

DNA Barcoding and ITS2 Secondary Structure Predictions in Taro (Colocasia esculenta L. Schott) from the North Eastern Hill Region of India

 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Experimental Site
2.2. Plant Sample Preparation and Total Genomic DNA Isolation
2.3. PCR Amplification and Purification
2.4. DNA Sequencing
2.5. Bioinformatics Analysis
2.6. RNA Secondary Structure Predictions
3. Results
3.1. PCR Amplification, Sequencing, and Multiple Sequence Alignment
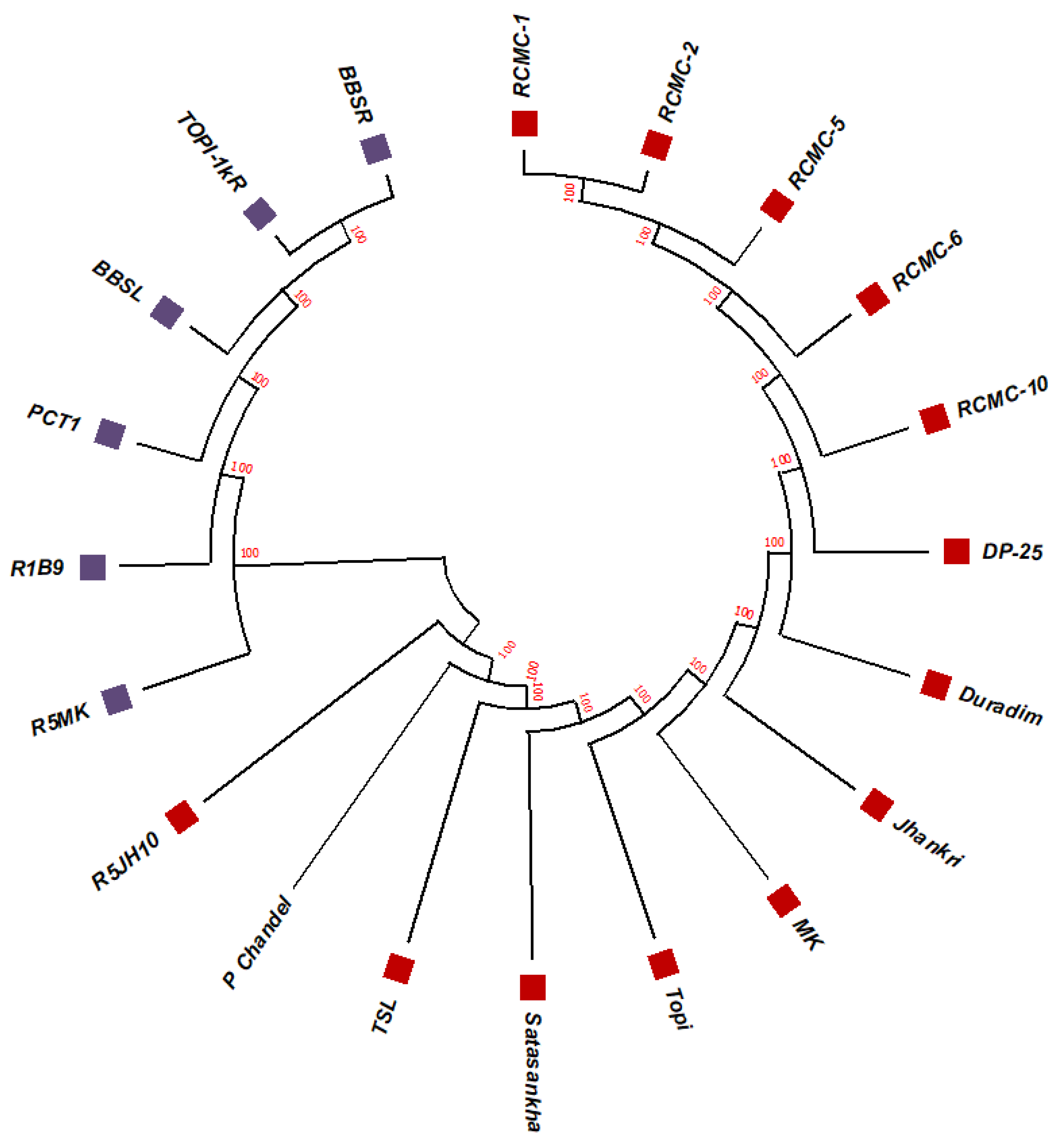
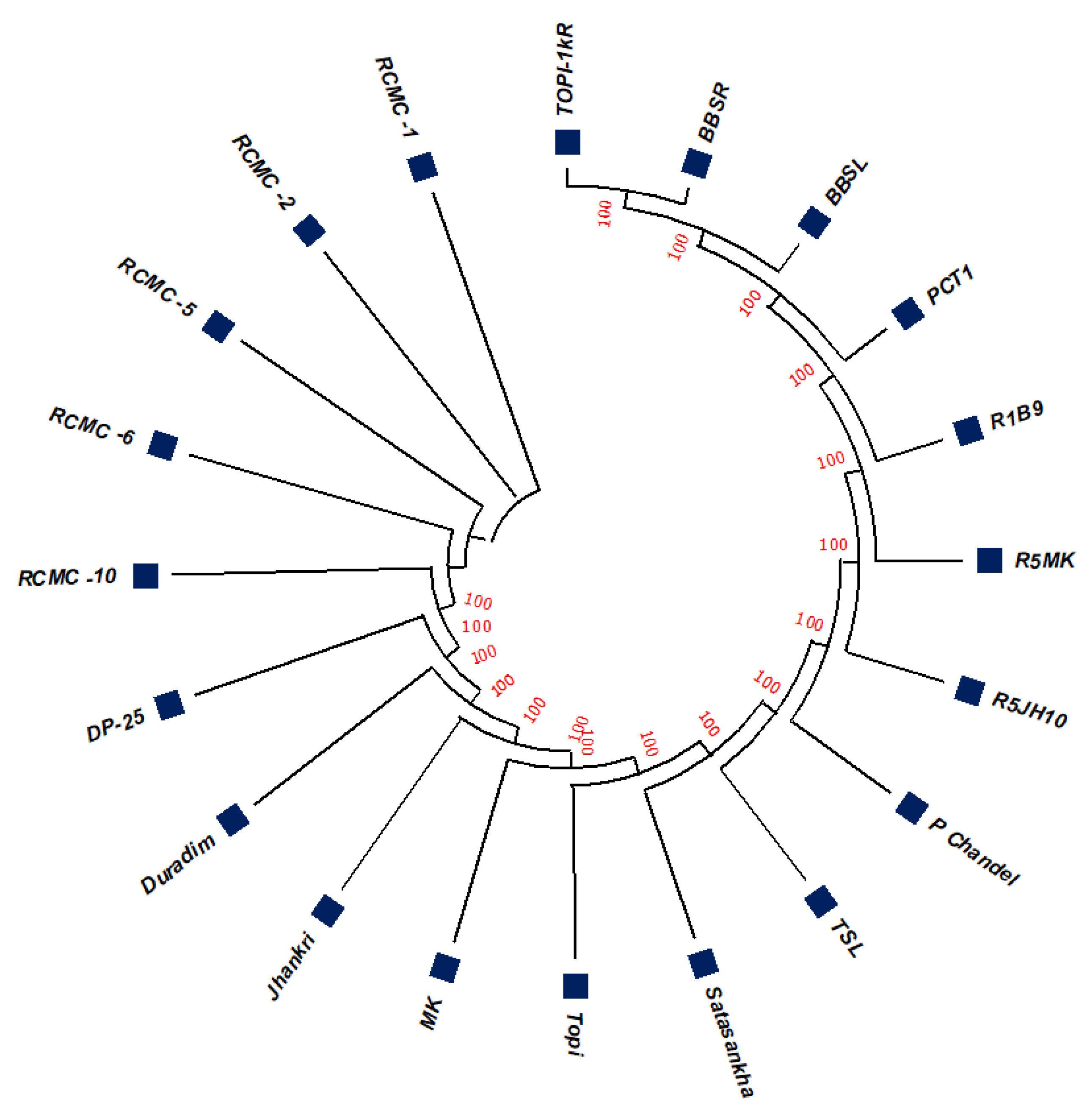
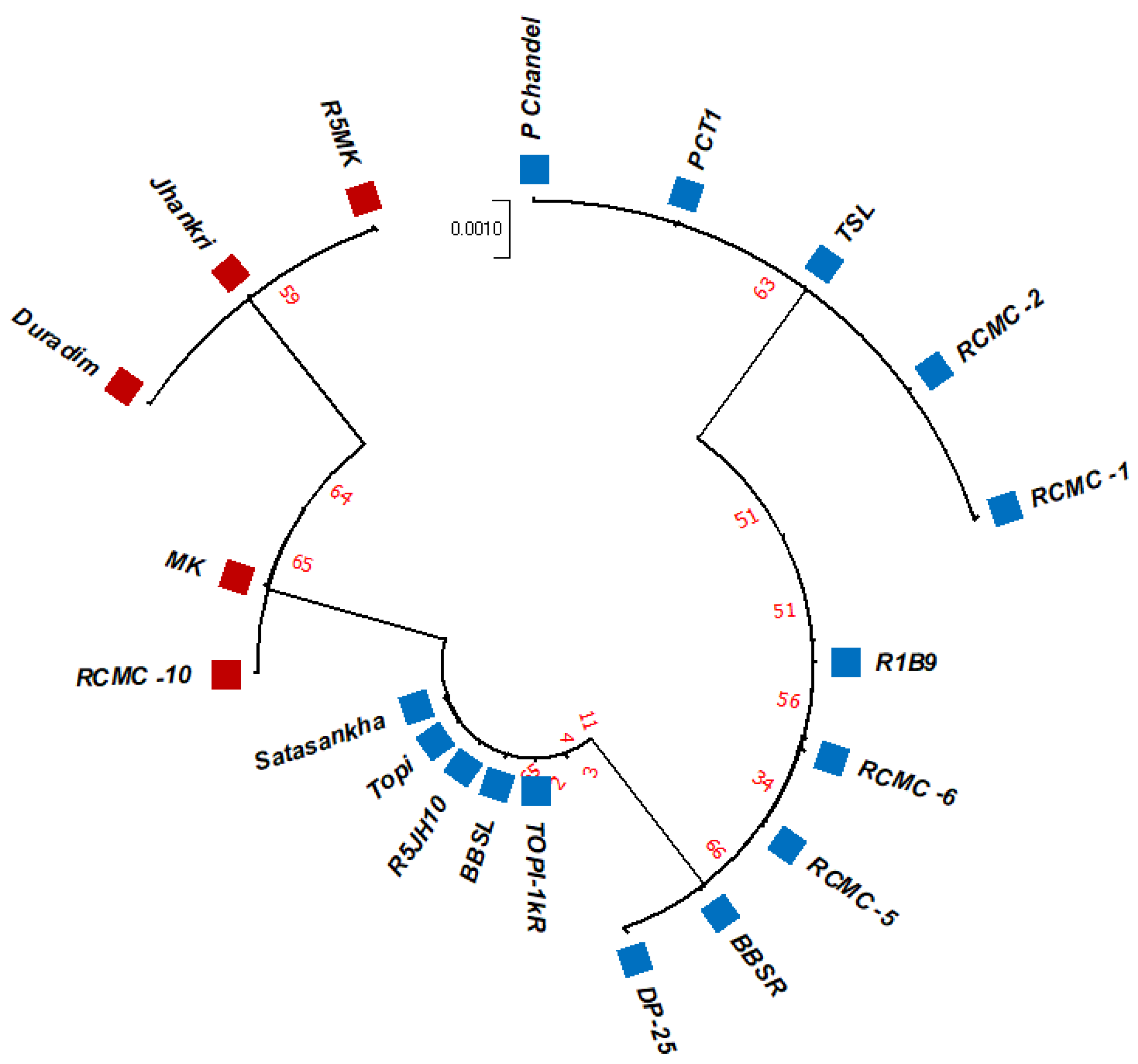
3.2. Phylogenetic Studies, Replacement Rate Matrices, and DNA Barcoding
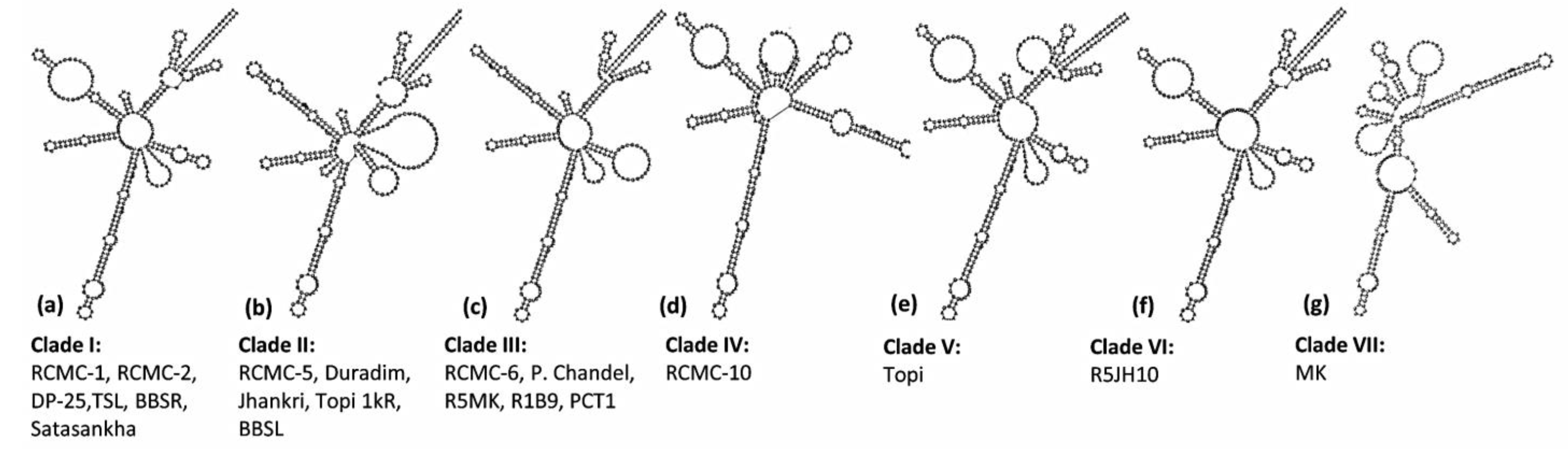
3.3. ITS2 Secondary Structure Predictions
4. Discussion
4.1. DNA Barcoding Using rbcL, matK, and ITS2 Genes
4.2. ITS2 Marker and RNA Secondary Structure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmed, I.; Lockhart, P.J.; Agoo, E.M.G.; Naing, K.W.; Nguyen, D.V.; Medhi, D.K.; Matthews, P.J. Evolutionary origins of taro (Colocasia esculenta) in Southeast Asia. Eco. Evo. 2020, 10, 13530–13543. [Google Scholar] [CrossRef] [PubMed]
- Matthews, P.J.; Ghanem, M.E. Perception gaps that may explain the status of taro (Colocasia esculenta) as an “orphan crop”. Plants People Planet 2020, 3, 99–112. [Google Scholar] [CrossRef]
- Mwamba, N.; Patrick, A.; Michael, P. Genetic diversity analysis in South African taro (Colocasia esculenta) accessions using molecular tools. Int. J. Genet. Mol. Bio. 2016, 8, 18–24. [Google Scholar] [CrossRef]
- Matthews, P. A possible tropical wildtype taro: Colocasia esculenta var. aquatilis. Indo. Pac. Prehis. Assn. Bull. 1991, 11, 69–81. [Google Scholar]
