
Influence of Catalytic Formulation and Operative Conditions on Coke Deposition over CeO2-SiO2 Based Catalysts for Ethanol Reforming





Abstract
1. Introduction
2. Experimental
2.1. Catalyst Preparation and Characterization
2.2. Catalyst Testing
3. Results and Discussion
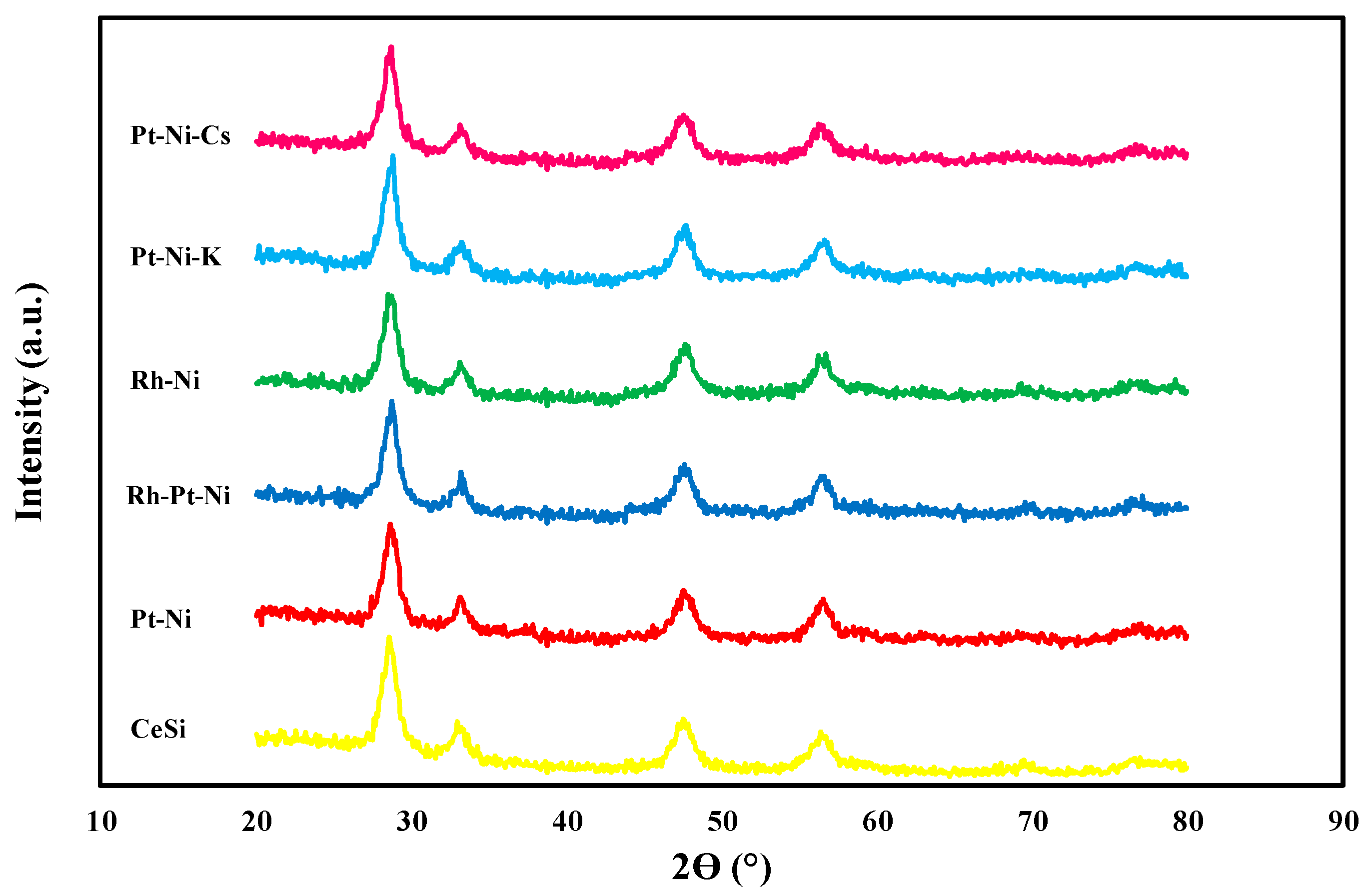
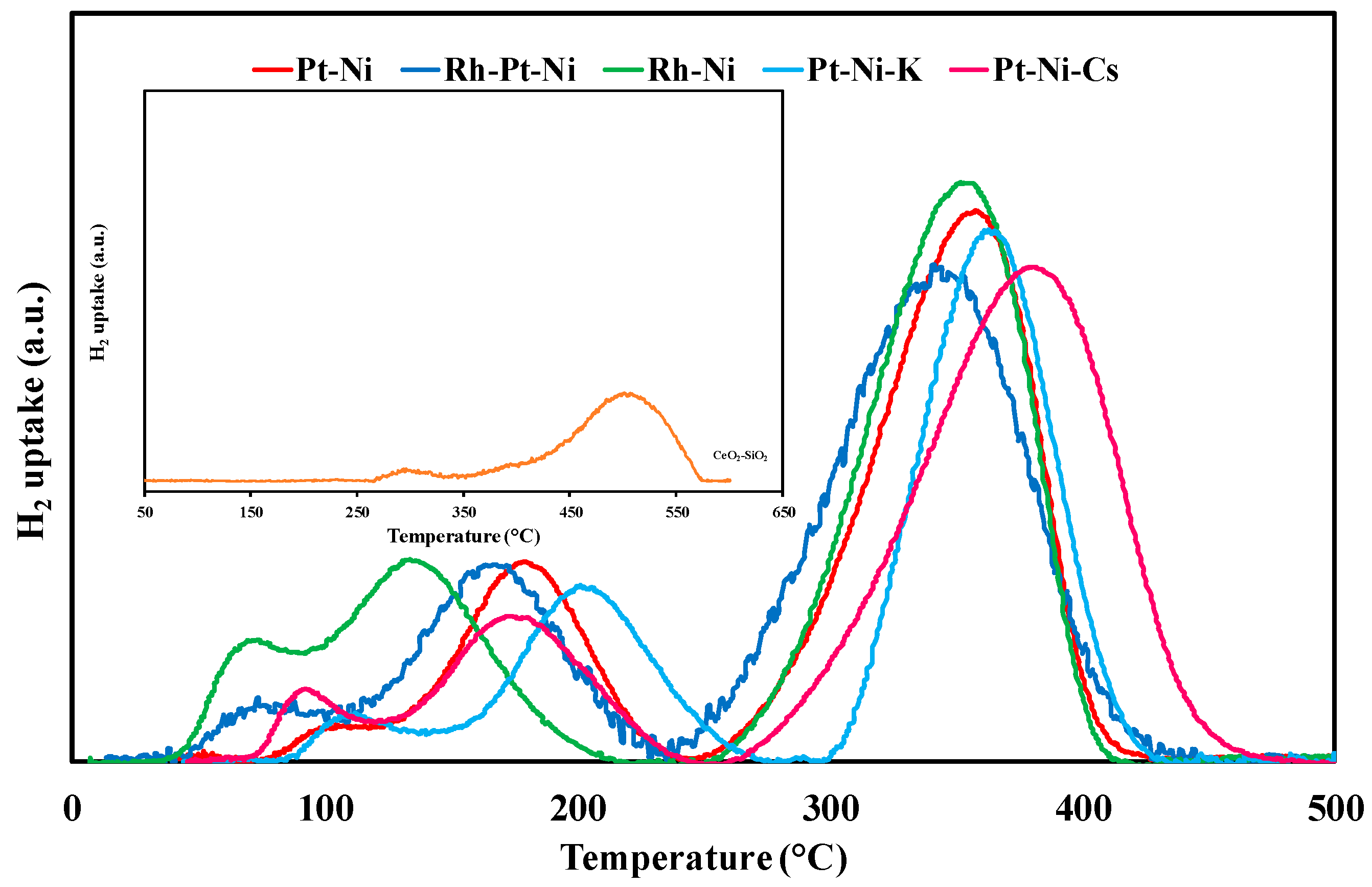
3.1. Textural/Structural Properties and H2-TPR Measurements
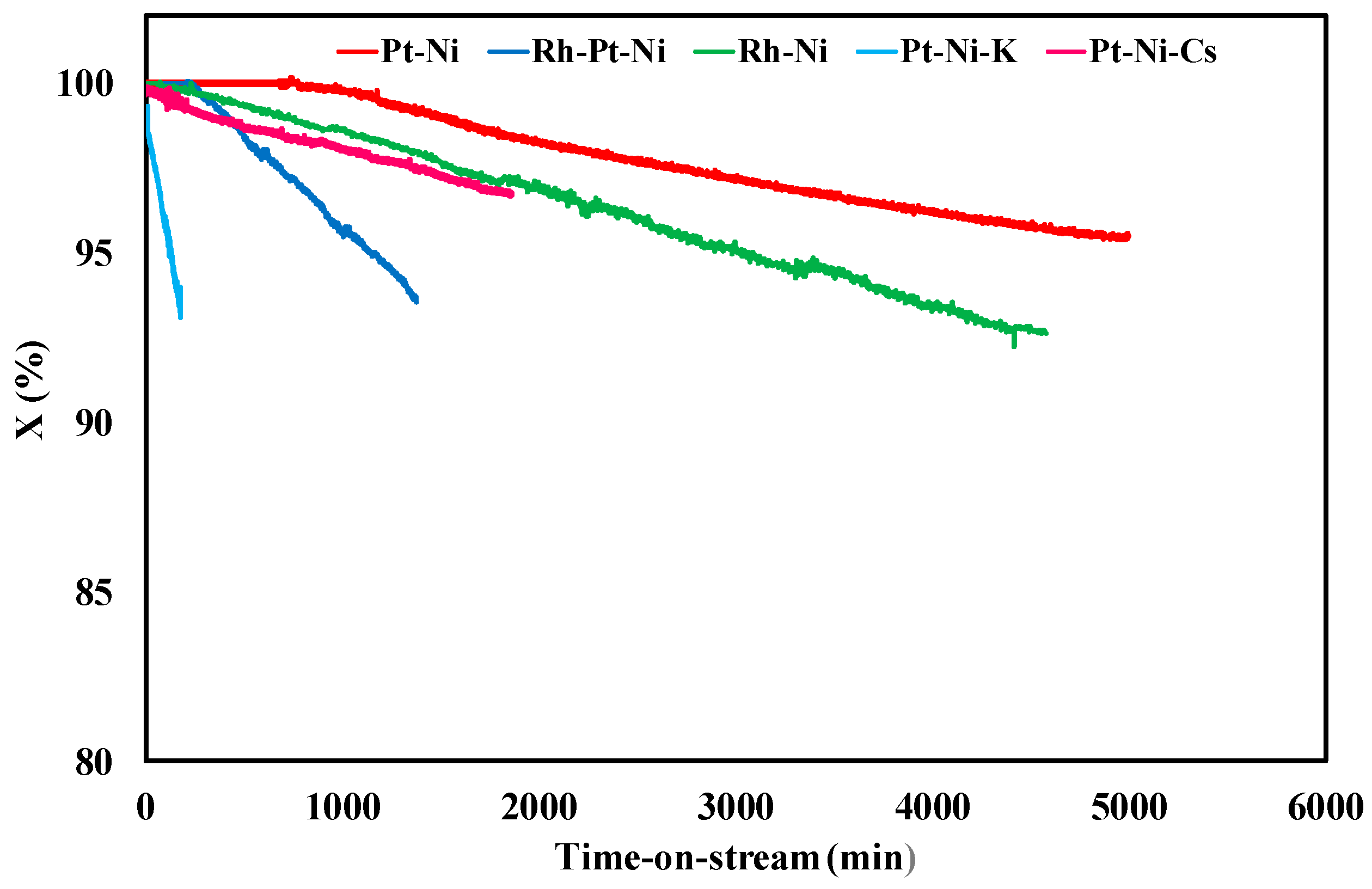
3.2. Ethanol Steam Reforming Tests: Effect of Catalytic Formulation
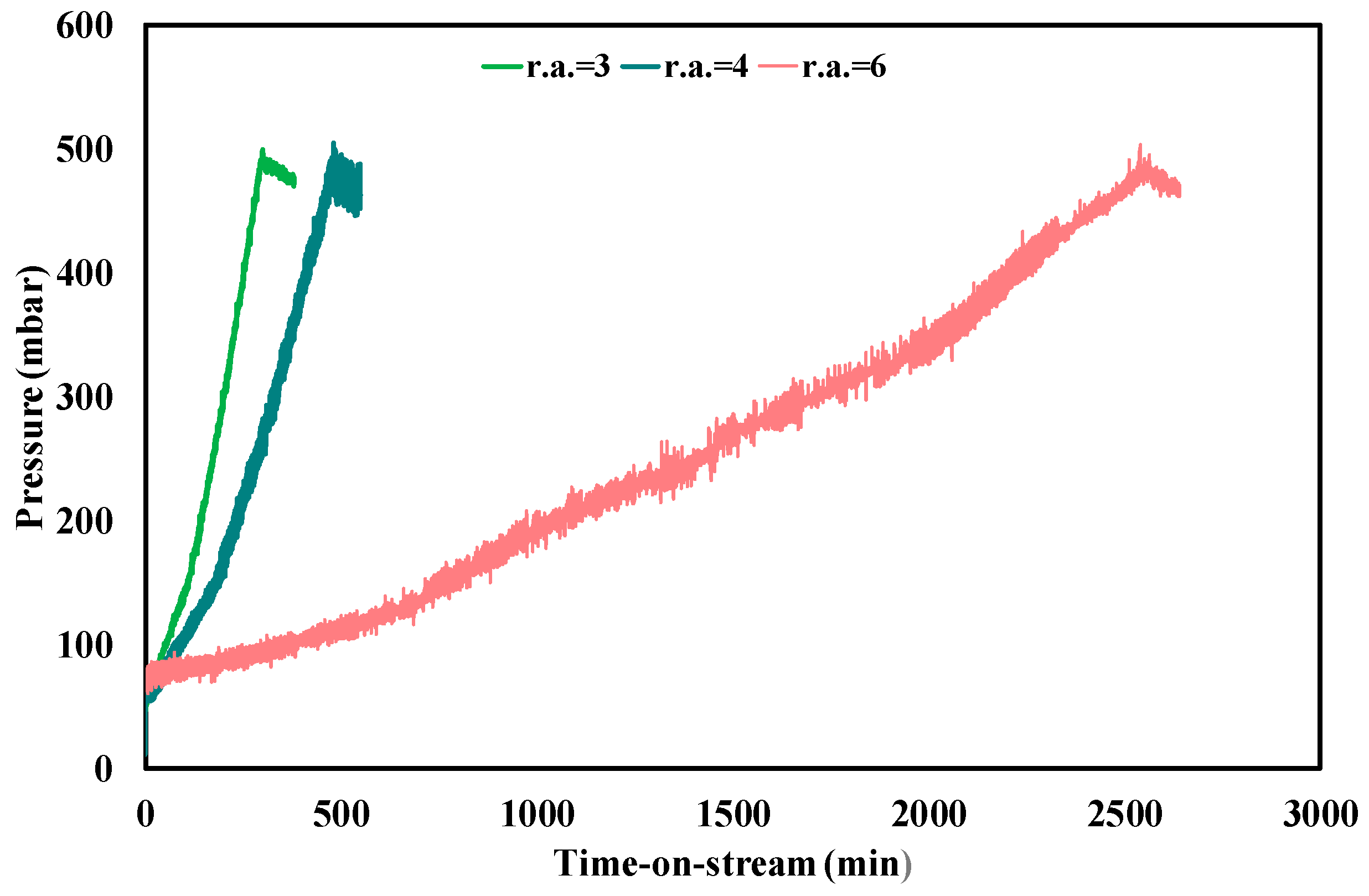
3.3. Ethanol Steam Reforming Tests: Effect of Operative Conditions
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Conte, M.; Di Mario, F.; Iacobazzi, A.; Mattucci, A.; Moreno, A.; Ronchetti, M. Hydrogen as future energy carrier: The ENEA point of view on technology and application prospects. Energies 2009, 2, 150. [Google Scholar] [CrossRef]
- Cifuentes, B.; Hernández, M.; Monsalve, S.; Cobo, M. Hydrogen production by steam reforming of ethanol on a RhPt/CeO2/SiO2 catalyst: Synergistic effect of the Si:Ce ratio on the catalyst performance. Appl. Catal. A Gen. 2016, 523, 283–293. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, Z.; Liu, L.; Wang, X. Heat recovery from high temperature slags: A review of chemical methods. Energies 2015, 8, 1917. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, X.; Chu, B.; Cao, C.; Jin, Y.; Cheng, Y. Catalytic performance of Ni catalyst for steam methane reforming in a micro-channel reactor at high pressure. Chem. Eng. Proc. Proc. Intensif. 2017, 118, 19–25. [Google Scholar] [CrossRef]
- Hu, L.; Lin, L.; Liu, S. Chemoselective hydrogenation of biomass-derived 5-hydroxymethylfurfural into the liquid biofuel 2, 5-dimethylfuran. Ind. Eng. Chem. Res. 2014, 53, 9969–9978. [Google Scholar] [CrossRef]
- Magdeldin, M.; Kohl, T.; De Blasio, C.; Järvinen, M.; Won Park, S.; Giudici, R. The BioSCWG project: Understanding the trade-offs in the process and thermal design of hydrogen and synthetic natural gas production. Energies 2016, 9, 838. [Google Scholar] [CrossRef]
