
Interleukin-33 Induces Neutrophil Extracellular Trap (NET) Formation and Macrophage Necroptosis via Enhancing Oxidative Stress and Secretion of Proatherogenic Factors in Advanced Atherosclerosis

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient and Control Groups
2.2. Neutrophil Extracellular Trap Formation
2.3. Monocyte Isolation, Macrophage Culture and Stimulation
2.4. Quantitative Polymerase Chain Reaction (qPCR)
2.5. Oxidative Stress Estimation
2.6. Immunofluorescence
2.7. Enzyme Linked Immunosorbent Assay (ELISA)
2.8. Statistical Analysis
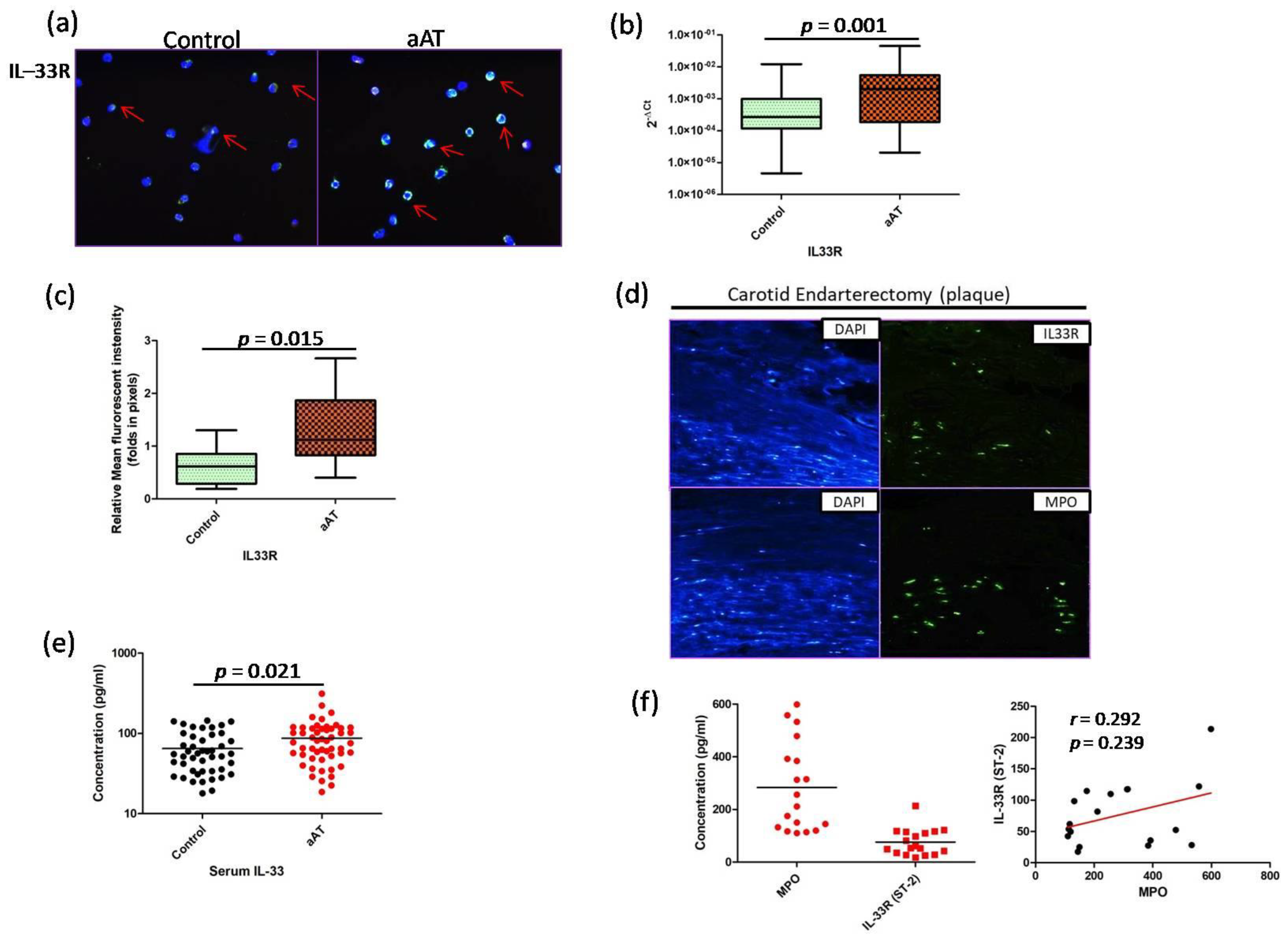
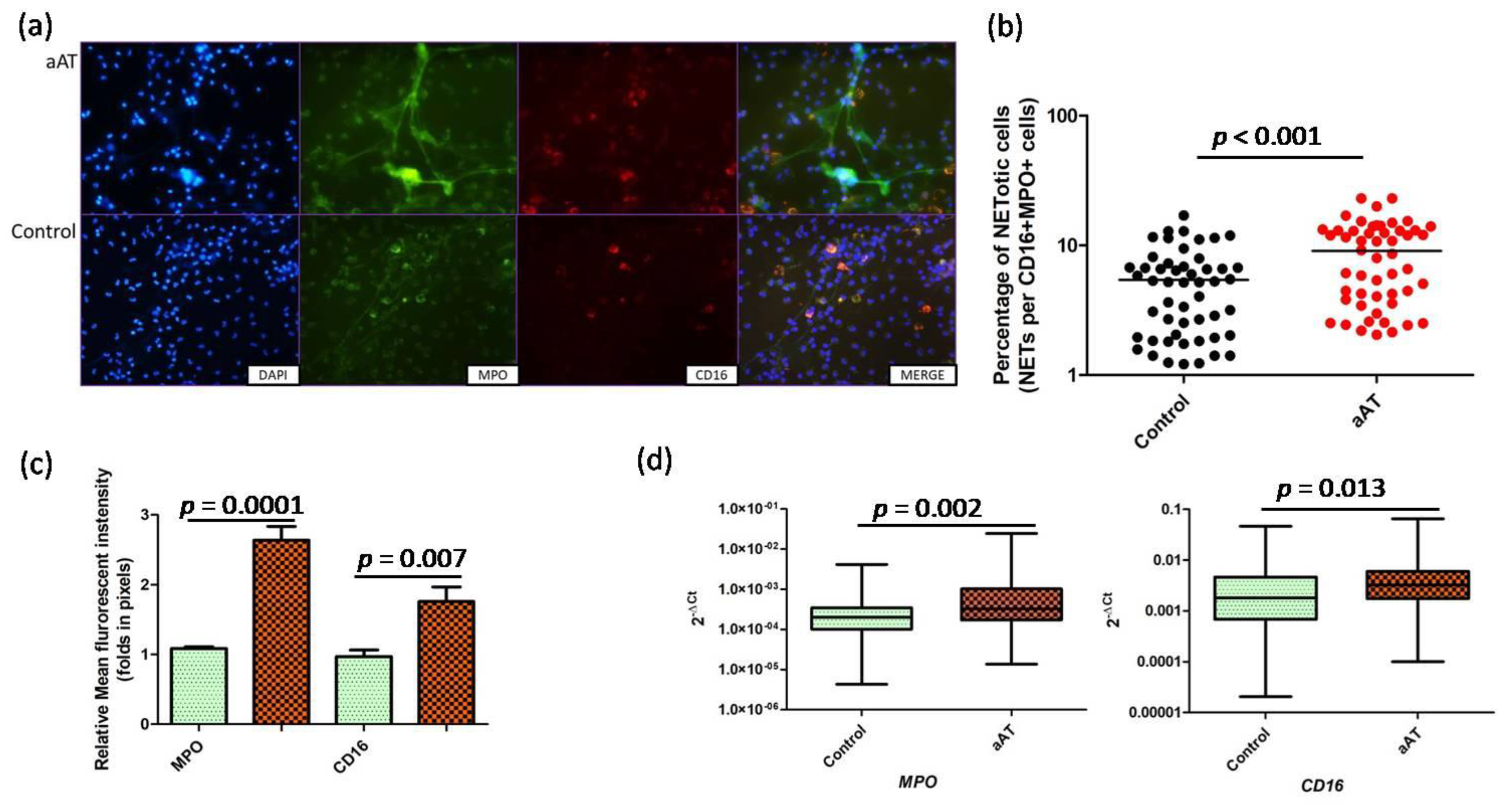
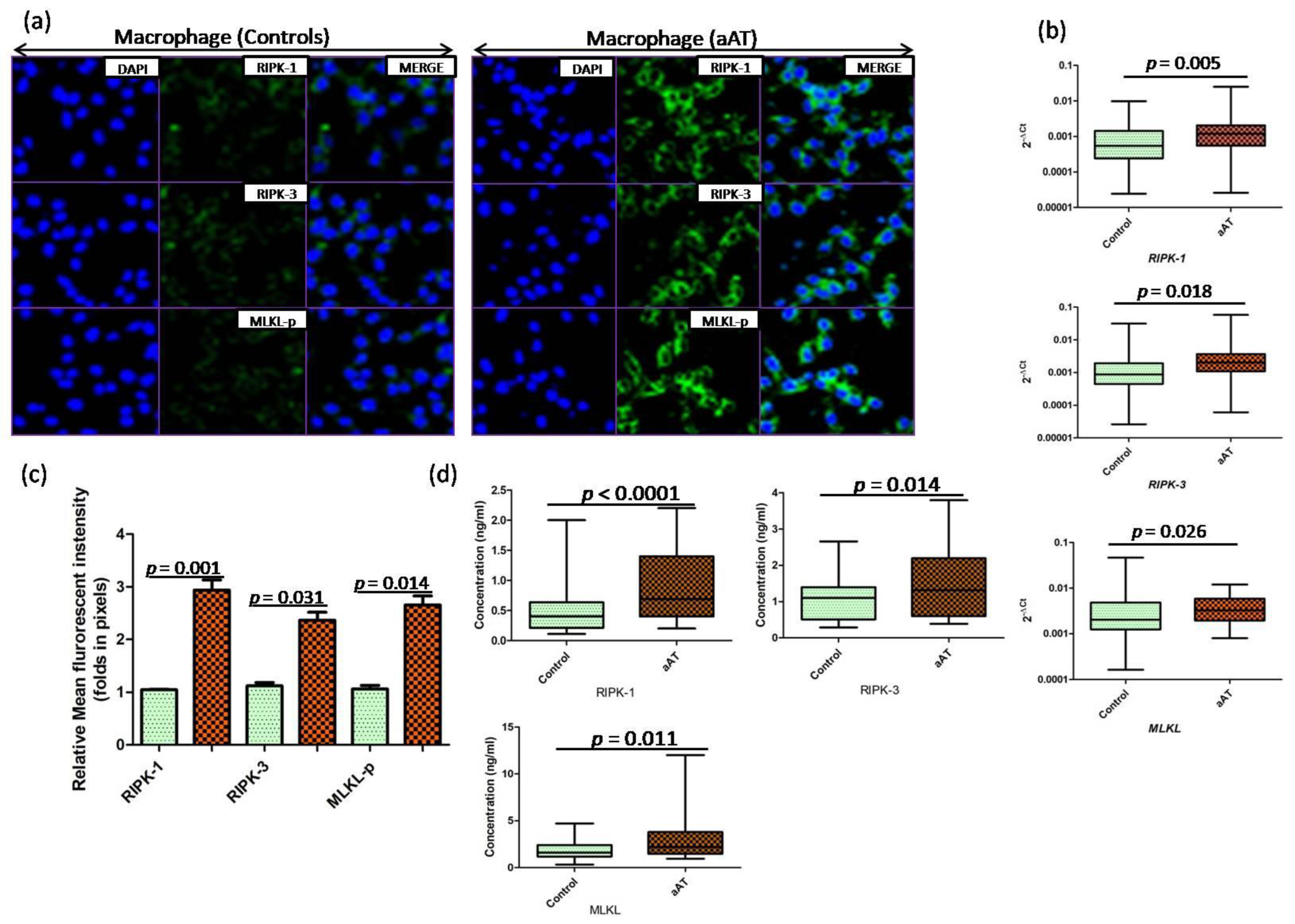
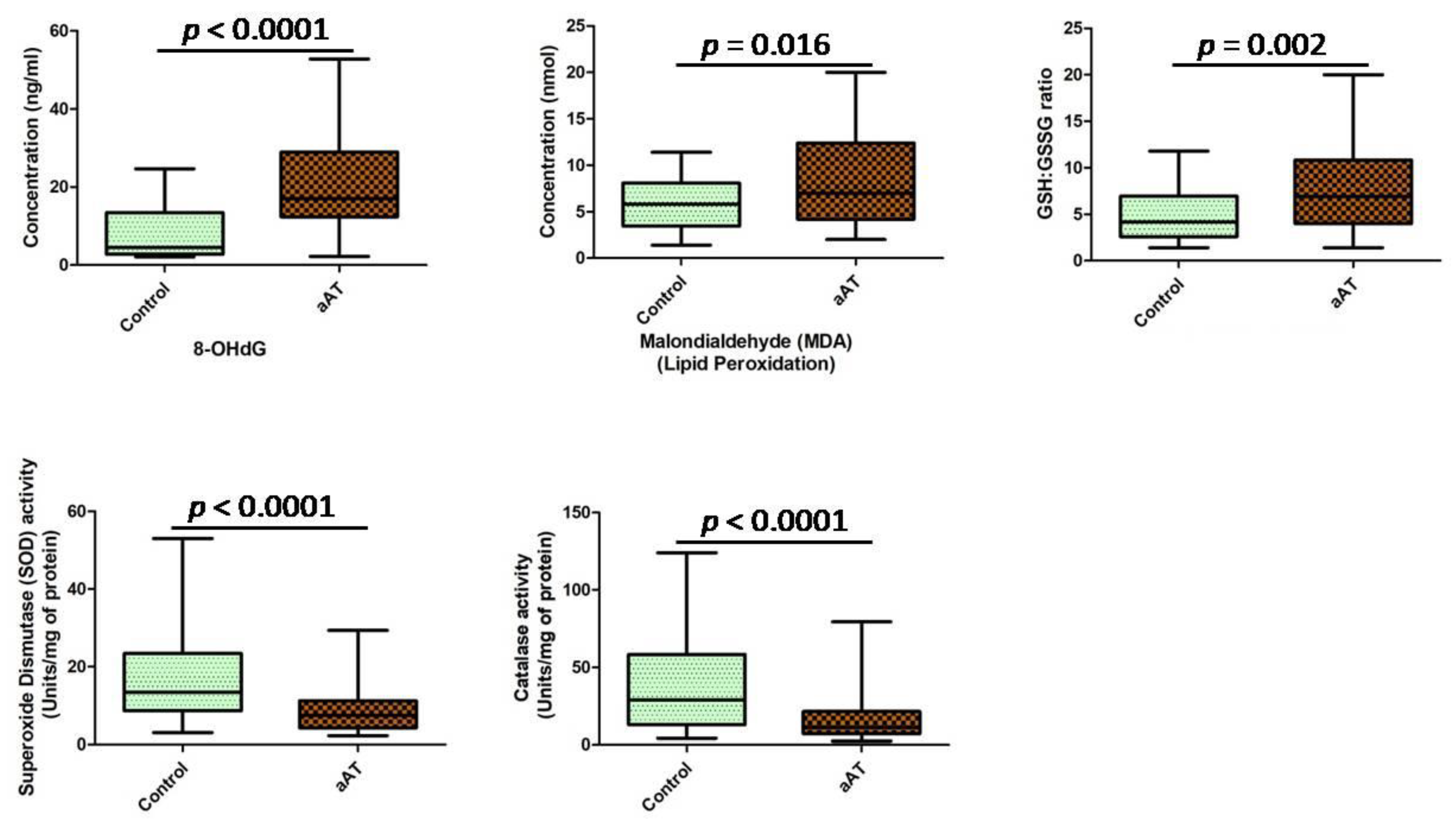
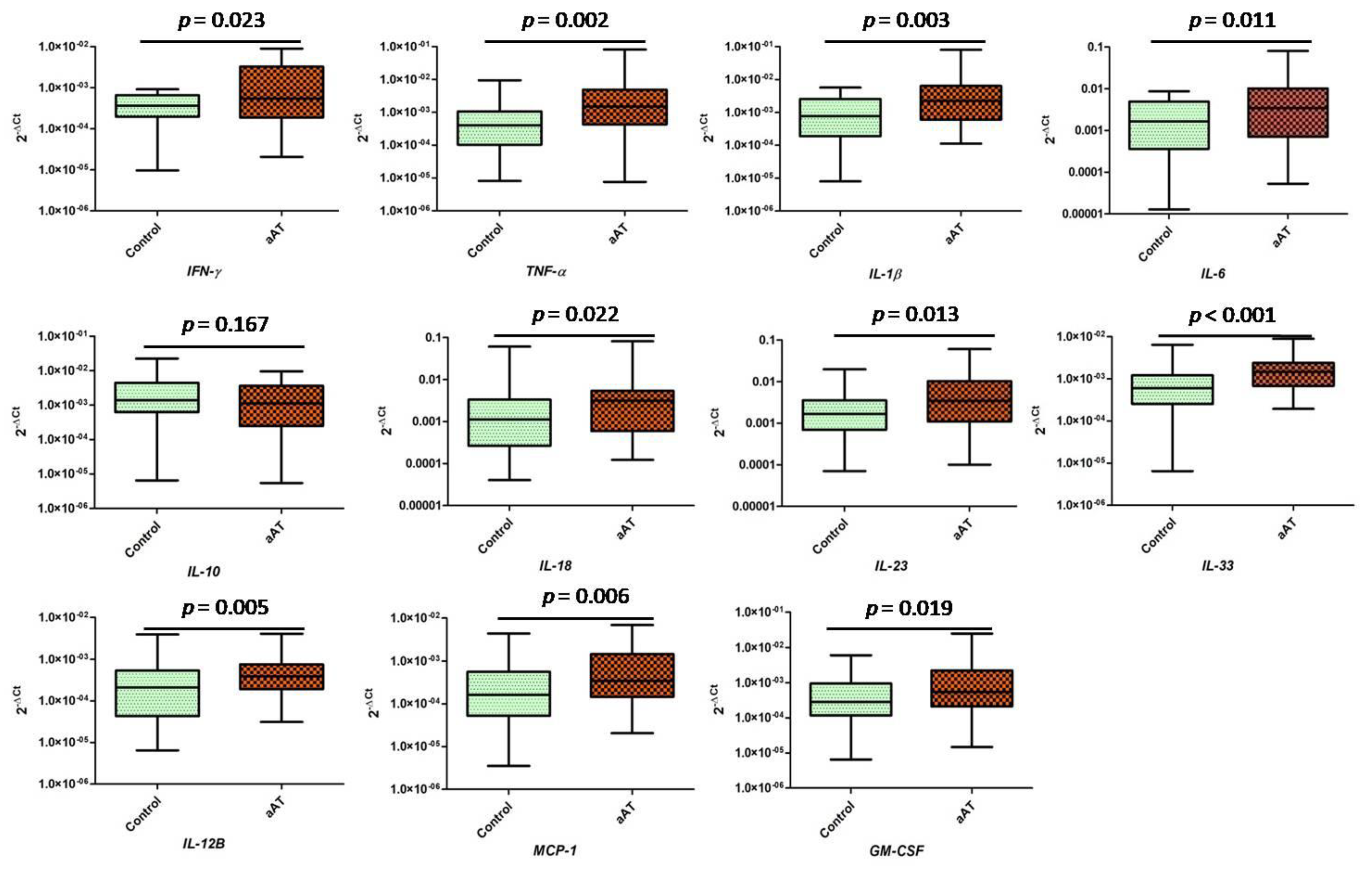
3. Results
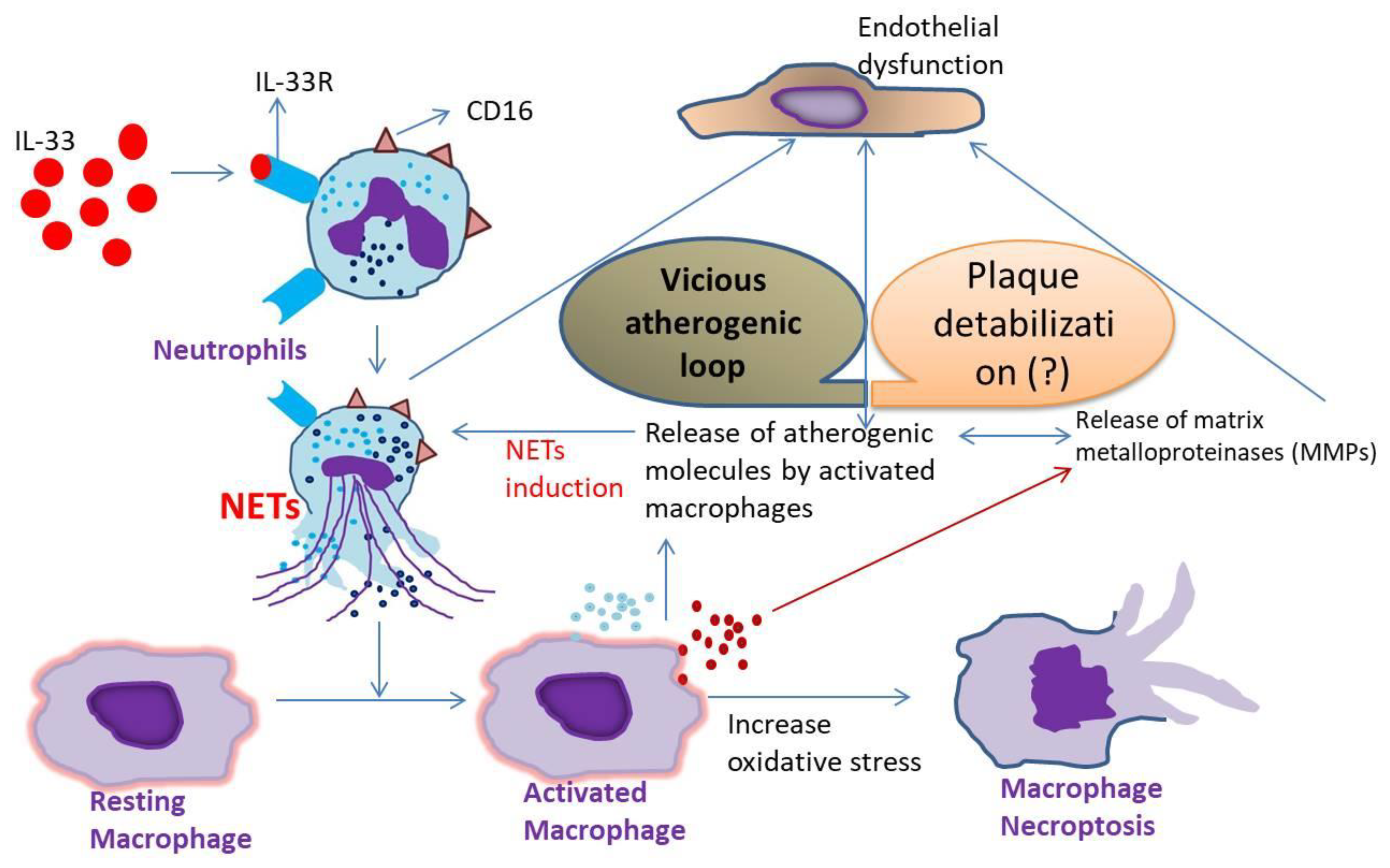
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Björkegren, J.L.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2021, 61, 194–211. [Google Scholar] [CrossRef]
- Klopf, J.; Brostjan, C.; Eilenberg, W.; Neumayer, C. Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. Int. J. Mol. Sci. 2021, 22, 559. [Google Scholar] [CrossRef]
- Nguyen, S.D.; Maaninka, K.; Mäyränpää, M.I.; Baumann, M.; Soliymani, R.; Lee-Rueckert, M.; Jauhiainen, M.; Kovanen, P.T.; Öörni, K. Neutrophilproteinase 3—An LDL- and HDL-proteolyzing enzyme with a potential to contribute to cholesterol accumulation in human atherosclerotic lesions. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2022, 1867, 159225. [Google Scholar] [CrossRef]