- Ivancic, A.; Lebot, V. The Genetics and Breeding of Taro; CIRAD: Montpellier, France, 2000. [Google Scholar]
- Chair, H.; Traore, R.E.; Duval, M.F.; Rivallan, R.; Mukherjee, A.; Aboagye, L.M. Genetic diversification and dispersal of Taro (Colocasia esculenta (L.) Schott). PLoS ONE 2016, 11, e0157712. [Google Scholar] [CrossRef] [PubMed]
- Mathews, P. Genetic diversity in taro and the preservation of culinary knowledge. Ethnobot. Res. Appl. 2004, 2, 55–71. [Google Scholar] [CrossRef]
- Anbalagan, T.; Deka, B.C.; Sivakumar, S.; Rangnamei, L.; Walling, N. Colocasia (Colocasia esculenta L.) in Northeast India. J. Innov. Agric. 2022, 9, 1–7. [Google Scholar] [CrossRef]
- Devi, Y.I.; Sahoo, M.R.; Mandal, J.; Dasgupta, M.; Prakash, N. Correlations between antioxidative enzyme activities and resistance to Phytophthora leaf blight in taro. J. Crop. Impro. 2020, 35, 250–263. [Google Scholar] [CrossRef]
- Ahmed, I.; Matthews, P.J.; Biggs, P.J.; Naeem, M.; McLenachan, P.A.; Lockhart, P.J. Identification of chloroplast genome loci suitable for high-resolution phylogeographic studies of Colocasia esculenta (L.) Schott (Araceae) and closely related taxa. Mol. Ecol. Resour. 2013, 13, 929–937. [Google Scholar] [CrossRef]
- Vinutha, K.B.; Devi, A.A.; Sreekumar, J. Morphological characterization of above ground characters of Taro (Colocasia esculenta (L.) Schott.) accessions from North East India. J. Root. Crop. 2015, 41, 3–11. [Google Scholar]
- Ochiai, T.; Tahara, M.; Yoshino, H. Phylogenetic relationships of taro and allied species based on restriction fragment length polymorphisms (RFLPs) of chloroplast DNA. Sci. Rep. Fac. Agri. 2000, 89, 15–21. [Google Scholar]
