2O3 | Continuous | 0.5 | 400 | 85 | 28 | 84 | 177 |
| Pd/C | Batch | 4 | 350 | 200 | 85 | 65 | 175 |
| Pd/C | Continuous | 4 | 340 | 140 | 64 | 48 | 178 |
| Pt/Al2O3-SiO2 | Continuous | 0.5 | 400 | 85 | 45 | 81 | 177 |
| Ru/Al2O3 | Batch | 4 | 350 | 200 | 78 | 36 | 175 |
| Ru/C | Continuous | 0.2 | 350-400 | 230 | 73 | 38 | 179 |
| Ru/C | Batch | 4 | 350 | 200 | 86 | 53 | 175 |
| Ru/TiO2 | Batch | 4 | 350 | 200 | 77 | 67 | 175 |
| Catalyst | Time, h | Temperature, °C | Pressure, bar | DOD 1, % | Yield of Oil, wt% | Ref. |
|---|---|---|---|---|---|---|
| GaHZSM-5 | 0.32 | 380 | 1 | n/a | 18 | 180 |
| H-Modernite | 0.56 | 330 | 1 | n/a | 17 | 181 |
| H-Y | 0.28 | 330 | 1 | n/a | 28 | 181 |
| HZSM-5 | 0.32 | 380 | 1 | 50 | 24 | 180 |
| HZSM-5 | 0.91 | 500 | 1 | 50 | 12 | 182 |
| MgAPO-36 | 0.28 | 370 | 1 | n/a | 16 | 183 |
| SAPO-11 | 0.28 | 370 | 1 | n/a | 20 | 183 |
| SAPO-5 | 0.28 | 370 | 1 | n/a | 22 | 183 |
| ZnHZSM-5 | 0.32 | 380 | 1 | n/a | 19 | 180 |
| Property | Petroleum Diesel European Standard | Petroleum Diesel USA Standard | Green Diesel European Standard | Biodiesel European Standard | Typical Fuel Properties | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EN590:2013+A1:2017 | ASTM D975-17a | EN15940:2016/A1:2018 | EN14214/A2:2018-A2 | |||||||||
| Min | Max | Min | Max | Min | Max | Min | Max | No.2 Petroleum Diesel | Biodiesel (FAME) | Green Diesel | FT Diesel | |
| Carbon (wt%) | - | - | - | - | - | - | - | - | 86.8 | 76.2 | 84.9 | |
| Hydrogen (wt%) | - | - | - | - | - | - | - | - | 13.2 | 12.6 | 15.1 | |
| Oxygen (wt%) | - | - | - | - | - | - | - | - | 0.0 | 11.2 | 0.0 | |
| Cetane Number (CN) | 51 | - | 40 | - | 70/51 | - | 51 | - | 44.5–67 | 45-55 | >70 | 74–80 |
| Cetane Index | 46 | - | 40 | - | - | - | - | - | ||||
| Lower Heating Value (LHV) MJ/kg | - | - | - | - | - | - | - | - | ≈43.1 | ≈37.2 | 43.7–44.5 | ≈43.5 |
| Density at 15 °C (kg/m3) | 820 | 845 | - | - | 765 780 | 800 810 | 860 | 890 | 796–841 | ≈880 | 770–790 | 774–782 |
| Polycyclic Aromatic Hydrocarbons (wt%) | - | 8 | - | - | - | - | - | - | 1.5–4.4 | <0.1 | <0.2 | |
| Aromaticity | - | - | - | 35 vol% | - | 1.1 wt% | - | - | 35 vol% max | 0 max | ||
| Sulfur Content (mg/kg) | - | 10 | - | 15 | - | 5 | - | 10 | 3.8–15 | <5 | <5 | |
| Manganese Content (mg/lt) | - | 2 | - | - | - | - | - | - | ||||
| Flash Point (°C) | 55 | - | 52/38 | - | 55 | - | 101 | - | 54–148 | 100–180 | >59 | ≈71 |
| Cloud Point (°C) | Down to −34 | - | - | Down to −34 | - | - | 10.5...−13 | −5...−34 | ||||
| Ash Content (wt%) | - | 0.01 | - | 0.01 | - | 0.01 | - | - | 0.01 | <0.001 | ||
| Water Content (mg/kg) | - | 200 | - | - | - | 200 | - | 500 | <200 | |||