- Sisinni, M.; Di Carlo, A.; Bocci, E.; Micangeli, A.; Naso, V. Hydrogen-rich gas production by sorption enhanced steam reforming of woodgas containing TAR over a commercial Ni Catalyst and calcined dolomite as CO2 sorbent. Energies 2013, 6, 3167. [Google Scholar] [CrossRef]
- Inayat, A.; Ahmad, M.M.; Yusup, S.; Mutalib, M.I.A. Biomass steam gasification with in-situ CO2 capture for enriched hydrogen gas production: A reaction kinetics modelling approach. Energies 2010, 3, 1472. [Google Scholar] [CrossRef]
- Tonezzer, M.; Dang, T.T.L.; Tran, Q.H.; Iannotta, S. Dual-selective hydrogen and ethanol sensor for steam reforming systems. Sens. Actuators B Chem. 2016, 236, 1011–1019. [Google Scholar] [CrossRef]
- Yasuda, M.; Kurogi, R.; Tsumagari, H.; Shiragami, T.; Matsumoto, T. New approach to fuelization of herbaceous lignocelluloses through simultaneous saccharification and fermentation followed by photocatalytic reforming. Energies 2014, 7, 4087. [Google Scholar] [CrossRef]
- Hari Krishna, S.; Chowdary, G.V. Optimization of simultaneous saccharification and fermentation for the production of ethanol from lignocellulosic biomass. J. Agric. Food Chem. 2000, 48, 1971–1976. [Google Scholar] [CrossRef]
- Dantas, S.C.; Resende, K.A.; Ávila-Neto, C.N.; Noronha, F.B.; Bueno, J.M.C.; Hori, C.E. Nickel supported catalysts for hydrogen production by reforming of ethanol as addressed by in situ temperature and spatial resolved XANES analysis. Int. J. Hydrogen Energy 2016, 41, 3399–3413. [Google Scholar] [CrossRef]
- Carvalho, F.L.S.; Asencios, Y.J.O.; Bellido, J.D.A.; Assaf, E.M. Bio-ethanol steam reforming for hydrogen production over Co3O4/CeO2 catalysts synthesized by one-step polymerization method. Fuel Proc. Technol. 2016, 142, 182–191. [Google Scholar] [CrossRef]
- Sharma, Y.C.; Kumar, A.; Prasad, R.; Upadhyay, S.N. Ethanol steam reforming for hydrogen production: Latest and effective catalyst modification strategies to minimize carbonaceous deactivation. Renew. Sustain. Energy Rev. 2017, 74, 89–103. [Google Scholar] [CrossRef]
