Boosting Hydrogen Production from Formic Acid over Pd Catalysts by Deposition of N-Containing Precursors on the Carbon Support



Abstract

1. Introduction
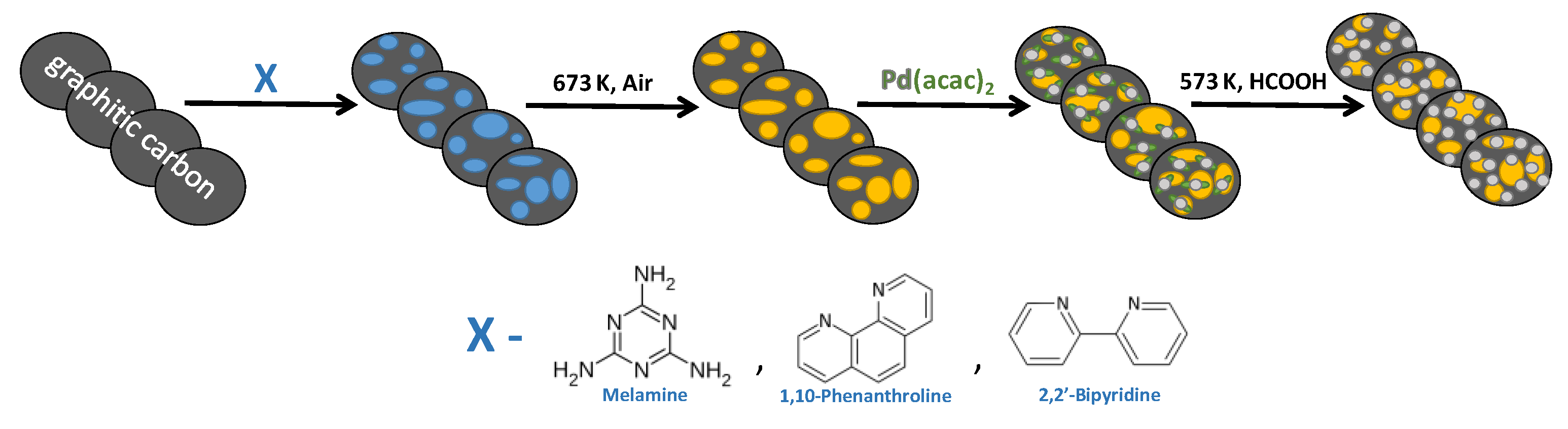
2. Materials and Methods
2.1. Synthesis of Supports