- Brinkmann, V.; Zychlinsky, A. Beneficial suicide: Why neutrophils die to make NETs. Nat. Rev. Genet. 2007, 5, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Zychlinsky, A. NETs: A new strategy for using old weapons. Trends Immunol. 2009, 30, 513–521. [Google Scholar] [CrossRef]
- Aulik, N.A.; Hellenbrand, K.M.; Czuprynski, C.J. Mannheimiahaemolytica and Its Leukotoxin Cause Macrophage Extracellular Trap Formation by Bovine Macrophages. Infect. Immun. 2012, 80, 1923–1933. [Google Scholar] [CrossRef]
- Yousefi, S.; Gold, J.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schmid, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J.; et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008, 14, 949–953. [Google Scholar] [CrossRef]
- von KöCkritz-Blickwede, M.; Goldmann, O.; Thulin, P.; Heinemann, K.; Norrby-Teglund, A.; Rohde, M.; Medina, E. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 2008, 111, 3070–3080. [Google Scholar] [CrossRef]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Swirski, F.K. Neutrophil-macrophage communication in inflammation and atherosclerosis. Science 2015, 349, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef] [PubMed]
- Quinn, K.L.; Henriques, M.; Tabuchi, A.; Han, B.; Yang, H.; Cheng, W.-E.; Tole, S.; Yu, H.; Luo, A.; Charbonney, E.; et al. Human Neutrophil Peptides Mediate Endothelial-Monocyte Interaction, Foam Cell Formation, and Platelet Activation. Arter. Thromb. Vasc. Biol. 2011, 31, 2070–2079. [Google Scholar] [CrossRef] [PubMed]
- Wantha, S.; Alard, J.-E.; Megens, R.T.; van der Does, A.M.; Döring, Y.; Drechsler, M.; Pham, C.T.; Wang, M.-W.; Wang, J.-M.; Gallo, R.L.; et al. Neutrophil-Derived Cathelicidin Promotes Adhesion of Classical Monocytes. Circ. Res. 2013, 112, 792–801. [Google Scholar] [CrossRef]