- Arslan, A.; Doğu, T. Effect of calcination/reduction temperature of Ni impregnated CeO2-ZrO2 catalysts on hydrogen yield and coke minimization in low temperature reforming of ethanol. Int. J. Hydrogen Energy 2016, 41, 16752–16761. [Google Scholar] [CrossRef]
- Cifuentes, B.; Valero, M.; Conesa, J.; Cobo, M. Hydrogen production by steam reforming of ethanol on Rh-Pt catalysts: Influence of CeO2, ZrO2, and La2O3 as Supports. Catalysts 2015, 5, 1872. [Google Scholar] [CrossRef]
- Yaakob, Z.; Bshish, A.; Ebshish, A.; Tasirin, S.; Alhasan, F. Hydrogen production by steam reforming of ethanol over nickel catalysts supported on sol gel made alumina: Influence of calcination temperature on supports. Materials 2013, 6, 2229. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M. Steam reforming of bio-ethanol to produce hydrogen over Co/CeO2 catalysts derived from Ce1-xCoxO2-y precursors. Catalysts 2016, 6, 26. [Google Scholar] [CrossRef]
- Kugai, J.; Velu, S.; Song, C. Low-temperature reforming of ethanol over CeO2-supported Ni-Rh bimetallic catalysts for hydrogen production. Catal. Lett. 2005, 101, 255–264. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Castaldo, F.; Ricca, A.; Boettge, D. Ethanol steam reforming over bimetallic coated ceramic foams: Effect of reactor configuration and catalytic support. Int. J. Hydrogen Energy 2015, 40, 12650–12662. [Google Scholar] [CrossRef]
- Zhou, L.; Guo, Y.; Kameyama, H.; Basset, J.-M. An anodic alumina supported Ni-Pt bimetallic plate-type catalysts for multi-reforming of methane, kerosene and ethanol. Int. J. Hydrogen Energy 2014, 39, 7291–7305. [Google Scholar] [CrossRef]
- Carrero, A.; Calles, J.A.; Vizcaíno, A.J. Effect of Mg and Ca addition on coke deposition over Cu–Ni/SiO2 catalysts for ethanol steam reforming. Chem. Eng. J. 2010, 163, 395–402. [Google Scholar] [CrossRef]
- Wang, S.; Lu, G.Q. Effects of promoters on catalytic activity and carbon deposition of Ni/γ-Al2O3 catalysts in CO2 reforming of CH4. J. Chem. Technol. Biotechnol. 2000, 75, 589–595. [Google Scholar] [CrossRef]
