Three categories of Hyperparathyroidism (HPT) are recognized. The primary HPT is due to hypersecretion of PTH by one or more parathyroid glands, as a result of benign parathyroid adenoma (85%), primary parathyroid hyperplasia (10–20%) and, more rarely, is attributed to parathyroid carcinoma (1%) [
2]. The secondary HPT is due mainly to chronic renal insufficiency, and most of the patients develop the disease at the time that hemodialysis starts [
3]. In this condition, abnormalities in the renal tubular absorption of phosphate results in reduced phosphate excretion, and renal synthesis of vitamin D is also impaired. The association of hyperphosphatemia and reduced vitamin D levels result in hypocalcemia and, consequently, oversecretion of PTH, resulting in parathyroid hyperplasia [
4]. After restoration of renal disease by transplantation, the hyperplastic glands should return to normal functional states. However, in a small percentage of patients it does not occur, resulting in the rarest form of HPT, the tertiary HPT [
3].
The aim of this study is to report a clinical case of ossifying fibroma in the mandible associated with HPT-JT syndrome, which was characterized clinically and radiographically, treated with surgical resection as well as mandibular reconstruction in a second surgical intervention, followed by implant placement.
1. Case Report
A 46-year-old Caucasian female presented to the Oral Surgery Department at Alameda County Medical Center–Highland General Hospital, complaining of dental pain in maxillary left region for 2 months. Her medical history was significant for hypertension and congestive heart insufficiency for which she takes enalapril, carvedilol, furosemide, and Klor-Con (Klor- Con
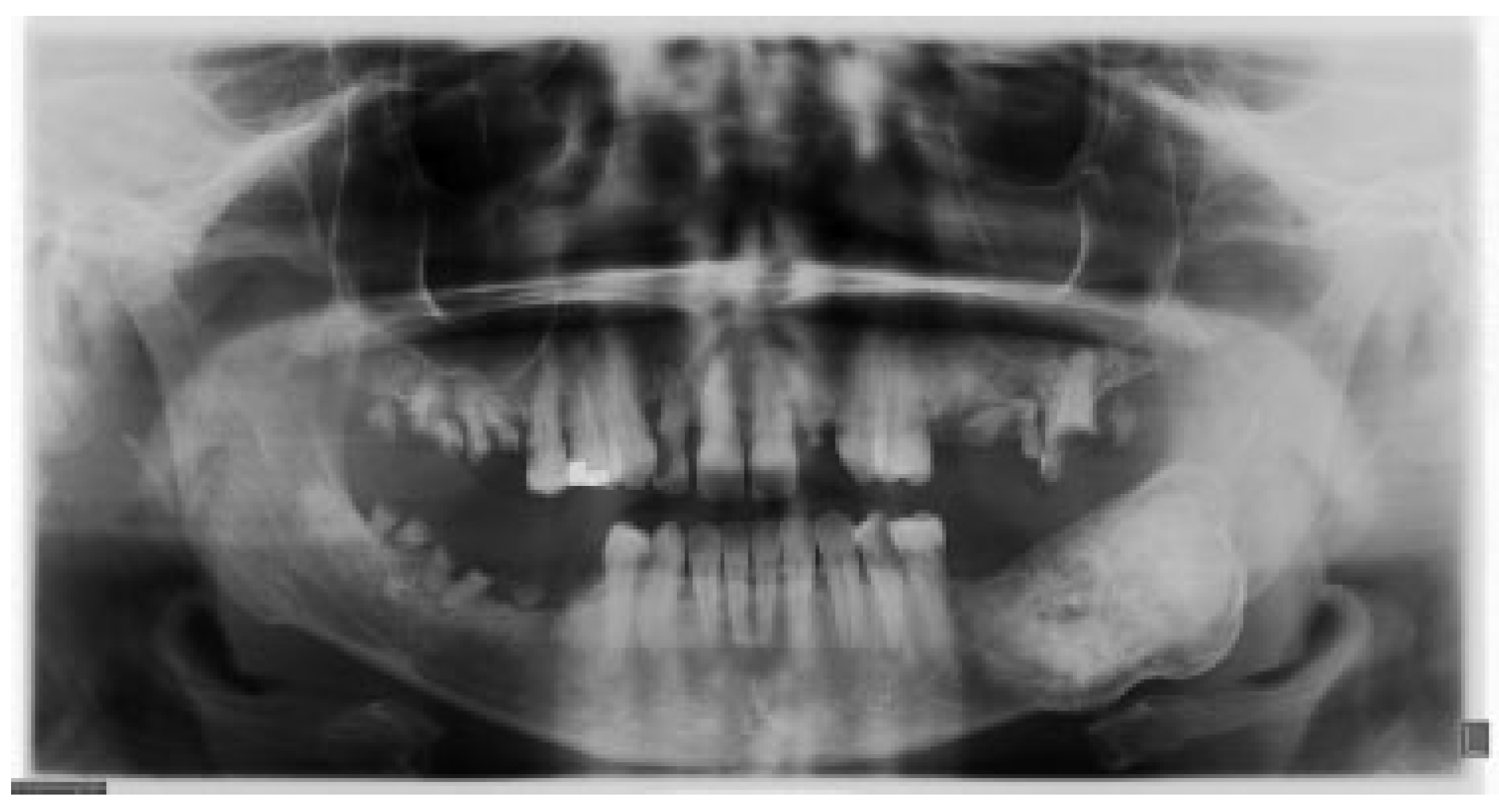
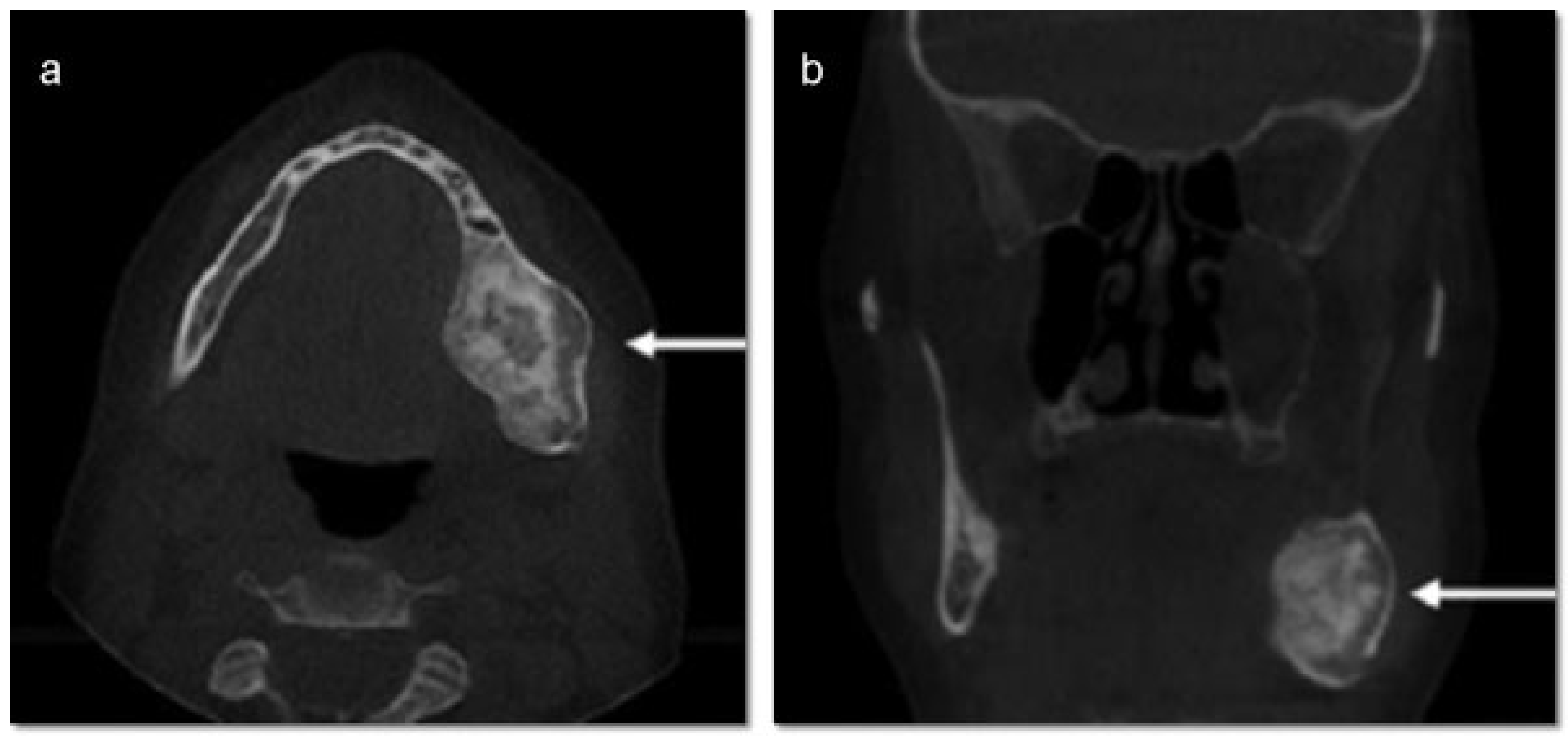
®, Upsher-Smith Laboratories, Inc., Morristown, NJ) (potassium chloride). She is allergic to codeine. She has a 10 pack- year history of smoking and a remote history of methamphetamine use. Her past surgical history is significant for parathyroid carcinoma, which resulted in primary HTP. She reported that time weight loss, hives, insomnia, fatigue, and bone pain before surgical excision performed 17 years ago. No uterine or kidney lesions were present. She also reported that her brother had a jaw tumor, not surgically treated, because the “jaw growth stopped.” Unfortunately, her brother does not live in the city, and a better investigation concerning his disease could not be performed. Extraoral examination revealed a small swelling in the left mandibular border. Intraorally, the patient presented with poor hygiene with multiple extensive dental caries and periodontal disease. An asymptomatic and firm posterior left mandibular swelling was also present, with buccal and lingual expansion. Radiographic and computed tomography (CT) scan examination revealed a 5-cm mixed radiolucent and radiopaque image in the left mandibular body with well-defined borders and base expansion (