- Liew, F.Y.; Pitman, N.I.; McInnes, I.B. Disease-associated functions of IL-33: The new kid in the IL-1 family. Nat. Rev. Immunol. 2010, 10, 103–110. [Google Scholar] [CrossRef]
- Martin, N.T.; Martin, M.U. Interleukin 33 is a guardian of barriers and a local alarmin. Nat. Immunol. 2016, 17, 122–131. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an Interleukin-1-like Cytokine that Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.-P. IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef]
- Hung, L.-Y.; Tanaka, Y.; Herbine, K.; Pastore, C.; Singh, B.; Ferguson, A.; Vora, N.; Douglas, B.; Zullo, K.; Behrens, E.M.; et al. Cellular context of IL-33 expression dictates impact on anti-helminth immunity. Sci. Immunol. 2020, 5, eabc6259. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Girard, J.-P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Uchida, A.M.; Lenehan, P.J.; Vimalathas, P.; Miller, K.C.; Valencia-Yang, M.; Qiang, L.; Canha, L.A.; Ali, L.R.; Dougan, M.; Garber, J.J.; et al. Tissue eosinophils express the IL-33 receptor ST2 and type 2 cytokines in patients with eosinophilic esophagitis. Allergy 2022, 77, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Neumann, K.; Schiller, B.; Tiegs, G. NLRP3 Inflammasome and IL-33: Novel Players in Sterile Liver Inflammation. Int. J. Mol. Sci. 2018, 19, 2732. [Google Scholar] [CrossRef]
- Eren, E.; Ellidag, H.Y.; Aydin, O.; Yilmaz, N. HDL functionality and crystal-based sterile inflammation in atherosclerosis. Clin. Chim. Acta 2015, 439, 18–23. [Google Scholar] [CrossRef]