- Rass-Hansen, J.; Christensen, C.H.; Sehested, J.; Helveg, S.; Rostrup-Nielsen, J.R.; Dahl, S. Renewable hydrogen: Carbon formation on Ni and Ru catalysts during ethanol steam-reforming. Green Chem. 2007, 9, 1016–1021. [Google Scholar] [CrossRef]
- Akiyama, M.; Oki, Y.; Nagai, M. Steam reforming of ethanol over carburized alkali-doped nickel on zirconia and various supports for hydrogen production. Catal. Today 2012, 181, 4–13. [Google Scholar] [CrossRef]
- Osorio-Vargas, P.; Campos, C.H.; Navarro, R.M.; Fierro, J.L.G.; Reyes, P. Improved ethanol steam reforming on Rh/Al2O3 catalysts doped with CeO2 or/and La2O3: Influence in reaction pathways including coke formation. Appl. Catal. A Gen. 2015, 505, 159–172. [Google Scholar] [CrossRef]
- Konsolakis, M.; Ioakimidis, Z.; Kraia, T.; Marnellos, G. Hydrogen production by ethanol steam reforming (ESR) over CeO2 supported transition metal (Fe, Co, Ni, Cu) catalysts: Insight into the structure-activity relationship. Catalysts 2016, 6, 9. [Google Scholar] [CrossRef]
- Tao, J.; Zhao, L.; Dong, C.; Lu, Q.; Du, X.; Dahlquist, E. Catalytic steam reforming of toluene as a model compound of biomass gasification tar using Ni-CeO2/SBA-15 catalysts. Energies 2013, 6, 3284. [Google Scholar] [CrossRef]
- Santander, J.A.; Tonetto, G.M.; Pedernera, M.N.; López, E. Ni/CeO2-MgO catalysts supported on stainless steel plates for ethanol steam reforming. Int. J. Hydrogen Energy 2017, 42, 9482–9492. [Google Scholar] [CrossRef]
- Parlett, C.M.A.; Aydin, A.; Durndell, L.J.; Frattini, L.; Isaacs, M.A.; Lee, A.F.; Liu, X.; Olivi, L.; Trofimovaite, R.; Wilson, K.; et al. Tailored mesoporous silica supports for Ni catalysed hydrogen production from ethanol steam reforming. Catal. Commun. 2017, 91, 76–79. [Google Scholar] [CrossRef]