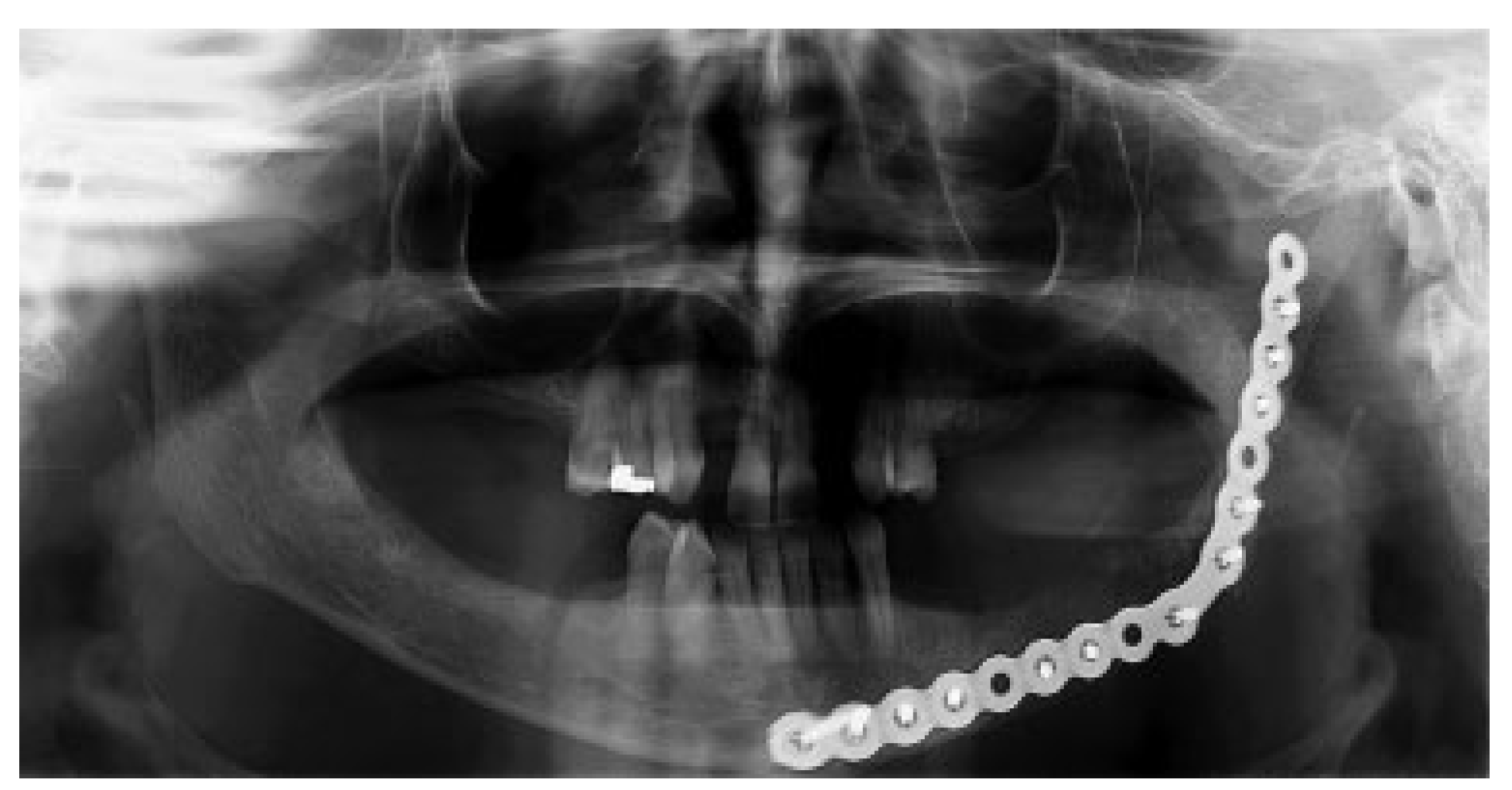
Figure 1 and
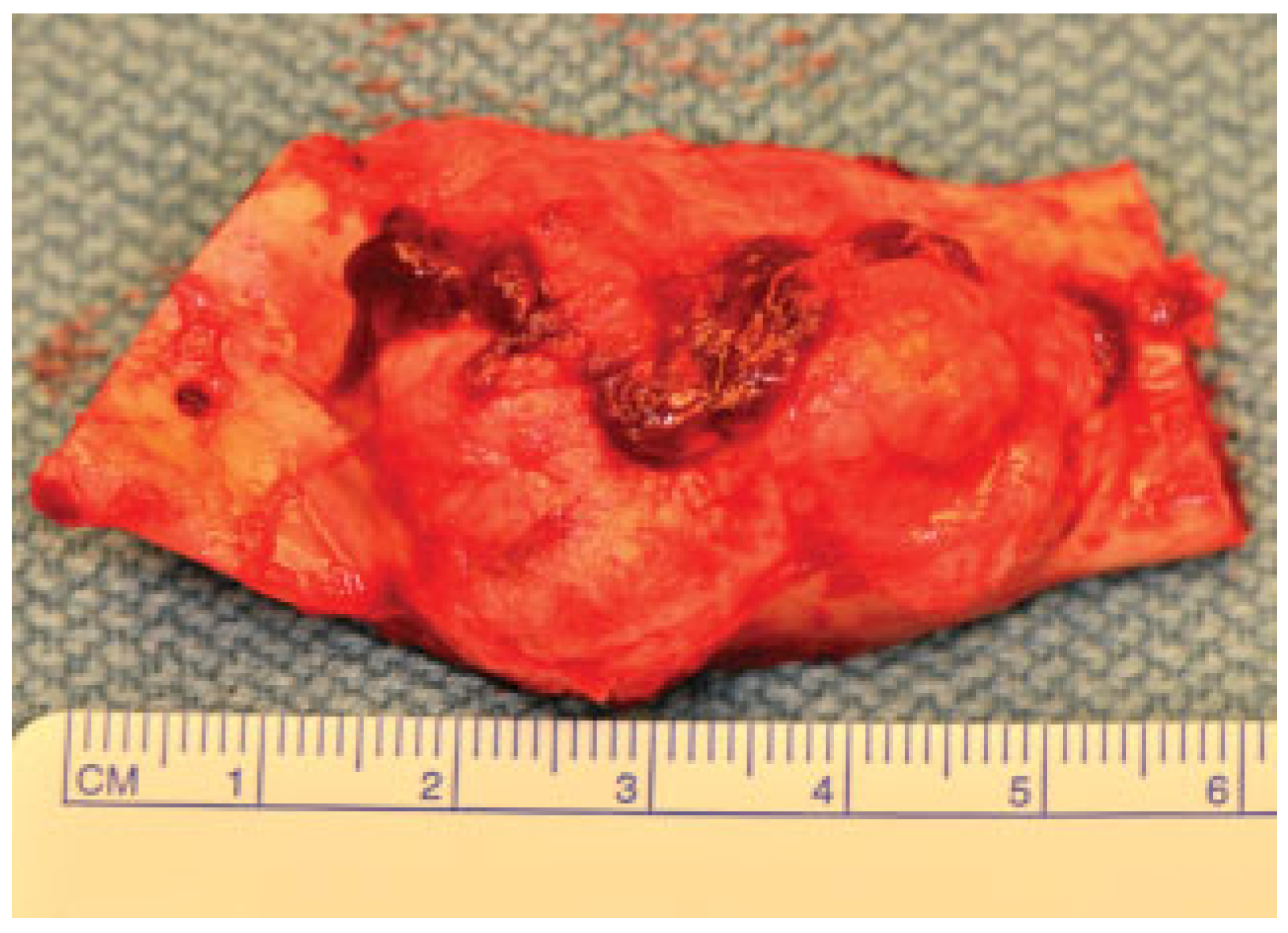
Figure 2a,b). An incisional biopsy was obtained with an intraoral approach which was diagnosed as a benign fibro-osseous lesion, consistent with ossifying fibroma. Initial laboratory analysis prior surgery showed a small increase in serum calcium of 10.5 mg/dL (8.4–10.2 mg/dL). Based on these findings and with the history of parathyroid carcinoma, the patient was referred to an endocrinologist. CT scan of the neck was unequivocal for recurrence and no correction of serum calcium was necessary. The association of ossifying fibroma with primary HPT due parathyroid carcinoma, with a family history of a similar jaw lesion, led to a high suspicion of HPT-JT syndrome. It was extensively discussed with the patient, but she refused to perform any genetic test. Our treatment planning initially included the extraction of the left inferior premolars, which had extensive caries lesions and mobility, performed before the lesion surgical removal. It would enable a better wound closure after resection. Three months later (2010 March), the ossifying fibroma was surgically resected with 5 mm margins (
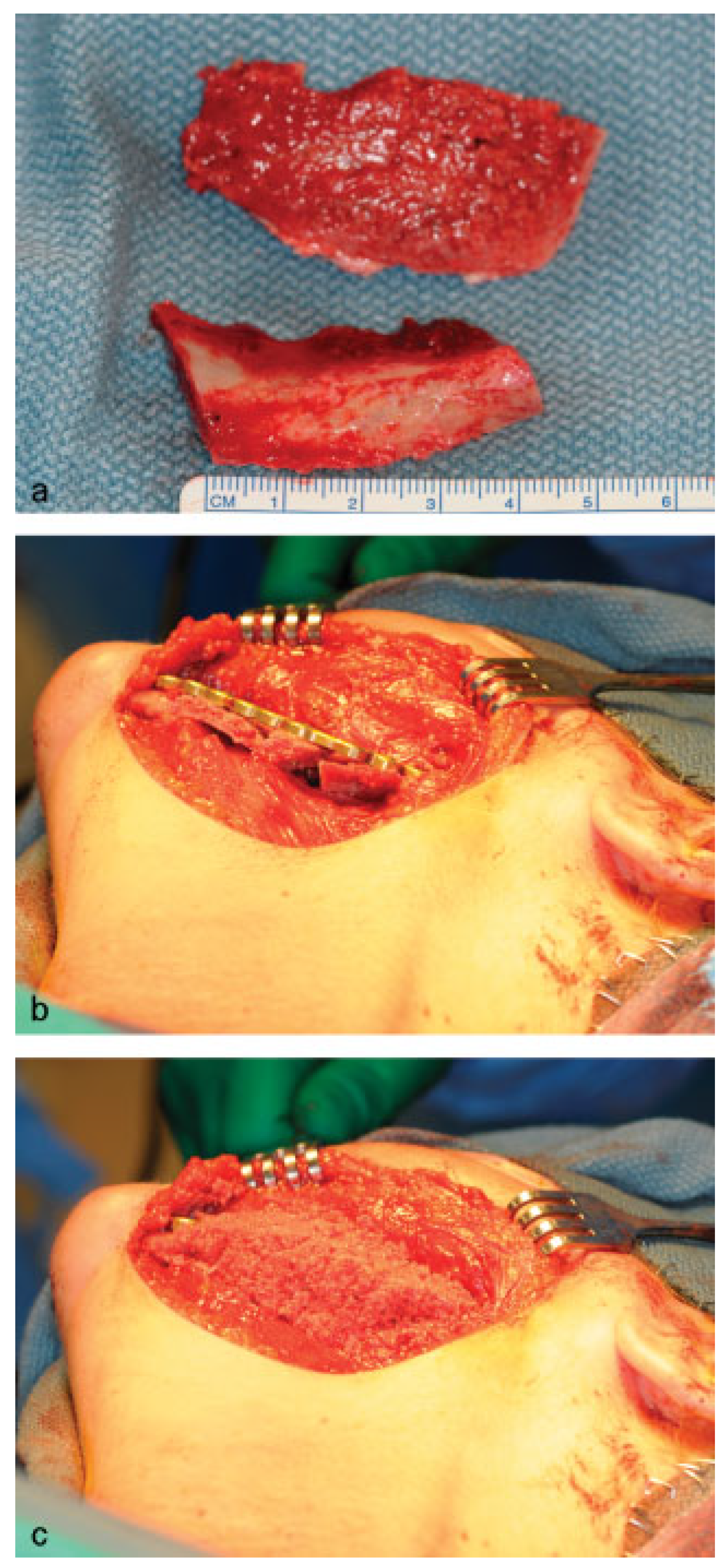
Figure 3) using an intraoral approach, and a 2.4-mm reconstruction plate was placed from the left mandibular ramus to the parasymphysis (