- Stankovic, M.; Ljujic, B.; Babic, S.; Maravic-Stojkovic, V.; Mitrovic, S.; Arsenijevic, N.; Radak, D.; Pejnovic, N.; Lukic, M.L. IL-33/IL-33R in various types of carotid artery atherosclerotic lesions. Cytokine 2019, 120, 242–250. [Google Scholar] [CrossRef]
- Miller, A.M.; Xu, D.; Asquith, D.L.; Denby, L.; Li, Y.; Sattar, N.; Baker, A.H.; McInnes, I.B.; Liew, F.Y. IL-33 reduces the development of atherosclerosis. J. Exp. Med. 2008, 205, 339–346. [Google Scholar] [CrossRef]
- Moore, K.J.; Tabas, I. The Cellular Biology of Macrophages in Atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef]
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 2019, 10, 306. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. J. Clin. Investig. 2019, 4, e128834. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ren, Z.; Xu, W.; Jiang, Z. Necroptosis in atherosclerosis. Clin. Chim. Acta 2022, 534, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Laube, B.; Abu Abed, U.; Goosmann, C.; Zychlinsky, A. Neutrophil Extracellular Traps: How to Generate and Visualize Them. J. Vis. Exp. 2010, 36, e1724. [Google Scholar] [CrossRef]
- Tembhre, M.; Parihar, A.; Sharma, V.; Sharma, A.; Chattopadhyay, P.; Gupta, S. Alteration in regulatory T cells and programmed cell death 1-expressing regulatory T cells in active generalized vitiligo and their clinical correlation. Br. J. Dermatol. 2015, 172, 940–950. [Google Scholar] [CrossRef]
- Sun, Y.; Pavey, H.; Wilkinson, I.; Fisk, M. Role of the IL-33/ST2 axis in cardiovascular disease: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0259026. [Google Scholar] [CrossRef] [PubMed]