- Hu, J.; Yu, C.; Bi, Y.; Wei, L.; Chen, J.; Chen, X. Preparation and characterization of Ni/CeO2-SiO2 catalysts and their performance in catalytic partial oxidation of methane to syngas. Chin. J. Catal. 2014, 35, 8–20. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Meloni, E.; Ricca, A. Oxidative steam reforming of ethanol on mesoporous silica supported PtNi/CeO2 catalysts. Int. J. Hydrogen Energy 2017, 42, 1598–1608. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Meloni, E.; Gallucci, F.; Ricca, A. Enhancing Pt-Ni/CeO2 performances for ethanol reforming by catalyst supporting on high surface silica. Catal. Today 2017, in press. [Google Scholar] [CrossRef]
- Qin, H.; Qian, X.; Meng, T.; Lin, Y.; Ma, Z. Pt/MOx/SiO2, Pt/MOx/TiO2, and Pt/MOx/Al2O3 Catalysts for CO Oxidation. Catalysts 2015, 5, 606. [Google Scholar] [CrossRef]
- Solsona, B.; Sanchis, R.; Dejoz, A.; García, T.; Ruiz-Rodríguez, L.; López Nieto, J.; Cecilia, J.; Rodríguez-Castellón, E. Total oxidation of propane using CeO2 and CuO-CeO2 catalysts prepared using templates of different nature. Catalysts 2017, 7, 96. [Google Scholar] [CrossRef]
- Chen, A.; Guo, H.; Song, Y.; Chen, P.; Lou, H. Recyclable CeO2-ZrO2 and CeO2-TiO2 mixed oxides based Pt catalyst for aqueous-phase reforming of the low-boiling fraction of bio-oil. Int. J. Hydrogen Energy 2017, 42, 9577–9588. [Google Scholar] [CrossRef]
- Hong, X.; Sun, Y.; Zhu, T.; Liu, Z. Pt-Au/MOx-CeO2 (M = Mn, Fe, Ti) catalysts for the co-oxidation of CO and H2 at room temperature. Molecules 2017, 22, 351. [Google Scholar] [CrossRef] [PubMed]
- Hérault, N.; Olivet, L.; Pirault-Roy, L.; Especel, C.; Vicerich, M.A.; Pieck, C.L.; Epron, F. Controlled preparation and characterization of Pt-Rh/Al2O3 bimetallic catalysts for reactions in reducing conditions. Appl. Catal. A Gen. 2016, 517, 81–90. [Google Scholar] [CrossRef]
- Atzori, L.; Cutrufello, M.G.; Meloni, D.; Cannas, C.; Gazzoli, D.; Monaci, R.; Sini, M.F. Highly active NiO-CeO2 catalysts for synthetic natural gas production by CO2 methanation. Catal. Today 2017, in press. [Google Scholar] [CrossRef]
- Cobo, M.; Pieruccini, D.; Abello, R.; Ariza, L.; Córdoba, L.F.; Conesa, J.A. Steam reforming of ethanol over bimetallic RhPt/La2O3: Long-term stability under favorable reaction conditions. Int. J. Hydrogen Energy 2013, 38, 5580–5593. [Google Scholar] [CrossRef]