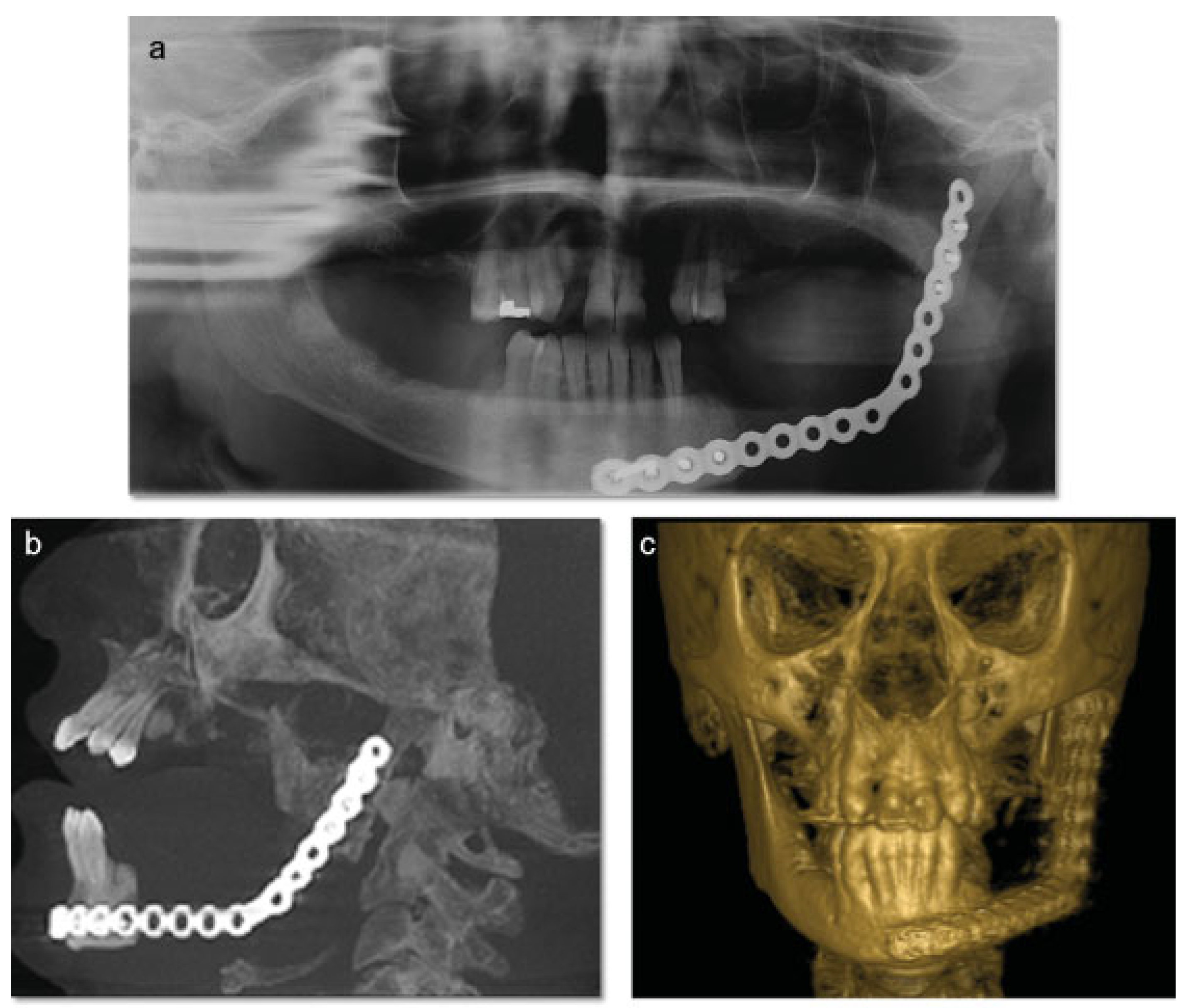
Figure 4a–c). The postoperative course was remarkable only for neurosensory disturbance in the left inferior alveolar nerve, which improved but was not completely resolved after 3 years of follow-up. Seven months later (2010 October), using an extraoral approach, the patient underwent a left mandibular reconstruction using her left posterior iliac crest, as well as the right anterior iliac crest. The corticocancellous blocks were secured with 2.4 mm monocortical screws, while cancellous bone chips were inserted between the block grafts (
Figure 5a–c). Although regular follow-ups were performed within the first 4 postoperative months, the patient recently returned to the oral surgery clinic for revaluation in 2012. Clinical and radiological examinations (
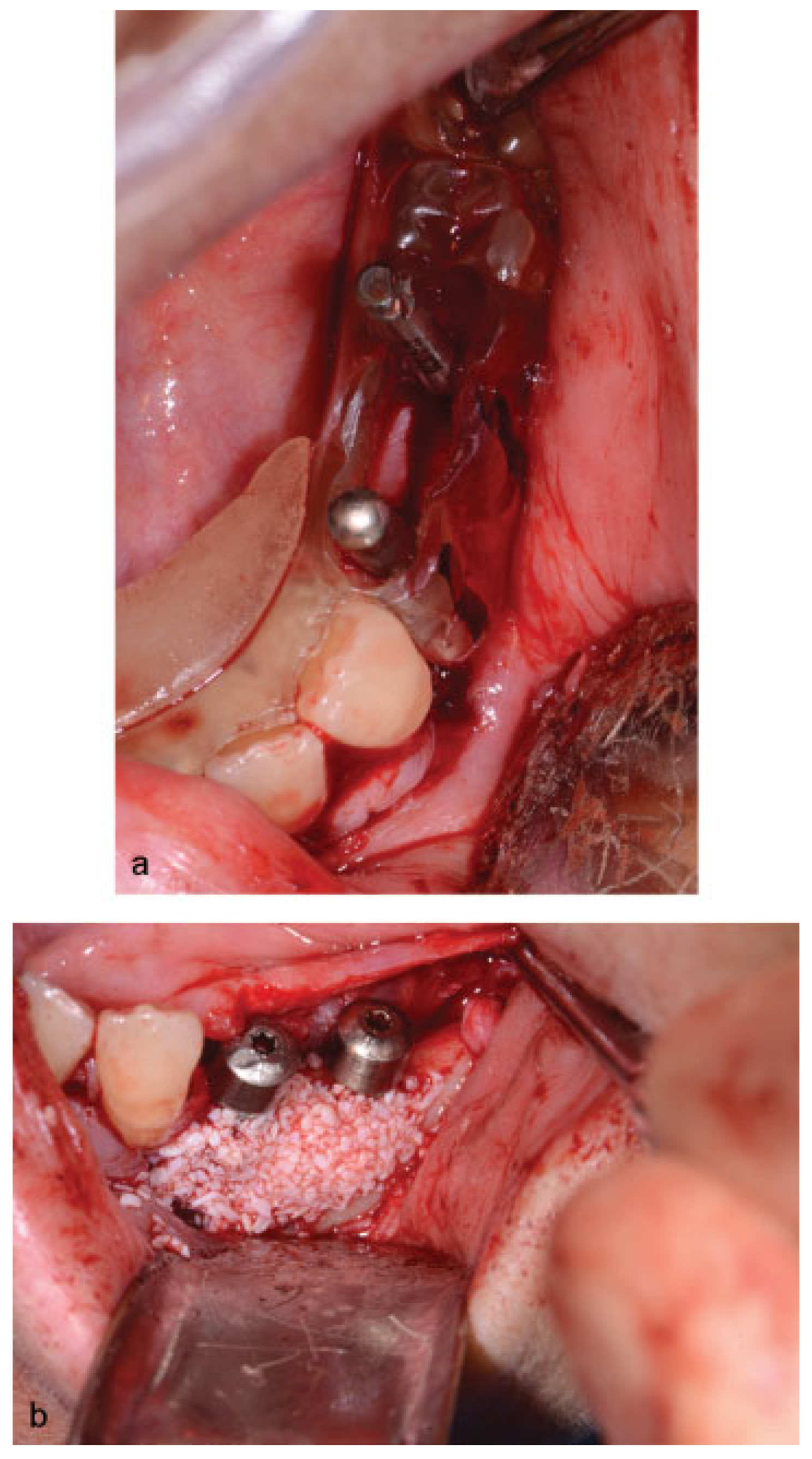
Figure 6) were performed, and oral rehabilitation with dental implants was planned (June 2012). The patient subsequently underwent a third surgical intervention, under local anesthetic and intravenous sedation. The remaining maxillary teeth were extracted, followed by osteoplasty and placement of four implants: two anterior axially placed and two posterior tilted placed. At the same operating time, monocortical screws were from the mandible and three implants were placed in the region of teeth 20, 21, and 28, and alloplastic bone graft was placed covering the exposed threads of the implants (