- Sahoo, M.R.; Naresh, P.; Kumari, M.; Acharya, G.C. Omics in leafy vegetables. In Omics in Horticultural Crops; Rout, G.R., Peter, K.V., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 281–302. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. B Bio. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Kress, W.J.; Erickson, D.L. DNA barcodes: Methods and protocols. Methods Mol. Biology 2012, 858, 3–8. [Google Scholar]
- Acharya, G.C.; Mohanty, S.; Dasgupta, M.; Sahu, S.; Singh, S.; Koundinya, A.V.V.; Kumari, M.; Naresh, P.; Sahoo, M.R. Molecular phylogeny, DNA barcoding, and ITS2 secondary structure predictions in the medicinally important Eryngium genotypes of East Coast Region of India. Genes 2022, 13, 1678. [Google Scholar] [CrossRef]
- Macke, T.J. RNAMotif, an RNA secondary structure definition, and search algorithm. Nucl. Acid. Res. 2001, 29, 4724–4735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tian, W.; Gao, Z.; Wang, G.; Zhao, H. Phylogenetic utility of rRNA ITS2 sequence-structure under functional constraint. Int. J. Mol. Sci. 2020, 21, 6395. [Google Scholar] [CrossRef] [PubMed]
- Rampersad, S.N. ITS1, 5.8S and ITS2 secondary structure modelling for intra-specific differentiation among species of the Colletotrichum gloeosporioidessensulato species complex. Springer Plus 2014, 3, 684. [Google Scholar] [CrossRef] [PubMed]
- CBOL Plant Working Group. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [CrossRef]
- Yang, L.; Tian, J.; Xu, L.; Zhao, X.; Song, Y.; Wang, D. Comparative chloroplast genomes of six Magnoliaceae species provide new insights into intergeneric relationships and phylogeny. Biology 2022, 11, 1279. [Google Scholar] [CrossRef]
- Kress, W.J.; Wurdack, K.J.; Zimmer, E.A.; Weigl, A.T.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [Google Scholar] [CrossRef] [PubMed]