| Carbon Residue on 10% Distillation (wt%) | - | 0.3 | - | 0.35 | - | 0.3 | - | 0.3 | <0.1 | |||
| Total Contamination (mg/kg) | - | 24 | - | - | - | 24 | 24 | <10 | ||||
| Water and Sediment (vol%) | - | - | - | 0.05 | - | 0.02 | - | - | ≤0.02 | |||
| Copper Strip Corrosion Rating | Class 1 | - | No. 3 | Class 1 | Class 1 | No.3 | Class 1 | Class 1 | ||||
| Fatty Acid Methyl Esters (FAME) (vol%) | - | 7 | - | 5 | - | 7 | 96.5 wt% | - | 0 | |||
| Oxidation Stability | 20 h | 25g/m3 | - | - | 20 h | 25 g/m3 | 6 h | - | 2–15 h | <25 g/m3 | ≈0.8 | |
| Lubricity, Wear Scar Diameter at 60 °C (μm) | - | 460 | - | 520 | - | 460 | - | - | 226–354 | >700 (additive required) | 420–570 (additive required) | |
| Viscosity at 40 °C (mm2/s) | 2 | 4.5 | 1.9/1.3 | 4.1/2.4 | 2 | 4.5 | 3.5 | 5.0 | 1.9–4.1 | 2.9–11 | 2–4 | 2–4.5 |
| Distillation | ||||||||||||
| vol% Recovered at 250 °C | - | <65 | - | - | - | <65 | - | - | - | |||
| vol% Recovered at 350 °C | 85 | - | - | - | 85 | - | - | - | - | |||
| Distillation Temperature (°C) | ||||||||||||
| 90 vol% Recovered | - | - | 282 | 282–338 | - | - | - | - | 279–360 | 282–338 | ≈305.8 | |
| 95 vol% Recovered | - | 360 | - | - | - | 360 | - | - | - | <320 | ≈312.2 | |
| Conductivity (pS/m) | - | - | 25 | - | 25 | - | - | ≥50 | ||||
| Acid Number (mg KOH/g) | - | - | - | 0.1 | - | 0.1 | - | 0.5 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Douvartzides, S.L.; Charisiou, N.D.; Papageridis, K.N.; Goula, M.A. Green Diesel: Biomass Feedstocks, Production Technologies, Catalytic Research, Fuel Properties and Performance in Compression Ignition Internal Combustion Engines. Energies 2019, 12, 809. https://doi.org/10.3390/en12050809
Douvartzides SL, Charisiou ND, Papageridis KN, Goula MA. Green Diesel: Biomass Feedstocks, Production Technologies, Catalytic Research, Fuel Properties and Performance in Compression Ignition Internal Combustion Engines. Energies. 2019; 12(5):809. https://doi.org/10.3390/en12050809
Chicago/Turabian StyleDouvartzides, Savvas L., Nikolaos D. Charisiou, Kyriakos N. Papageridis, and Maria A. Goula. 2019. "Green Diesel: Biomass Feedstocks, Production Technologies, Catalytic Research, Fuel Properties and Performance in Compression Ignition Internal Combustion Engines" Energies 12, no. 5: 809. https://doi.org/10.3390/en12050809
APA StyleDouvartzides, S. L., Charisiou, N. D., Papageridis, K. N., & Goula, M. A. (2019). Green Diesel: Biomass Feedstocks, Production Technologies, Catalytic Research, Fuel Properties and Performance in Compression Ignition Internal Combustion Engines. Energies, 12(5), 809. https://doi.org/10.3390/en12050809