Figure 7a,b). A temporary superior and inferior prostheses were subsequently placed, and a period of 6 months for implant osseointegration before final prosthesis rehabilitation is being observed. The patient is currently followed regularly and, after 3 years of surgical resection, no evidenceof tumor recurrence is seen (
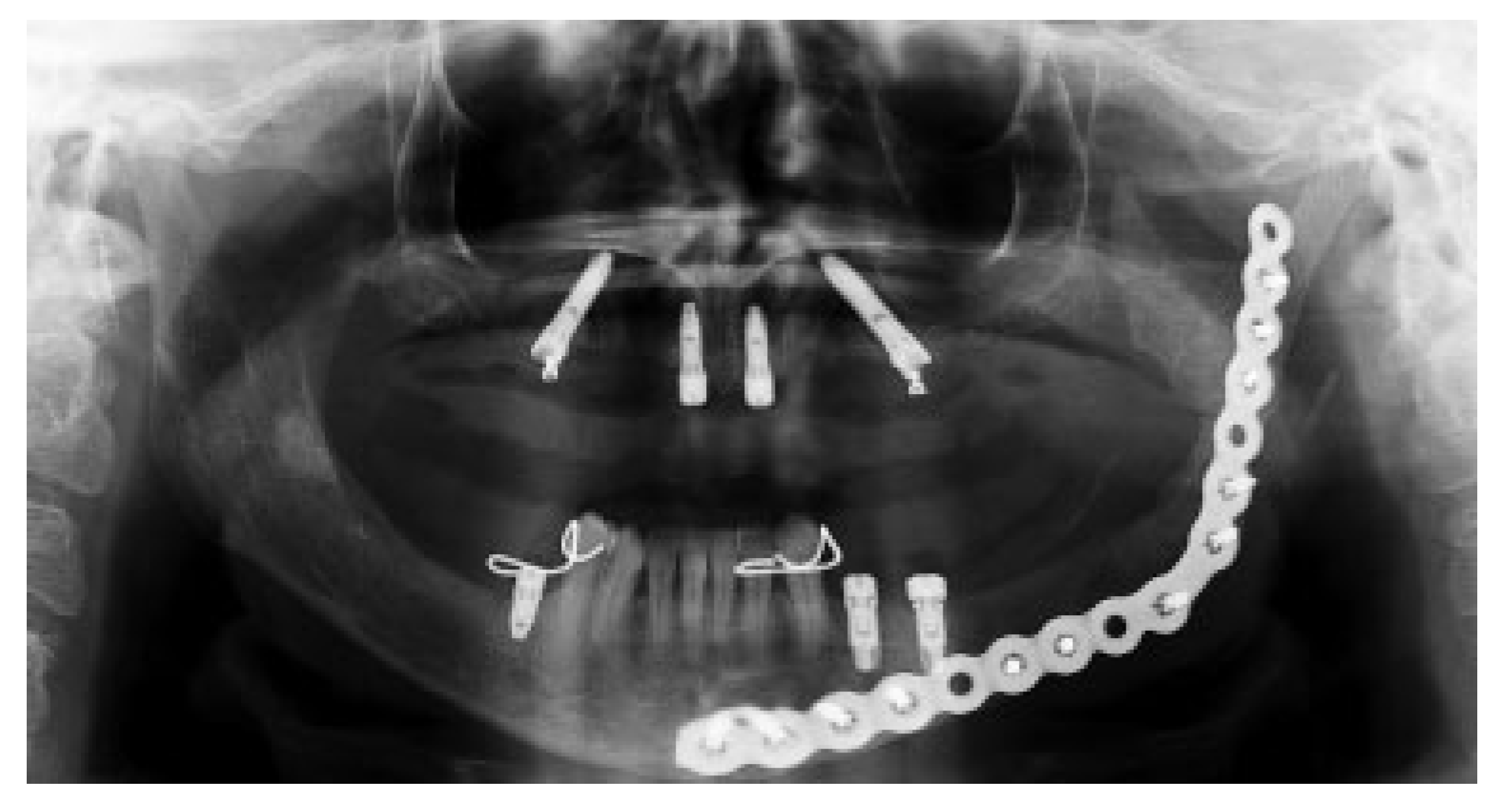
Figure 8). Therefore, no evidence of parathyroid carcinoma recurrence is seen after 21 years of endocrinological follow-up.
2. Discussion
The HTP-JT is an inherited syndrome with an autosomal dominant pattern, clinically and genetically distinct from other endocrine neoplasia syndromes [
8]. The pathogenesis of this hereditary condition has been associated with an endocrine tumor gene, the
HRPT2, mapped to chromosome 1– 1q21-q31 [
9]. It is a tumor suppressor gene that encodes a predicted protein of 531 amino acids, named parafibromin due to its involvement in the development of parathyroid tumors and ossifying fibromas [
10]. Despite these insights, the precise mechanisms in which the loss of that protein function leads to parathyroid cell transformation and other manifestations of HPT-JT syndrome remain to be understood [
2].