- Ghali, R.; Altara, R.; Louch, W.E.; Cataliotti, A.; Mallat, Z.; Kaplan, A.; Zouein, F.A.; Booz, G.W. IL-33 (Interleukin 33)/sST2 Axis in Hypertension and Heart Failure. Hypertension 2018, 72, 818–828. [Google Scholar] [CrossRef]
- McCarthy, P.C.; Phair, I.R.; Greger, C.; Pardali, K.; McGuire, V.A.; Clark, A.; Gaestel, M.; Arthur, J.S.C. IL-33 regulates cytokine production and neutrophil recruitment via the p38 MAPK-activated kinases MK2/3. Immunol. Cell Biol. 2019, 97, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Vecchié, A.; Abbate, A.; Montecucco, F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells 2020, 9, 231. [Google Scholar] [CrossRef]
- Demyanets, S.; Konya, V.; Kastl, S.P.; Kaun, C.; Rauscher, S.; Niessner, A.; Pentz, R.; Pfaffenberger, S.; Rychli, K.; Lemberger, C.E.; et al. Interleukin-33 Induces Expression of Adhesion Molecules and Inflammatory Activation in Human Endothelial Cells and in Human Atherosclerotic Plaques. Arter. Thromb. Vasc. Biol. 2011, 31, 2080–2089. [Google Scholar] [CrossRef]
- Olejarz, W.; Łacheta, D.; Kubiak-Tomaszewska, G. Matrix Metalloproteinases as Biomarkers of Atherosclerotic Plaque Insta-bility. Int. J. Mol. Sci. 2020, 21, 3946. [Google Scholar] [CrossRef] [PubMed]
- Buckley, M.L.; Williams, J.O.; Chan, Y.H.; Laubertová, L.; Gallagher, H.; Moss, J.W.E.; Ramji, D.P. The Interleukin-33-Mediated Inhibition of Expression of Two Key Genes Implicated in Atherosclerosis in Human Macrophages Requires MAP Kinase, Phosphoinositide 3-Kinase and Nuclear Factor-κB Signaling Pathways. Sci. Rep. 2019, 9, 11317. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Parameters | Advanced Atherosclerotic Patients | Healthy Controls |
|---|---|---|
| Total number of subjects | ||