- Cuenoud, P.V.; Savolainen, W.; Chatrou, L.W.; Powell, M.; Grayer, R.J.; Chase, M.W. Molecular phylogenetics of Caryophyllales based on nuclear 18S rDNA and plastid rbcL, atpBand matK DNA sequences. Americ. J. Bot. 2002, 89, 132–144. [Google Scholar] [CrossRef]
- Hilu, K.W.; Borsch, T.; Muller, K.; Soltis, D.E.; Soltis, P.S.; Savolainen, V.; Chase, M.W.; Powell, M.P.; Alice, L.A.; Evans, R.; et al. Angiosperm phylogeny based on matK sequence information. Am. J. Bot. 2003, 90, 1758–1776. [Google Scholar] [CrossRef]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and using a plant DNA barcode. PLoS ONE 2011, 5, e19254. [Google Scholar] [CrossRef]
- Cowan, R.S.; Fay, M.F. Challenges in the DNA barcoding of plant material. Mol. Bio. 2012, 862, 23–33. [Google Scholar]
- Armenise, L.; Simeone, M.C.; Piredda, R.; Schirone, B. Validation of DNA barcoding as an efficient tool for taxon identification and detection of species diversity in Italian conifers. Europ. J. For. Res. 2012, 131, 1337–1353. [Google Scholar] [CrossRef]
- Singh, S.D.R.; Singh, F.F.; Naresh, K.; Damodaran, V.; Srivastava, R.C. Diversity of 21 Taro (Colocasia esculenta (L.) Schott) accessions of Andaman Islands. Gen. Resou. Crop. Evo. 2012, 59, 821–829. [Google Scholar] [CrossRef]
- Nunes, R.S.C.; Del Aguila, E.; Paschoalin, V.; da Silva, J. DNA barcoding assessment of the genetic diversity of varieties of taro, Colocasia esculenta (L.) Schott in Brazil. In Breeding and Genetic Engineering: The Biology and Biotechnology Research; iConcept Press Ltd.: Hong Kong, China, 2014. [Google Scholar]