In primary HTP, laboratory analysis indicates increased PTH secretion and hypercalcemia. Patients with this disorder may suffer from symptoms of hypercalcemia, and may manifest consequences of excessive PTH action including bone pain, increased bone resorption, osteopenia, fractures, hypercalciuria, and nephrolithiasis. Bone densitometry indicates severe bone loss (cortical bone), mainly in the femoral neck [
2].
Although previous publications reported the association of HPT with jaw lesions in members of a same family, suggesting the existence of a clinically distinct syndrome [
11,
12,
13], the HPT-JT syndrome was first documented by Jackson et al. [
14] The author reported multiple cases of HPT and jaw tumors in two generations of the same family, which progressed after surgical correction of the hypercalcemia. This autosomal disorder has a reduced penetrance in females [
15], most commonly found in the third decade of life, while it is unlikely to present before 7 years of age [
16], and carries a serious risk of morbidity and mortality due to severe hypercalcemia. Although some clinical reports were published, the incidence and prevalence of this entity remain unknown [
8]. In the case report presented here, the patient was female and was 29 years old when the diagnosis of primary HPT was performed. It is speculated that the jaw lesion may have been presented at the time of diagnosis even though the lesion was not clinically detected until almost two decades later, when the patient sought for dental care.
The essential features of this syndrome are characterized by neoplastic and/or cystic lesions in two main organs: the parathyroid glands and the jaws, although some families have also kidneys’ abnormalities. The tumors found in the parathyroid glands are usually adenomas, although malignancy can also be present [
17], with a markedly increased risk of parathyroid carcinoma (10–20%) versus 1% in nonsyndromic primary HPT [
2]. The renal lesions include hamartomas, Wilms tumor, renal cysts, and polycystic kidney disease [
17]. In women, uterine tumors can also be seen in up to 75%, both benign and malignant [
18]. The jaw tumors, which are found in the maxilla and in the mandible, can precede the development of hypercalcemia by several decades [
16]. It has been described as nonodontogenic fibro-osseous pathology, originate from periodontal ligament [
19], compound of fibrous tissue with varying amounts of calcified tissue within the lesion, resembling bone, cementum [
20]. Therefore, it can be designated as cementifying fibroma, ossifying fibroma, cemento-ossifying fibroma, and ossifying jaw fibroma [
17] and, for this reason, radiographically the lesions can be completely radiolucent, radiolucent with variable degrees of radiopacity, or totally radiopaque. Thus, the differential diagnoses involves radiolucent lesions, as ameloblastoma, cystic lesions, and giant cell granuloma; mixed lesions as cementomas, myxoma, and adenomatoid odontogenic tumor; and radiopaque lesions, as fibrous dysplasia [
20].