2.1.1. g-C3N4
2.1.2. Mel/C
2.1.3. Phen/C and Bpy/C
2.2. Synthesis of Pd Catalysts
2.3. Characterization
2.4. Catalytic Measurements
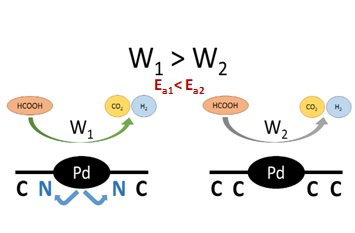
3. Results and Discussion
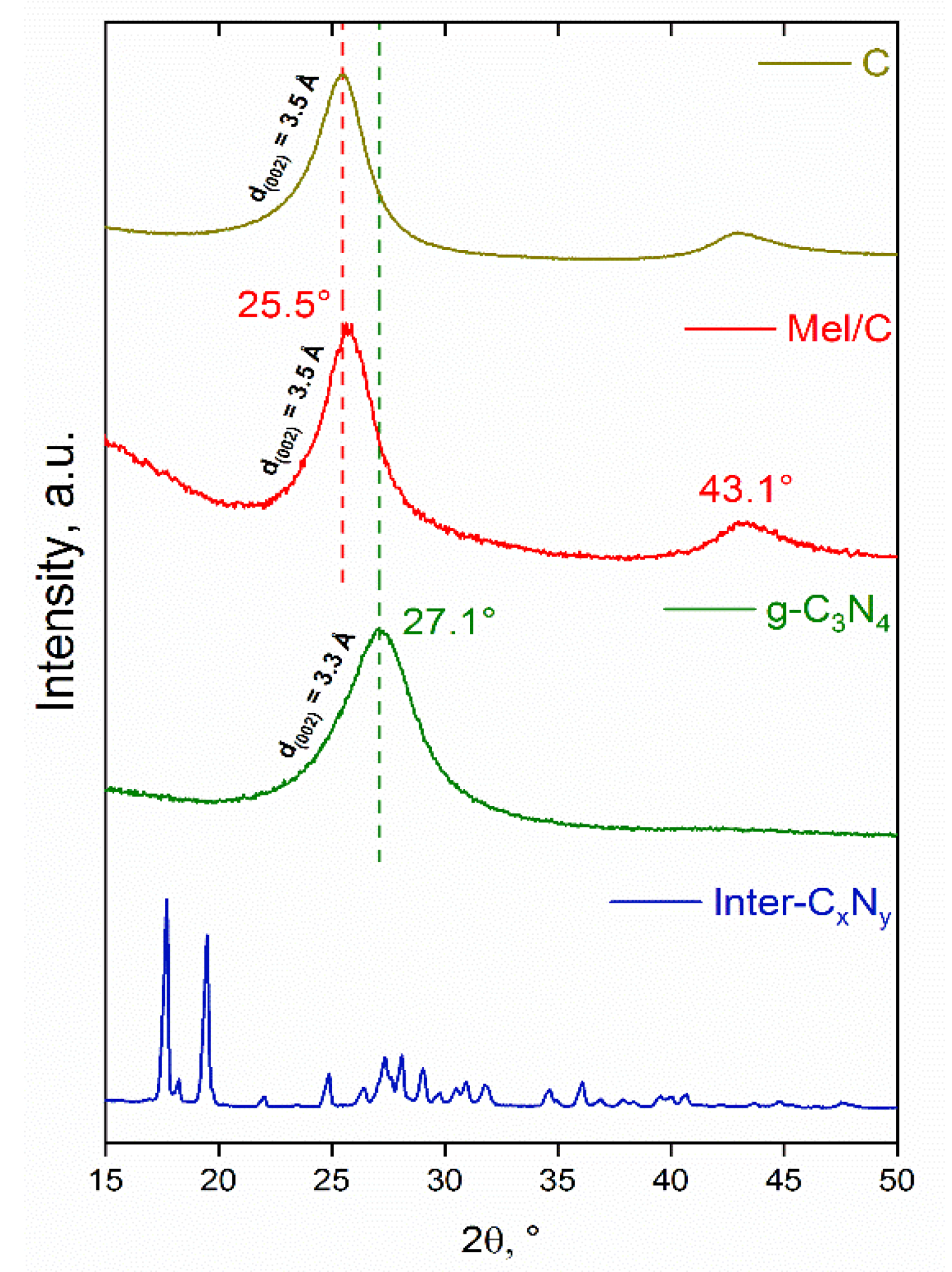