- Al-Juhani, W.S.; Khalik, K.N.A. Identification and phylogenetics study of Arabis alpina L. From the Kingdom of Saudi Arabia. Pak. J. Bot. 2021, 53, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Bio. Evo. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; HönerzuSiederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algo. Mol. Bio. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xiao, W.; Tong, T.; Li, Y.; Zhang, M.; Lin, X.; Guo, X. The specific DNA barcodes based on chloroplast genes for species identification of Orchidaceae plants. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Kreike, C.M.; Van-Eck, H.J.; Lebot, V. Genetic diversity of taro, Colocasia esculenta (L.) Schott, in Southeast Asia and the Pacific. Theor. Appl. Genet. 2004, 109, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Bosmali, I.; Lagiotis, G.; Haider, N.; Osathanunkul, M.; Biliaderis, C.; Madesis, P. DNA-based identification of Eurasian Vicia species using chloroplast and nuclear DNA barcodes. Plants 2022, 11, 947. [Google Scholar] [CrossRef] [PubMed]
- Ude, G.N.; Igwe, D.O.; McCormick, J.; Ozokonkwo-Alor, O.; Harper, J.; Ballah, D.; Aninweze, C.; Chosen, O.; Okoro, M.; Ene, C.; et al. Genetic diversity and DNA barcoding of yam accessions from Southern Nigeria. Am. J. Plant Sci. 2019, 10, 179–207. [Google Scholar] [CrossRef][Green Version]
- Ankenbrand, M.J.; Keller, A.; Wolf, M.; Schultz, J.; Förster, F. ITS2 database V: Twice as much. Mol. Biol. Evol. 2015, 32, 3030–3032. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Fu, Y.; Sharma, G.; Mathews, D.H. TurboFold II: RNA structural alignment and secondary structure prediction informed by multiple homologs. Nucl. Acid. Res. 2017, 45, 11570–11581. [Google Scholar] [CrossRef]
- Seemann, S.E.; Mirza, A.H.; Hansen, C.; Bang-Berthelsen, C.H.; Garde, C.; Christensen-Dalsgaard, M.; Torarinsson1, E.; Yao, Z.Z.; Workman, C.T.; Pociot, F. The identification and functional annotation of RNA structures conserved in vertebrates. Genome Res. 2017, 27, 1371–1383. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Primers | Sequences (5′ to 3′) | Amplicon Size | Amplification Success Rate |