| Age in years (Mean ± standard deviation (SD)) | n = 52 (58.9 ± 21.4) | n = 52 (55.5 ± 22.5) |
| Number of Male (Age in years, Mean ± SD) = | n = 43 (58.6 ± 25.8) | n = 42 (52.7 ± 21.2) |
| Number of Female (Age in years, Mean ± SD) = | n = 9 (61.6 ± 26.3) | n = 10 (48.70 ± 22.4) |
| Hypertension | n = 21 (Male = 16; Female= 5) | None |
| Diabetes Mellitus | n = 16 (Male = 12; Female= 4) | None |
| Family history of cardiovascular diseases | n = 12 (Male = 10; Female= 2) | None |
| Percentage of carotid stenosis (>70%) | n = 52 | None (No symptoms of angina and any other cardiovascular diseases, normal electrocardiogram) |
| Cholesterol (mg/dL) | 123.1 ± 58.5 | 113.4 ± 28.2 |
| Triglyceride | 148 ± 67.3 | 111 ± 39.1 |
| LDL | 82.8 ± 27.6 | 44.2 ± 21.6 |
| VLDL | 16.8 ± 8.9 | 14.2 ± 7.9 |
| HDL | 35.5 ± 8.1 | 57.3 ± 28.5 |
| Genes | Gene Accession Number | Forward Primers (5′-3′) Reverse Primers (5′-3′) |
|---|---|---|
| β-ACTIN | NM_001101 | GCGTGACATTAAGGAGAAG GAAGGAAGGCTGGAAGAG |
| IFN-γ | NM_000619 | GCAGAGCCAAATTGTCTCCT ATGCTCTTCGACCTCGAAAC |
| TNF-α | NM_000594 | CCATCAGAGGGCCTGTACCT GTGGGTGAGGAGTACATGGG |
| IL-1β | NM_000576 | CCAAACCTCTTCGAGGCACA AGCCATCATTTCACTGGCGA |
| IL-6 | NM_001371096 | CCACCGGGAACGAAAGAGAA TCTCCTGGGGGTATTGTGGA |
| IL-10 | NM_000572 | TCTCCGAGATGCCTTCAGCAGA TAGCATCTCGGCTGGACTTCGA |
| IL-12B | NM_002187 | TCCCTGGTTTTTCTGGCATCT CATTTCTCCAGGGGCATCCG |
| IL-18 | NM_001386420 | GCTGAAGATGATGAAAACCTGGA GAGGCCGATTTCCTTGGTCA |
| IL-23 | NM_016584 | CCCAAGGACTCAGGGACAAC TGGAGGCTGCGAAGGATTTT |
| IL-33 | NM_033439 | AATCAGGTGACGGTGTTG ACACTCCAGGATCAGTCTTG |
| IL-33R (ST-2/IL1RL1) | NM_016232 | ATGTTCTGGATTGAGGCCAC GACTACATCTTCTCCAGGTAGCAT |