3.1. Characterization of the Supports
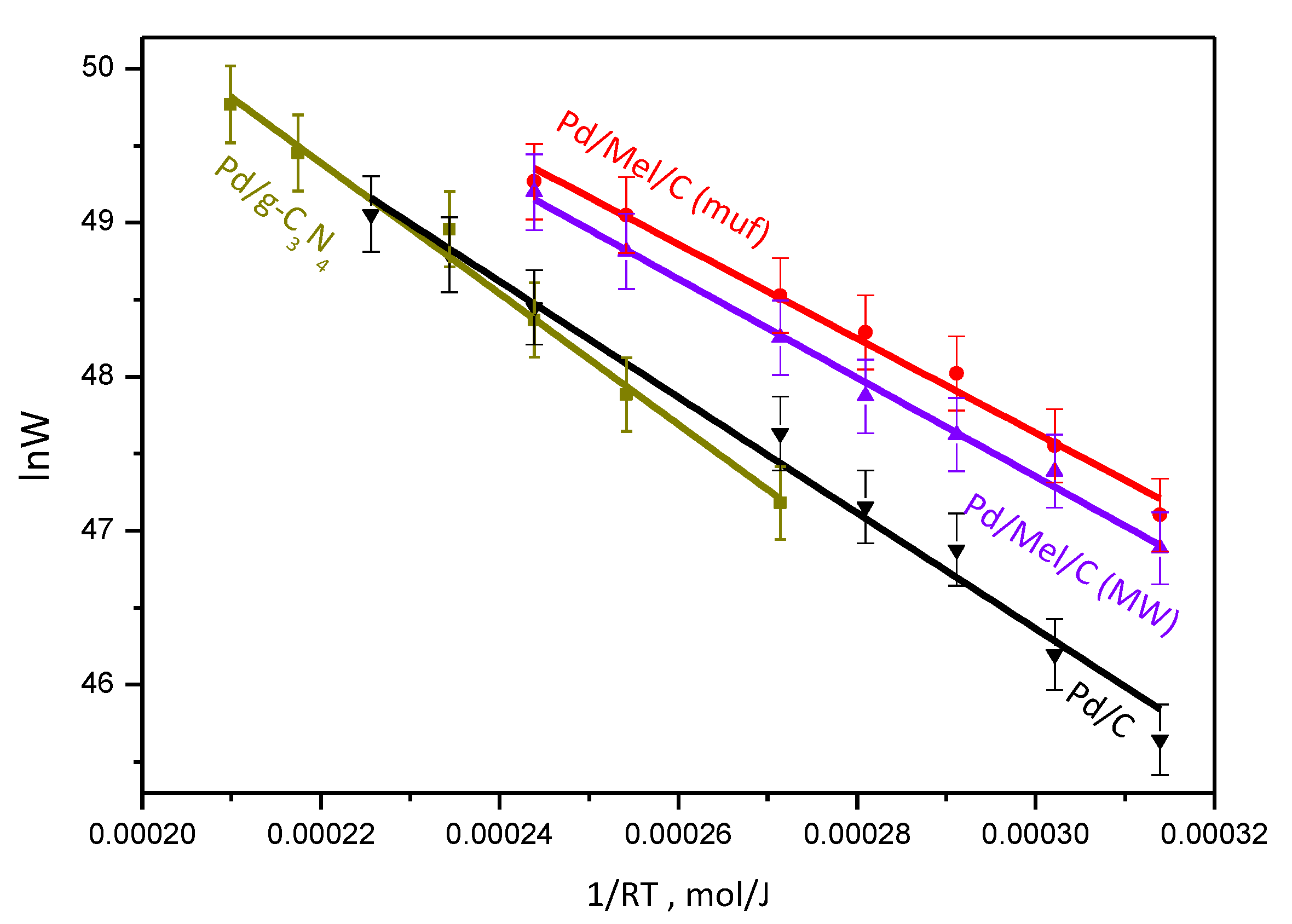
3.2. Catalytic Activity and H2 Selectivity
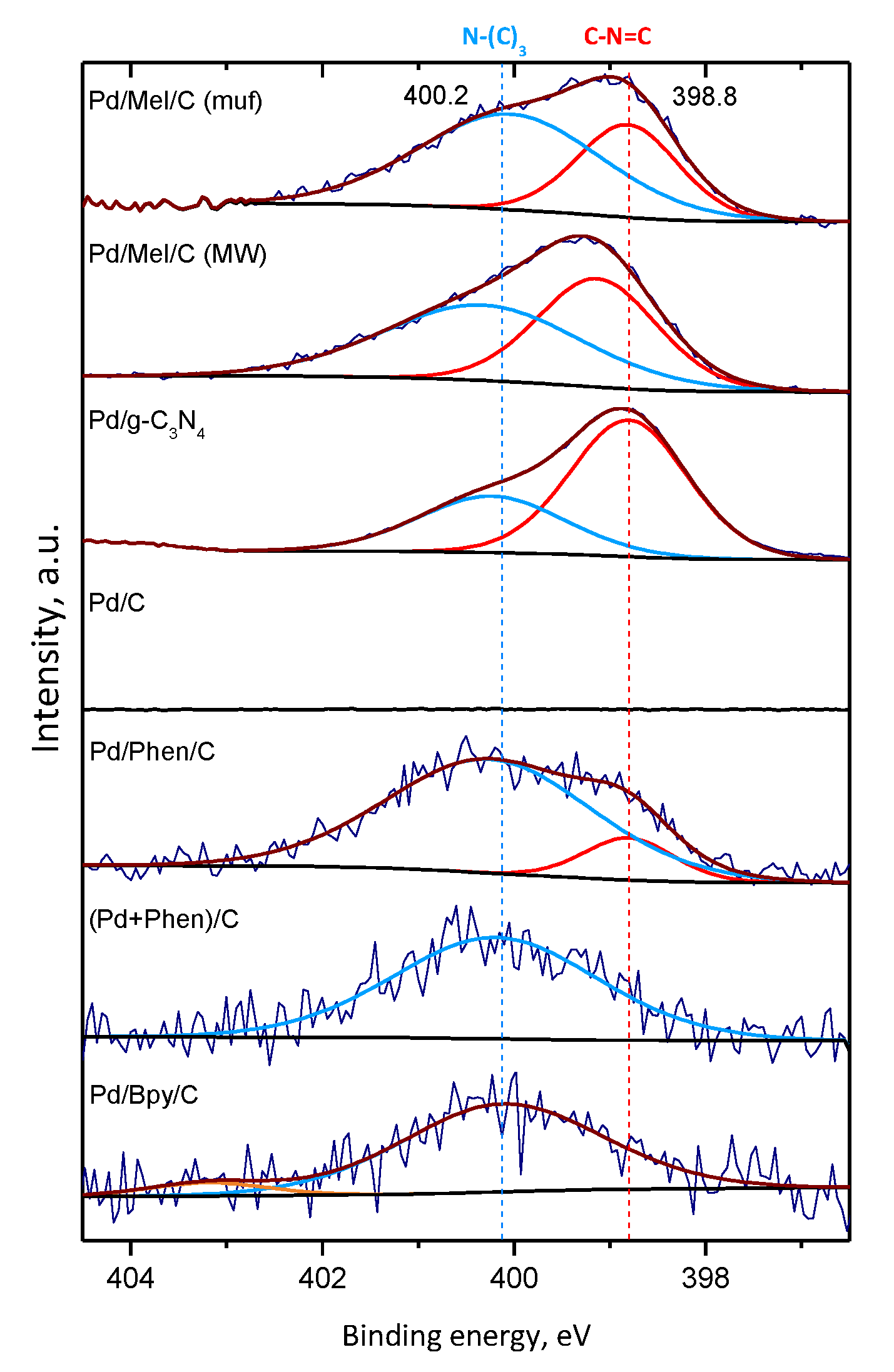
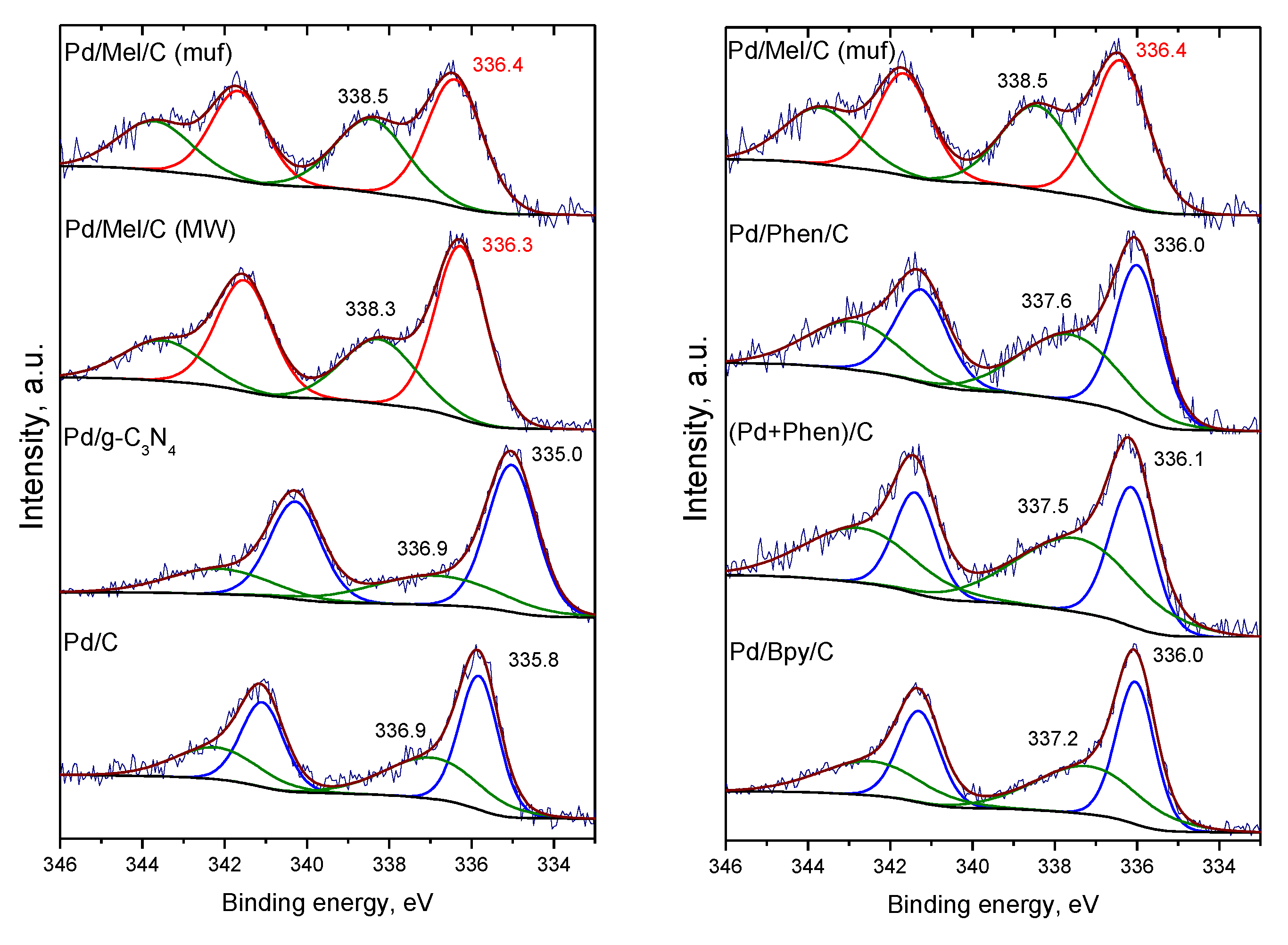
3.3. Characterization of the Pd Catalysts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- US Energy Information Administration. International Energy Outlook 2016. 2016; No. 0484. Available online: www.eia.gov/outlooks/ieo/pdf/0484(2016).pdf (accessed on 14 October 2019).
