
Multi-Stage Temporal Convolution Network for COVID-19 Variant Classification


 , ,
, ,  , ,
, ,  ,
,
Abstract
1. Introduction
- Currently, many methods for classifying miRNAs rely on manual feature extraction to be successful. The two main types of methods used for analyzing pre-miRNAs, either focusing on their spatial or sequential structure, are ineffective. We propose a temporal convolution neural network to learn spatiotemporal relationships and accurately identify different variants of COVID-19.
- A sequence is represented by both labels and encoding, which keep track of nucleotide positions within the sequence. We convert this information into a numerical description of the nucleotide position within the sequence.
- We conduct a detailed ablation study on both deep learning architectures as well as machine learning algorithms for classifying DNA sequences based on a deep learning architecture.
- The proposed framework is validated on challenging COVID-19 sequences and achieves state-of-the-art results for classification.
2. Literature Review
3. Proposed Methodology
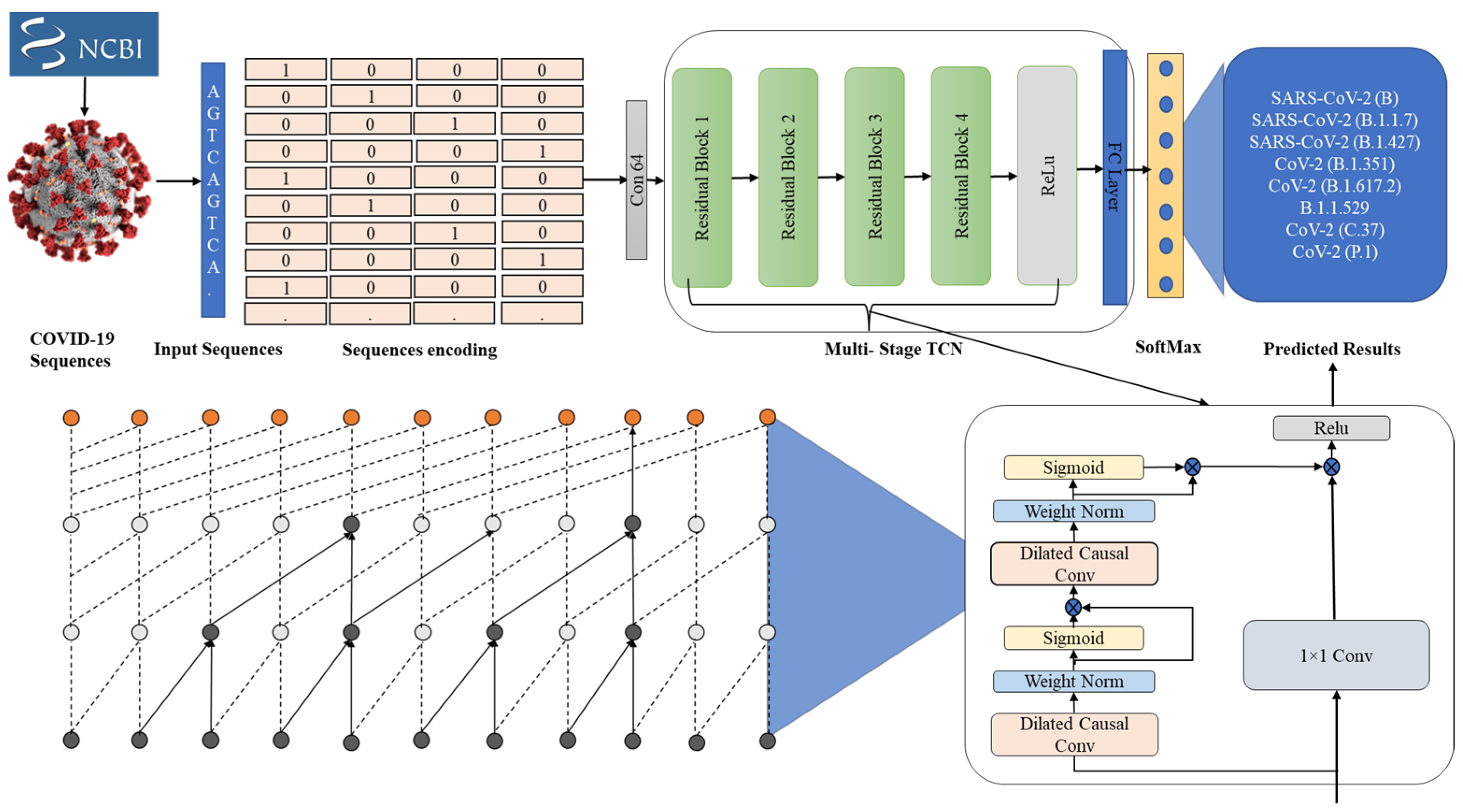
3.1. Genomic Sequence Data Collection
3.2. Genomic Sequence Data Preprocessing
3.3. Temporal Sequential Learning Mechanism for COVID-19 Variants
3.4. Proposed Model’s Objectives
4. Experimental Results
4.1. Method Evaluation
4.2. Model Implementation
4.3. Dataset
4.4. Results and Discussion
4.5. Comparison with State of the Art
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chan, J.F.-W.; Yuan, S.; Kok, K.-H.; To, K.K.-W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.-Y.; Poon, R.W.-S. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Paraskevis, D.; Kostaki, E.G.; Magiorkinis, G.; Panayiotakopoulos, G.; Sourvinos, G.; Tsiodras, S. Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect. Genet. Evol. 2020, 79, 104212. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by the novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Liu, X.; Wang, X.-J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genom. 2020, 47, 119. [Google Scholar] [CrossRef]