|---|---|---|---|---|
| matK | matK XF matK 5R | TAATTTACGATCAATTCATTC GTTCTAGCACAAGAAAGTCG | 1000 bp | 90–95% |
| rbcL | rbcLa F rbcLa R | ATGTCACCACAAACAGAGACTAAAGC GTAAAATCAAGTCCACCRCG | 600 bp | 90% |
| ITS2 | ITS2 S2F ITS2 S3R | ATGCGATA CTTGGTGTGAATTATAGAAT GACGCTTCTCCAGACTACAAT | 300–400 bp | 80–85% |
| Sequences | Base Contents (%) | |||||
|---|---|---|---|---|---|---|
| A | T | G | C | AT | GC | |
| matK | 30.75 | 37.56 | 12.56 | 19.13 | 68.31 | 31.69 |
| rbcL | 25.80 | 33.41 | 20.28 | 20.51 | 59.20 | 40.80 |
| ITS2 | 19.52 | 11.44 | 34.78 | 34.26 | 30.96 | 69.04 |
| Voucher Name | matK and rbcL | ITS2 | Species | |||||
|---|---|---|---|---|---|---|---|---|
| Percent Identity | E Value | Accession No. (matK) | Accession No. (rbcL) | Percent Identity | E Value | Accession No. (ITS2) | ||
| RCMC-1 | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.05% | 3 × 10−156 | MK961250.1 | C. esculenta |
| RCMC-2 | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.05% | 3 × 10−156 | MK961250.1 | C. esculenta |
| RCMC-5 | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.37% | 7 × 10−158 | MK961250.1 | C. esculenta |
| RCMC-6 | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.37% | 7 × 10−158 | MK961250.1 | C. esculenta |
| RCMC-10 | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.36% | 2 × 10−157 | MK961250.1 | C. esculenta |
| DP-25 | 100% | 0.0 | LT995105.1 | MH270468.1 | 100.00% | 1 × 10−160 | MK961250.1 | C. esculenta |
| Duradim | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.04% | 1 × 10−155 | MK961250.1 | C. esculenta |