- Zhong, H.; Iguchi, M.; Chatterjee, M.; Himeda, Y.; Xu, Q.; Kawanami, H. Formic Acid-Based Liquid Organic Hydrogen Carrier System with Heterogeneous Catalysts. Adv. Sust. Syst. 2018, 2, 1700161. [Google Scholar] [CrossRef]
- Boddien, A.; Federsel, C.; Sponholz, P.; Mellmann, D.; Jackstell, R.; Junge, H.; Laurenczy, G.; Beller, M. Towards the Development of a Hydrogen Battery. Energy Environ. Sci. 2012, 5, 8907–8911. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Ross, J.R.H. Towards Sustainable Production of Formic Acid. ChemSusChem 2018, 11, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.; Brunner, B.; Jess, A.; Wasserscheid, P.; Albert, J. Biomass Oxidation to Formic Acid in Aqueous Media Using Polyoxometalate Catalysts—Boosting Fa Selectivity by in-Situ Extraction. Energy Environ. Sci. 2015, 8, 2985–2990. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Ross, J.R.H. Heterogeneous Catalysts for Hydrogenation of CO2 and Bicarbonates to Formic Acid and Formates. Catal. Rev. 2018, 60, 566–593. [Google Scholar] [CrossRef]
- Germain, J.; Hradil, J.; Fréchet, J.M.J.; Svec, F. High Surface Area Nanoporous Polymers for Reversible Hydrogen Storage. Chem. Mater. 2006, 18, 4430–4435. [Google Scholar] [CrossRef]
- Hull, J.F.; Himeda, Y.; Wang, W.-H.; Hashiguchi, B.; Periana, R.; Szalda, D.J.; Muckerman, J.T.; Fujita, E. Reversible Hydrogen Storage Using CO2 and a Proton-Switchable Iridium Catalyst in Aqueous Media under Mild Temperatures and Pressures. Nat. Chem. 2012, 4, 383–388. [Google Scholar] [CrossRef]
- Koroteev, V.O.; Bulushev, D.A.; Chuvilin, A.L.; Okotrub, A.V.; Bulusheva, L.G. Nanometer-Sized MoS2 Clusters on Graphene Flakes for Catalytic Formic Acid Decomposition. ACS Catal. 2014, 4, 3950–3956. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Chuvilin, A.L.; Sobolev, V.I.; Stolyarova, S.G.; Shubin, Y.V.; Asanov, I.P.; Ishchenko, A.V.; Magnani, G.; Riccò, M.; Okotrub, A.V.; et al. Copper on Carbon Materials: Stabilization by Nitrogen Doping. J. Mat. Chem. A 2017, 5, 10574–10583. [Google Scholar] [CrossRef]
- Wang, Q.; Tsumori, N.; Kitta, M.; Xu, Q. Fast Dehydrogenation of Formic Acid over Palladium Nanoparticles Immobilized in Nitrogen-Doped Hierarchically Porous Carbon. ACS Catal. 2018, 8, 12041–12045. [Google Scholar] [CrossRef]
- Navlani-García, M.; Mori, K.; Salinas-Torres, D.; Kuwahara, Y.; Yamashita, H. New Approaches toward the Hydrogen Production from Formic Acid Dehydrogenation over Pd-Based Heterogeneous Catalysts. Front. Mat. 2019, 6. [Google Scholar] [CrossRef]
- Podyacheva, O.; Bulushev, D.; Suboch, A.; Svintsitskiy, D.; Lisitsyn, A.; Modin, E.; Chuvilin, A.; Gerasimov, E.; Sobolev, V.; Parmon, V. Highly Stable Single-Atom Catalyst with Ionic Pd Active Sites Supported on N-Doped Carbon Nanotubes for Formic Acid Decomposition. ChemSusChem 2018, 11, 3724–3727. [Google Scholar] [CrossRef] [PubMed]