| GM-CSF | NM_000758 | AATGTTTGACCTCCAGGAGCC TCTGGGTTGCACAGGAAGTTT |
| MCP-1 | NM_002982 | GACCATTGTGGCCAAGGAGA TTGGGTTTGCTTGTCCAGGT |
| MPO | NM_000250 | TTTGACAACCTGCACGATGAC CGGTTGTGCTCCCGAAGTAA |
| RIPK1 | NM_003804 | GGAGACTAGGTGGCAGGGT CCAGTTCTGCACTCTCCAGG |
| RIPK3 | NM_006871 | TGGCCCCAGAACTGTTTGTT GGATCCCGAAGCTGTAGACG |
| MLKL | NM_152649 | GAGGGCACTGGACAGAAACA ACTCTGCTGACTGTACCGGA |
| CD16 | NM_000569 | ATCTTCAAGCAGGGAAGCCC TGTTGCTTTGCTGTGAGGGA |
| NLRP1 | NM_033004 | GGACCAGTATCGAGAGCAGC GAGGTGAGGATGGGTCTCCT |
| NLRP3 | NM_183395 | CCTGAGCAGCCTCATCAGAA GCAAGTGCTGCAGTTTCTCC |
| NLRC4 | NM_001199138 | GAACTCGAGGCCTCACTGAA GGGCTCGGCTATTGTCCTTT |
| AIM2 | NM_004833 | TAGGTTATTTGGGCATGCTCTC ACAACTTTGGGATCAGCCTCC |
| TIMP1 | NM_003254 | GGGGACACCAGAAGTCAACC GGGTGTAGACGAACCGGATG |
| TIMP2 | NM_003255 | CAGCTTTGCTTTATCCGGGC ATGCTTAGCTGGCGTCACAT |
| TIMP3 | NM_000362 | ATGGCAAGATGTACACGGGG ATGCAGGCGTAGTGTTTGGA |
| TIMP4 | NM_003256 | CTGCCTCCCAAACCCCATTA ACATTCGCCATTTCTCCCCT |
| MMP-1 | NM_00242 | CTGTTCTGGGGTGTGGTGTC GGGCCACTATTTCTCCGCTT |
| MMP-2 | NM_001127891 | TGTGTTGTCCAGAGGCAATG ATCACTAGGCCAGCTGGTTG |
| MMP-3 | NM_002422 | AAAGACAGGCACTTTTGGCG CTTCATATGCGGCATCCACG |
| MMP-7 | NM_002423 | TACCCATTTGATGGGCCAGG AGACTGCTACCATCCGTCCA |
| MMP-9 | NM_004994 | TTCCAAACCTTTGAGGGCGA CTGTACACGCGAGTGAAGGT |
| MMP-12 | NM_002426 | AACCAACGCTTGCCAAATCC TTTCCCACGGTAGTGACAGC |
| MMP-13 | NM_002427 | GCACTTCCCACAGTGCCTAT AGTTCTTCCCTTGATGGCCG |