- Carrero, A.; Calles, J.; García-Moreno, L.; Vizcaíno, A. Production of renewable hydrogen from glycerol steam reforming over bimetallic Ni-(Cu,Co,Cr) catalysts supported on SBA-15 Silica. Catalysts 2017, 7, 55. [Google Scholar] [CrossRef]
- Ocsachoque, M.; Pompeo, F.; Gonzalez, G. Rh–Ni/CeO2-Al2O3 catalysts for methane dry reforming. Catal. Today 2011, 172, 226–231. [Google Scholar] [CrossRef]
- Xiong, H.; Motchelaho, M.A.; Moyo, M.; Jewell, L.L.; Coville, N.J. Effect of group I alkali metal promoters on Fe/CNT catalysts in Fischer-Tropsch synthesis. Fuel 2015, 150, 687–696. [Google Scholar] [CrossRef]
- Aboud, M.; ALOthman, Z.; Habila, M.; Zlotea, C.; Latroche, M.; Cuevas, F. Hydrogen storage in pristine and d10-block metal-anchored activated carbon made from local wastes. Energies 2015, 8, 3578. [Google Scholar] [CrossRef]
- Mortola, V.B.; Damyanova, S.; Zanchet, D.; Bueno, J.M.C. Surface and structural features of Pt/CeO2-La2O3-Al2O3 catalysts for partial oxidation and steam reforming of methane. Appl. Catal. B Environ. 2011, 107, 221–236. [Google Scholar] [CrossRef]
- Mamontov, G.V.; Grabchenko, M.V.; Sobolev, V.I.; Zaikovskii, V.I.; Vodyankina, O.V. Ethanol dehydrogenation over Ag-CeO2/SiO2 catalyst: Role of Ag-CeO2 interface. Appl. Catal. A Gen. 2016, 528, 161–167. [Google Scholar] [CrossRef]
- de Caprariis, B.; de Filippis, P.; Palma, V.; Petrullo, A.; Ricca, A.; Ruocco, C.; Scarsella, M. Rh, Ru and Pt ternary perovskites type oxides BaZr(1 − x)MexO3 for methane dry reforming. Appl. Catal. A Gen. 2016, 517, 47–55. [Google Scholar] [CrossRef]
- Corella, J.; Toledo, J.M.; Molina, G. Steam gasification of coal at low-medium (600−800 °C) temperature with simultaneous CO2 capture in fluidized bed at atmospheric pressure: The effect of inorganic species. 1. Literature review and comments. Ind. Eng. Chem. Res. 2006, 45, 6137–6146. [Google Scholar] [CrossRef]