- Zacharska, M.; Bulusheva, L.G.; Lisitsyn, A.S.; Beloshapkin, S.; Guo, Y.; Chuvilin, A.L.; Shlyakhova, E.V.; Podyacheva, O.Y.; Leahy, J.J.; Okotrub, A.V.; et al. Factors Influencing the Performance of Pd/C Catalysts in the Green Production of Hydrogen from Formic Acid. ChemSusChem 2017, 10, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Bulushev, D.A.; Zacharska, M.; Shlyakhova, E.V.; Chuvilin, A.L.; Guo, Y.; Beloshapkin, S.; Okotrub, A.V.; Bulusheva, L.G. Single Isolated Pd2+ Cations Supported on N-Doped Carbon as Active Sites for Hydrogen Production from Formic Acid Decomposition. ACS Catal. 2016, 6, 681–691. [Google Scholar] [CrossRef]
- Arrigo, R.; Hävecker, M.; Wrabetz, S.; Blume, R.; Lerch, M.; McGregor, J.; Parrott, E.P.J.; Zeitler, J.A.; Gladden, L.F.; Knop-Gericke, A.; et al. Tuning the Acid/Base Properties of Nanocarbons by Functionalization Via Amination. J. Am. Chem. Soc. 2010, 132, 9616–9630. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Sobolev, V.I.; Pirutko, L.V.; Starostina, A.V.; Asanov, I.P.; Modin, E.; Chuvilin, A.L.; Gupta, N.; Okotrub, A.V.; Bulusheva, L.G. Hydrogen Production from Formic Acid over Au Catalysts Supported on Carbon: Comparison with Au Catalysts Supported on SiO2 and Al2O3. Catalysts 2019, 9, 376. [Google Scholar] [CrossRef]
- Bi, Q.-Y.; Lin, J.-D.; Liu, Y.-M.; He, H.-Y.; Huang, F.-Q.; Cao, Y. Dehydrogenation of Formic Acid at Room Temperature: Boosting Palladium Nanoparticle Efficiency by Coupling with Pyridinic-Nitrogen-Doped Carbon. Angew. Chem. Int. Ed. 2016, 55, 11849–11853. [Google Scholar] [CrossRef]
- Li, Z.; Li, J.; Liu, J.; Zhao, Z.; Xia, C.; Li, F. Palladium Nanoparticles Supported on Nitrogen-Functionalized Active Carbon: A Stable and Highly Efficient Catalyst for the Selective Hydrogenation of Nitroarenes. ChemCatChem 2014, 6, 1333–1339. [Google Scholar] [CrossRef]
- Gribov, E.N.; Kuznetsov, A.N.; Golovin, V.A.; Krasnikov, D.V.; Kuznetsov, V.L. Effect of Modification of Multi-Walled Carbon Nanotubes with Nitrogen-Containing Polymers on the Electrochemical Performance of Pt/CNT Catalysts in PEMFC. Mater. Renew. Sustain. Energy 2019, 8, 7. [Google Scholar] [CrossRef]
- Salinas-Torres, D.; Navlani-García, M.; Mori, K.; Kuwahara, Y.; Yamashita, H. Nitrogen-Doped Carbon Materials as a Promising Platform toward the Efficient Catalysis for Hydrogen Generation. Appl. Catal. A 2018, 571, 25–41. [Google Scholar] [CrossRef]
- Cai, J.; Bennici, S.; Shen, J.; Auroux, A. Influence of N Addition in Mesoporous Carbons Used as Supports of Pt, Pd and Ru for Toluene Hydrogenation and Iron Oxide for Benzene Oxidation. React. Kinet. Mech. Catal. 2015, 115, 263–282. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Zacharska, M.; Lisitsyn, A.S.; Podyacheva, O.Y.; Hage, F.S.; Ramasse, Q.M.; Bangert, U.; Bulusheva, L.G. Single Atoms of Pt-Group Metals Stabilized by N-Doped Carbon Nanofibers for Efficient Hydrogen Production from Formic Acid. ACS Catal. 2016, 6, 3442–3451. [Google Scholar] [CrossRef]