This lesion can be confused with the classical hyperpar-athyroid brown tumor, which may occur almost anywhere in the human skeleton [
9], although the occurrence in the maxilla [
21,
22,
23] and in the mandible has also been reported [
24,
25,
26]. They are histologically indistinguishable from central giant cell granuloma, a giant cell lesion uniquely seen in the jaws. The identification of this pathology should always prompt clinicians to assess serum calcium and PTH levels to determine whether an underlying biochemical abnormality exists [
27]. However, the bone lesions specific to HPT-JT are restricted to the maxilla and mandible [
9], and they are histologically distinct lesions, especially in the absence of giant cells [
14]. They have a fibro-osseous appearance, with radiological and pathological characteristics most consistent with ossifying fibroma, with a tendency of occurring in multiple locations [
17]. Moreover, while hyperparathyroid brown tumors tend to slowly resolve after correction of the endocrine disease [
28,
29,
30], the same does not occur when the pathology is related to HPT-JT [
14]. In our case report, the patient presented with parathyroid carcinoma, the rarest form of primary HPT, and jaw tumor. No renal or uterine abnormalities were present.
In terms of treatment, complete excision is recommended although recurrences are uncommon, even after enucleation or curettage [
27]. However, due to the mandibular base involvement, large buccal and lingual bone expansion and extensive mobility of the premolars in this case report prompted the decision of surgical resection of the lesion and placement of a 2.4-mm reconstruction plate from the left mandibular ramus to the parasymphysis to preserve the mandibular contour. Mandibular reconstruction with posterior and anterior iliac crest was performed in a second surgical intervention due the possibility of lesion recurrence, even uncommon.
Several organs and systems are involved in the clinical manifestations of primary HPT. In general, the patients present with weight loss, polydipsia, and neurological findings, such as depression, lethargy, weakness, recent memory loss, confusion, and insomnia. Renal involvement includes colic, hematuria, and polyuria, while gastrointestinal problems are characterized by constipation, anorexia, nausea, and emesis. Furthermore, hypertension, nephrocalcinosis, arthritis, peptic ulcer disease, and pancreatitis can also occur [
1]. Concerning biochemical investigations, the patient presents with hypercalcemia and hypophosphatemia, increased serum levels of alkaline phosphatase and PTH, and increased 24-hour urinary calcium estimation [
16]. As the calcium serum measurement has been part of routine laboratory screening in the United States and other parts of the world, the diagnoses is often made in individuals with few or no symptom [
2]. In the present case, the slightly increased serum calcium levels in the routine preoperative exams allowed us to better investigate the past medical history of the patient and, thus, we could achieve a working diagnosis of HPT-JT syndrome. The onset of HPT was at the age of 29, when a diagnosis of parathyroid carcinoma was performed. HPT was revealed by weight loss, weakness, bone and joint pain, and insomnia. Unfortunately, we did not have access to the patient’s medical records at the time of HPT correction, but the biochemical values were within the normal limits 17 years later, during our surgical intervention. The small elevation in serum calcium levels did not require pharmacologic correction.
A large number of genetic studies have been performed on familial HPT resulting in a better understanding of its pathogenesis and etiology [
9,
10,
16]. Direct sequencing of the full coding region of the
HRPT2 gene is the standard approach to DNA mutational testing in the diagnosis of HPT-JT syndrome. However, a negative result in a potential proband is not conclusive and even typical HPT-JT kindred can have a false-negative result in 30% of cases. Thus, when clinical suspicion of this condition is present, other exams can be valuable, such as jaw imaging and renal ultrasound. A negative DNA test provides more relevant information in an asymptomatic relative in a family with a known specific mutation, since its absence indicates that the individual did not inherit the predisposing allele. These tests can play a paramount role not only to patients’ diagnoses and treatment but also important and sometimes life-saving contributions to patients’ family management [
2]. Moreover, the diagnosis of HPT-JT is clinically important because of the chance of malignant development [
17]. Unfortunately, our patient refused to perform the DNA test. As the disease could not be confirmed genetically, our diagnosis was made based on the association of primary HPT due hyperparathyroid carcinoma and jaw tumor. Moreover, the possible involvement of her brother suggested a familial trait of the disease. Concerning the follow-up period in syndromic kindred, biochemical screening for serum ionized calcium is recommended at 15 years of age. For jaw lesions, panoramic radiographs are advisable once every 3 years at minimal and annual abdominal ultrasounds or CT scans should be performed to evaluate possible recurrences of kidney and uterine tumors. Given the risk of recurrence of parathyroid carcinoma, annual examinations should be performed [
31].
The diagnosis of HPT-JT is clinically important, especially because of the possible involvement of other family members and the chance of malignant disease developing [
17]. The case reported here presented a left ossifying fibroma and primary HPT due to parathyroid carcinoma. Although the genetic test had not been performed and the chance of occurrence does exist, this clinical picture was consistent with HPT-JT diagnosis, which knowledge is paramount for its identification and its correct treatment. As the jaw tumor is not an isolated lesion, multidisciplinary interaction should be performed for the diagnoses and treatment, including an endocrinologist, an oral surgeon, a head and neck surgeon (for surgical removal of parathyroid lesion), and an oncologist (for radio- therapy and chemotherapy treatment). Patients with kidney and uterine abnormalities should be referred to a nephrologist and gynecologist. Furthermore, a close follow-up due to the possibility of parathyroid disease recurrence and the potential of malignancy. Moreover, familial screening should be encouraged in the suspicion of this single entity, as an early diagnosis can improve the treatment outcomes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}