- Hou, T.; Yu, B.; Zhang, S.; Xu, T.; Wang, D.; Cai, W. Hydrogen production from ethanol steam reforming over Rh/CeO2 catalyst. Catal. Commun. 2015, 58, 137–140. [Google Scholar] [CrossRef]
- Zhao, X.; Lu, G. Improving catalytic activity and stability by in-situ regeneration of Ni-based catalyst for hydrogen production from ethanol steam reforming via controlling of active species dispersion. Int. J. Hydrogen Energy 2016, 41, 13993–14002. [Google Scholar] [CrossRef]
- de Lima, S.M.; da Cruz, I.O.; Jacobs, G.; Davis, B.H.; Mattos, L.V.; Noronha, F.B. Steam reforming, partial oxidation, and oxidative steam reforming of ethanol over Pt/CeZrO2 catalyst. J. Catal. 2008, 257, 356–368. [Google Scholar] [CrossRef]
- Trimm, D.L. Coke formation and minimisation during steam reforming reactions. Catal. Today 1997, 37, 233–238. [Google Scholar] [CrossRef]
- Coll, R.; Salvadó, J.; Farriol, X.; Montané, D. Steam reforming model compounds of biomass gasification tars: Conversion at different operating conditions and tendency towards coke formation. Fuel Proc. Technol. 2001, 74, 19–31. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SiO2 (wt %) | CeO2 (wt %) | Ni (wt %) | Pt (or Rh) (wt %) | SSA (m2∙g−1) | d (Å) |
|---|---|---|---|---|---|---|
| CeSi | 68.9 | 31.1 | - | - | 254 | 78 |
| Pt-Ni | 65.3 | 30.3 | 3.4 | 1 | 255 | 73 |
| Rh-Pt-Ni | 65.3 | 30.3 | 3.4 | 1 | 197 | 68 |
| Rh-Ni | 65 | 30.3 | 3.6 | 1.1 | 223 | 73 |
| Pt-Ni-K | 65.3 | 30.2 | 3.5 | 1 | 155 | 68 |
| Pt-Ni-Cs | 65.3 | 30.3 | 3.3 | 1.1 | 203 | 69 |
| Sample | T (°C) | H2 Uptake (µmol/gcat) | ||
|---|---|---|---|---|
| Experimental | Total Experimental | Total Expected | ||
| CeO2-SiO2 | - | 300 | - | - |
| - | 448 | - | - | |
| - | 509 | 298 | 6210 | |
| Pt-Ni | 109 | 119 (PtOx) | - | - |
| 179 | 943 (PtOx) | - | - | |
| 334 | 1032 (NiO) | - | - | |
| 366 | 641 (NiO) | 2735 | 2012 | |
| Rh-Pt-Ni | 70 | 95 (RhOx) | - | - |
| 96 | 154 (PtOx) | - | - | |
| 167 | 1029 (PtOx) | - | - | |
| 315 | 836 (NiO) | - | - | |
| 357 | 967 (NiO) | 3081 | 2109 | |
| Rh-Ni | 69 | 286 (RhOx) | - | - |
| 134 | 1184 (RhOx) | - | - | |
| 339 | 1189 (NiO) | - | - | |