- Lee, J.H.; Ryu, J.; Kim, J.Y.; Nam, S.W.; Han, J.H.; Lim, T.H.; Gautam, S.; Chae, K.H.; Yoon, C.W. Carbon Dioxide Mediated, Reversible Chemical Hydrogen Storage Using a Pd Nanocatalyst Supported on Mesoporous Graphitic Carbon Nitride. J. Mater. Chem. A 2014, 2, 9490–9495. [Google Scholar] [CrossRef]
- Oh, T.H. A Formic Acid Hydrogen Generator Using Pd/C3N4 Catalyst for Mobile Proton Exchange Membrane Fuel Cell Systems. Energy 2016, 112, 679–685. [Google Scholar] [CrossRef]
- Tahir, M.; Cao, C.; Butt, F.K.; Butt, S.; Idrees, F.; Ali, Z.; Aslam, I.; Tanveer, M.; Mahmood, A.; Mahmood, N. Large Scale Production of Novel G-C3N4 Micro Strings with High Surface Area and Versatile Photodegradation Ability. CrystEngComm 2014, 16, 1825–1830. [Google Scholar] [CrossRef]
- Material Safety Data Sheet. Melamine. Available online: https://fscimage.fishersci.com/msds/96668.htm (accessed on 14 October 2019).
- Fenelonov, V.B.; Likholobov, V.A.; Derevyankin, A.Y.; Mel’gunov, M.S. Porous Carbon Materials Prepared from C1–C3 Hydrocarbons. Catal. Today 1998, 42, 341–345. [Google Scholar] [CrossRef]
- Chernousov, Y.D.; Shebolaev, I.V.; Ivannikov, V.I.; Ikryanov, I.M.; Bolotov, V.A.; Tanashev, Y.Y. An Apparatus for Performing Chemical Reactions under Microwave Heating of Reagents. Instrum. Exp. Tech. 2019, 62, 289–294. [Google Scholar] [CrossRef]
- Vella-Zarb, L.; Braga, D.; Orpen, A.G.; Baisch, U. The Influence of Hydrogen Bonding on the Planar Arrangement of Melamine in Crystal Structures of Its Solvates, Cocrystals and Salts. CrystEngComm 2014, 16, 8147–8159. [Google Scholar] [CrossRef]
- Papailias, I.; Giannakopoulou, T.; Todorova, N.; Demotikali, D.; Vaimakis, T.; Trapalis, C. Effect of Processing Temperature on Structure and Photocatalytic Properties of G-C3N4. Appl. Surf. Sci. 2015, 358, 278–286. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Xia, T.; Ju, H.; Zhang, K.; Long, R.; Xu, Q.; Wang, C.; Song, L.; Zhu, J.; et al. Implementing Metal-to-Ligand Charge Transfer in Organic Semiconductor for Improved Visible-near-Infrared Photocatalysis. Adv. Mater. 2016, 28, 6959–6965. [Google Scholar] [CrossRef]
- Thomas, A.; Fischer, A.; Goettmann, F.; Antonietti, M.; Müller, J.-O.; Schlögl, R.; Carlsson, J.M. Graphitic Carbon Nitride Materials: Variation of Structure and Morphology and Their Use as Metal-Free Catalysts. J. Mater. Chem. 2008, 18, 4893–4908. [Google Scholar] [CrossRef]
- Vorobyeva, E.; Chen, Z.; Mitchell, S.; Leary, R.K.; Midgley, P.; Thomas, J.M.; Hauert, R.; Fako, E.; Lopez, N.; Perez-Ramirez, J. Tailoring the Framework Composition of Carbon Nitride to Improve the Catalytic Efficiency of the Stabilised Palladium Atoms. J. Mater. Chem. A 2017, 5, 16393–16403. [Google Scholar] [CrossRef]
- Chesnokov, V.V.; Kriventsov, V.V.; Malykhin, S.E.; Svintsitskiy, D.A.; Podyacheva, O.Y.; Lisitsyn, A.S.; Richards, R.M. Nature of Active Palladium Sites on Nitrogen Doped Carbon Nanofibers in Selective Hydrogenation of Acetylene. Diam. Relat. Mater. 2018, 89, 67–73. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Xie, Z.; Yi, Y.; Wowsnick, G.; Sun, L.L.; Hermann, K.E.; Friedrich, M.; Kast, P.; Hävecker, M.; et al. Nature of the N–Pd Interaction in Nitrogen-Doped Carbon Nanotube Catalysts. ACS Catal. 2015, 5, 2740–2753. [Google Scholar] [CrossRef]