| Jhankri | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.36% | 2 × 10−157 | MK961250.1 | C. esculenta |
| MK | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.36% | 2 × 10−157 | MK961250.1 | C. esculenta |
| Topi | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.68% | 5 × 10−159 | MK961250.1 | C. esculenta |
| Satasankha | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.68% | 5 × 10−159 | MK961250.1 | C. esculenta |
| TSL | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.05% | 3 × 10−156 | MK961250.1 | C. esculenta |
| P Chandel | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.05% | 3 × 10−156 | MK961250.1 | C. esculenta |
| R5JH10 | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.68% | 5 × 10−159 | MK961250.1 | C. esculenta |
| R5MK | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.04% | 1 × 10−155 | MK961250.1 | C. esculenta |
| R1B9 | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.05% | 3 × 10−156 | MK961250.1 | C. esculenta |
| PCT1 | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.05% | 3 × 10−156 | MK961250.1 | C. esculenta |
| BBSL | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.68% | 5 × 10−159 | MK961250.1 | C. esculenta |
| TOPI-1kR | 100% | 0.0 | LT995105.1 | MH270468.1 | 99.68% | 5 × 10−159 | MK961250.1 | C. esculenta |
| BBSR | 100% | 0.0 | LT995105.1 | MH270468.1 | 100.00% | 1 × 10−160 | MK961250.1 | C. esculenta |
| matK | |||||
|---|---|---|---|---|---|
| matK | From/To | A | T | G | C |
| A | - | 12.3839 | 5.8154 | 4.9319 | |
| T | 10.2083 | - | 5.8096 | 4.9368 | |
| G | 10.2083 | 12.3715 | - | 4.9368 | |
| C | 10.1981 | 12.3839 | 5.8154 | - | |
| rbcL | |||||
| rbcL | From/To | A | T | G | C |
| A | - | 9.6984 | 7.2905 | 7.2164 | |
| T | 9.1319 | - | 7.2832 | 7.2236 | |