| 360 | 550 (NiO) | 3209 | 2287 | |
| Pt-Ni-K | 116 | 179 (PtOx) | - | - |
| 204 | 925 (NiO) | - | - | |
| 363 | 1367 (NiO) | 2471 | 2012 | |
| Pt-Ni-Cs | 95 | 151 (PtOx) | - | - |
| 174 | 742 (PtOx) | - | - | |
| 353 | 1024 (NiO) | - | - | |
| 393 | 771 (NiO) | 2688 | 2012 | |
| Sample | Y (%) | tplugging (min) | CFR (gcoke,oxidized·gcat−1·gc,fed−1·h−1) | CGR (gcoke,gasified·gcat−1·h−1) |
|---|---|---|---|---|
| Pt-Ni | 26.5 | 310 | 0.0030 | 0.035 |
| Rh-Pt-Ni | 23.9 | 1300 | 0.00084 | 0.033 |
| Rh-Ni | 23.2 | 4900 | 0.000065 | 0.035 |
| Pt-Ni-K | 20.1 | 200 | 0.017 | 0.037 |
| Pt-Ni-Cs | 24.7 | 1600 | 0.00039 | 0.036 |
| Test Condition | tplugging (min) | CFR (gcoke,oxidized·gcat−1·gc,fed−1·h−1) | CGR (gcoke,gasified·gcat−1·h−1) |
|---|---|---|---|
| f.r. = 3, T = 450 °C | 310 | 0.0030 | 0.035 |
| f.r. = 4, T = 450 °C | 480 | 0.0013 | 0.035 |
| f.r. = 6, T = 450 °C | 2550 | 0.00011 | 0.033 |
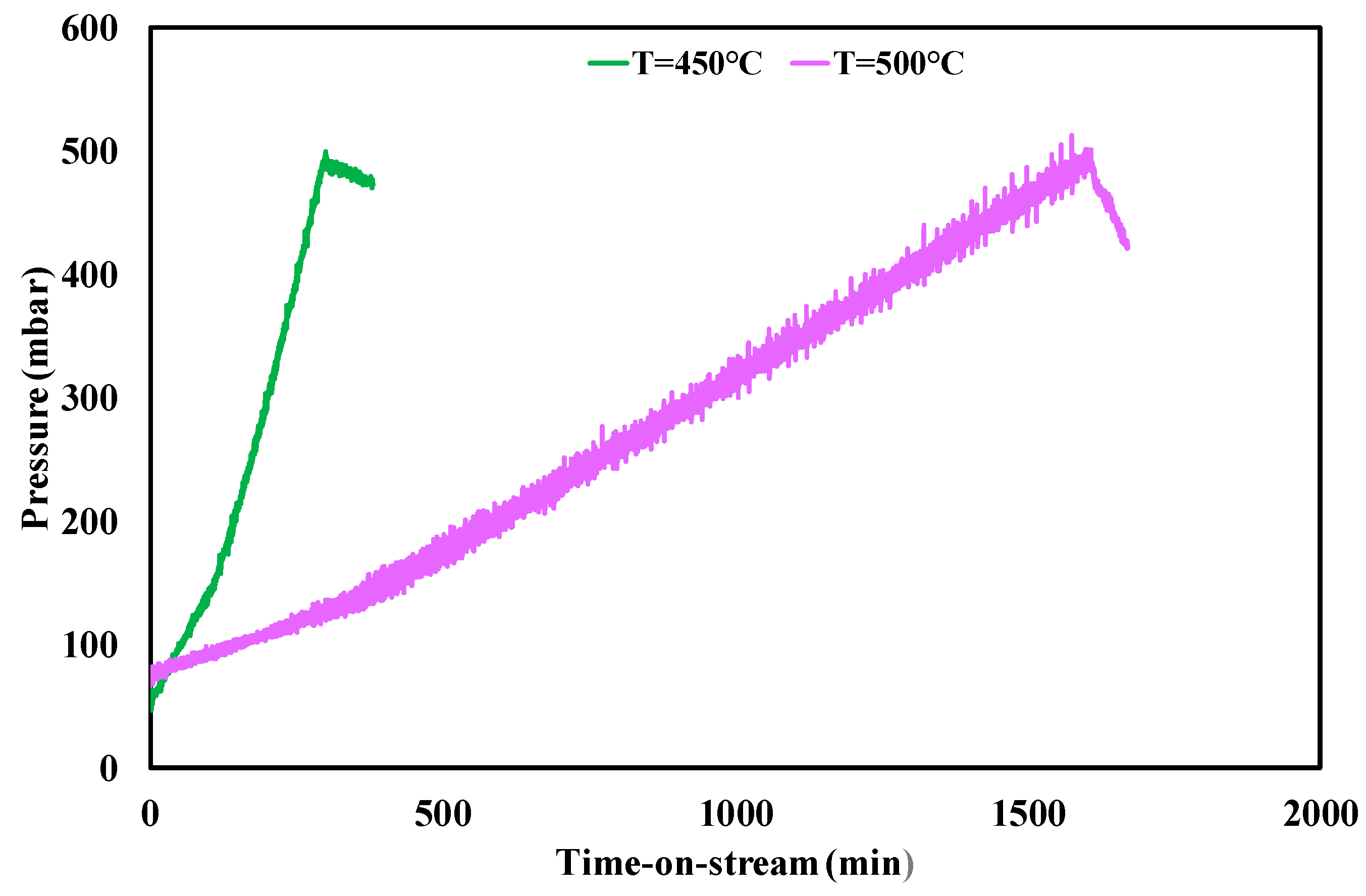
| f.r. = 3, T = 500 °C | 1600 | 0.00025 | 0.25 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palma, V.; Ruocco, C.; Meloni, E.; Ricca, A. Influence of Catalytic Formulation and Operative Conditions on Coke Deposition over CeO2-SiO2 Based Catalysts for Ethanol Reforming. Energies 2017, 10, 1030. https://doi.org/10.3390/en10071030
Palma V, Ruocco C, Meloni E, Ricca A. Influence of Catalytic Formulation and Operative Conditions on Coke Deposition over CeO2-SiO2 Based Catalysts for Ethanol Reforming. Energies. 2017; 10(7):1030. https://doi.org/10.3390/en10071030
Chicago/Turabian StylePalma, Vincenzo, Concetta Ruocco, Eugenio Meloni, and Antonio Ricca. 2017. "Influence of Catalytic Formulation and Operative Conditions on Coke Deposition over CeO2-SiO2 Based Catalysts for Ethanol Reforming" Energies 10, no. 7: 1030. https://doi.org/10.3390/en10071030
APA StylePalma, V., Ruocco, C., Meloni, E., & Ricca, A. (2017). Influence of Catalytic Formulation and Operative Conditions on Coke Deposition over CeO2-SiO2 Based Catalysts for Ethanol Reforming. Energies, 10(7), 1030. https://doi.org/10.3390/en10071030