- Stonkus, O.A.; Kibis, L.S.; Yu. Podyacheva, O.; Slavinskaya, E.M.; Zaikovskii, V.I.; Hassan, A.H.; Hampel, S.; Leonhardt, A.; Ismagilov, Z.R.; Noskov, A.S.; et al. Palladium Nanoparticles Supported on Nitrogen-Doped Carbon Nanofibers: Synthesis, Microstructure, Catalytic Properties, and Self-Sustained Oscillation Phenomena in Carbon Monoxide Oxidation. ChemCatChem 2014, 6, 2115–2128. [Google Scholar] [CrossRef]
- Brun, M.; Berthet, A.; Bertolini, J.C. XPS, AES and Auger Parameter of Pd and PdO. J. Electron Spectrosc. Relat. Phenom. 1999, 104, 55–60. [Google Scholar] [CrossRef]
- Kibis, L.S.; Titkov, A.I.; Stadnichenko, A.I.; Koscheev, S.V.; Boronin, A.I. X-ray Photoelectron Spectroscopy Study of Pd Oxidation by RF Discharge in Oxygen. Appl. Surf. Sci. 2009, 255, 9248–9254. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Support | Support Surface Area, m2/g | Mean Particle Size, nm (TEM) | Ea, kJ/mol | H2 Selectivity (50 % conv.) % |
|---|---|---|---|---|---|
| Pd/g-C3N4 | g-C3N4 | 8.1 | 2.6 ± 0.3 | 39 ± 1 | >98 |
| Pd/Mel/C (muf) | Mel/C (muf) | 86.5 | 2.0 ± 0.3 | 32 ± 1 | >98 |
| Pd/Mel/C (MW) | Mel/C (MW) | 42.0 | 2.3 ± 0.5 | 32 ± 1 | 95.7 |
| Pd/Phen/C | Phen/C | 301 | – | 43 ± 1 | 90.1 |
| (Pd + Phen)/C | C | 348 | – | 42 ± 1 | 92.1 |
| Pd/Bpy/C | Bpy/C | 228 | – | 45 ± 1 | 94.8 |
| Pd/C | C | 348 | 2.3 ± 0.3 | 46 ± 2 | 93.4 |
| Sample | Pd/g-C3N4 | Pd/Mel/C (muf) | Pd/Mel/C (MW) | Pd/Phen/C | (Pd+Phen)/C | Pd/Bpy/C | Pd/C |
|---|---|---|---|---|---|---|---|
| Ntot, at % | 46.0 | 4.5 | 8.9 | 1.35 | 0.57 | 0.48 | 0 |
| 0.66 | 0.35 | 0.46 | 0.14 | 0 | 0 | – |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golub, F.S.; Beloshapkin, S.; Gusel’nikov, A.V.; Bolotov, V.A.; Parmon, V.N.; Bulushev, D.A. Boosting Hydrogen Production from Formic Acid over Pd Catalysts by Deposition of N-Containing Precursors on the Carbon Support. Energies 2019, 12, 3885. https://doi.org/10.3390/en12203885
Golub FS, Beloshapkin S, Gusel’nikov AV, Bolotov VA, Parmon VN, Bulushev DA. Boosting Hydrogen Production from Formic Acid over Pd Catalysts by Deposition of N-Containing Precursors on the Carbon Support. Energies. 2019; 12(20):3885. https://doi.org/10.3390/en12203885
Chicago/Turabian StyleGolub, Fedor S., Sergey Beloshapkin, Artem V. Gusel’nikov, Vasily A. Bolotov, Valentin N. Parmon, and Dmitri A. Bulushev. 2019. "Boosting Hydrogen Production from Formic Acid over Pd Catalysts by Deposition of N-Containing Precursors on the Carbon Support" Energies 12, no. 20: 3885. https://doi.org/10.3390/en12203885
APA StyleGolub, F. S., Beloshapkin, S., Gusel’nikov, A. V., Bolotov, V. A., Parmon, V. N., & Bulushev, D. A. (2019). Boosting Hydrogen Production from Formic Acid over Pd Catalysts by Deposition of N-Containing Precursors on the Carbon Support. Energies, 12(20), 3885. https://doi.org/10.3390/en12203885