| G | 9.1319 | 9.6887 | - | 7.2236 | |
| C | 9.1227 | 9.6984 | 7.2905 | - | |
| ITS2 | |||||
| ITS2 | From/To | A | T | G | C |
| A | - | 0.5673 | 1.7239 | 18.0123 | |
| T | 1.0720 | - | 45.5929 | 1.7201 | |
| G | 1.0720 | 15.0027 | - | 1.7201 | |
| C | 11.2258 | 0.5673 | 1.7239 | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devi, M.P.; Dasgupta, M.; Mohanty, S.; Sharma, S.K.; Hegde, V.; Roy, S.S.; Renadevan, R.; Kumar, K.B.; Patel, H.K.; Sahoo, M.R. DNA Barcoding and ITS2 Secondary Structure Predictions in Taro (Colocasia esculenta L. Schott) from the North Eastern Hill Region of India. Genes 2022, 13, 2294. https://doi.org/10.3390/genes13122294
Devi MP, Dasgupta M, Mohanty S, Sharma SK, Hegde V, Roy SS, Renadevan R, Kumar KB, Patel HK, Sahoo MR. DNA Barcoding and ITS2 Secondary Structure Predictions in Taro (Colocasia esculenta L. Schott) from the North Eastern Hill Region of India. Genes. 2022; 13(12):2294. https://doi.org/10.3390/genes13122294
Chicago/Turabian StyleDevi, Mayengbam Premi, Madhumita Dasgupta, Sansuta Mohanty, Susheel Kumar Sharma, Vivek Hegde, Subhra Saikat Roy, Rennya Renadevan, Kinathi Bipin Kumar, Hitendra Kumar Patel, and Manas Ranjan Sahoo. 2022. "DNA Barcoding and ITS2 Secondary Structure Predictions in Taro (Colocasia esculenta L. Schott) from the North Eastern Hill Region of India" Genes 13, no. 12: 2294. https://doi.org/10.3390/genes13122294
APA StyleDevi, M. P., Dasgupta, M., Mohanty, S., Sharma, S. K., Hegde, V., Roy, S. S., Renadevan, R., Kumar, K. B., Patel, H. K., & Sahoo, M. R. (2022). DNA Barcoding and ITS2 Secondary Structure Predictions in Taro (Colocasia esculenta L. Schott) from the North Eastern Hill Region of India. Genes, 13(12), 2294. https://doi.org/10.3390/genes13122294