| MMP-14 | NM_004995 | CCGATGTGGTGTTCCAGACA TCGTATGTGGCATACTCGCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tembhre, M.K.; Sriwastva, M.K.; Hote, M.P.; Srivastava, S.; Solanki, P.; Imran, S.; Lakshmy, R.; Sharma, A.; Jaiswal, K.; Upadhyay, A.D. Interleukin-33 Induces Neutrophil Extracellular Trap (NET) Formation and Macrophage Necroptosis via Enhancing Oxidative Stress and Secretion of Proatherogenic Factors in Advanced Atherosclerosis. Antioxidants 2022, 11, 2343. https://doi.org/10.3390/antiox11122343
Tembhre MK, Sriwastva MK, Hote MP, Srivastava S, Solanki P, Imran S, Lakshmy R, Sharma A, Jaiswal K, Upadhyay AD. Interleukin-33 Induces Neutrophil Extracellular Trap (NET) Formation and Macrophage Necroptosis via Enhancing Oxidative Stress and Secretion of Proatherogenic Factors in Advanced Atherosclerosis. Antioxidants. 2022; 11(12):2343. https://doi.org/10.3390/antiox11122343
Chicago/Turabian StyleTembhre, Manoj Kumar, Mukesh Kumar Sriwastva, Milind Padmakar Hote, Shikha Srivastava, Priyanka Solanki, Shafaque Imran, Ramakrishnan Lakshmy, Alpana Sharma, Kailash Jaiswal, and Ashish Datt Upadhyay. 2022. "Interleukin-33 Induces Neutrophil Extracellular Trap (NET) Formation and Macrophage Necroptosis via Enhancing Oxidative Stress and Secretion of Proatherogenic Factors in Advanced Atherosclerosis" Antioxidants 11, no. 12: 2343. https://doi.org/10.3390/antiox11122343
APA StyleTembhre, M. K., Sriwastva, M. K., Hote, M. P., Srivastava, S., Solanki, P., Imran, S., Lakshmy, R., Sharma, A., Jaiswal, K., & Upadhyay, A. D. (2022). Interleukin-33 Induces Neutrophil Extracellular Trap (NET) Formation and Macrophage Necroptosis via Enhancing Oxidative Stress and Secretion of Proatherogenic Factors in Advanced Atherosclerosis. Antioxidants, 11(12), 2343. https://doi.org/10.3390/antiox11122343