- Ozturk, T.; Talo, M.; Yildirim, E.A.; Baloglu, U.B.; Yildirim, O.; Acharya, U.R. Automated detection of COVID-19 cases using deep neural networks with X-ray images. Comput. Biol. Med. 2020, 121, 103792. [Google Scholar] [CrossRef]
- Yan, Q.; Weeks, D.E.; Xin, H.; Swaroop, A.; Chew, E.Y.; Huang, H.; Ding, Y.; Chen, W. Deep-learning-based prediction of late age-related macular degeneration progression. Nat. Mach. Intell. 2020, 2, 141–150. [Google Scholar] [CrossRef]
- Koohi-Moghadam, M.; Wang, H.; Wang, Y.; Yang, X.; Li, H.; Wang, J.; Sun, H. Predicting disease-associated mutation of metal-binding sites in proteins using a deep learning approach. Nat. Mach. Intell. 2019, 1, 561–567. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, M.; Du, X.; Villaruz, A.E.; Diep, B.A.; Wang, D.; Song, Y.; Tian, Y.; Hu, J.; Yu, F.; Lu, Y. MRSA epidemic linked to a quickly spreading colonization and virulence determinant. Nat. Med. 2012, 18, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A. Minimum information about an uncultivated virus genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef] [PubMed]
- Alimadadi, A.; Aryal, S.; Manandhar, I.; Munroe, P.B.; Joe, B.; Cheng, X. Artificial Intelligence and Machine Learning to Fight COVID-19; American Physiological Society Bethesda: Rockville, MD, USA, 2020; Volume 52, pp. 200–202. [Google Scholar]
- Randhawa, G.S.; Hill, K.A.; Kari, L. MLDSP-GUI: An alignment-free standalone tool with an interactive graphical user interface for DNA sequence comparison and analysis. Bioinformatics 2020, 36, 2258–2259. [Google Scholar] [CrossRef]
- Zeng, H.; Edwards, M.D.; Liu, G.; Gifford, D.K. Convolutional neural network architectures for predicting DNA-protein binding. Bioinformatics 2016, 32, i121–i127. [Google Scholar] [CrossRef]
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamani, A.; Telenti, A. A primer on deep learning in genomics. Nat. Genet. 2019, 51, 12–18. [Google Scholar] [CrossRef]
- Phan, D.; Ngoc, G.N.; Lumbanraja, F.R.; Faisal, M.R.; Abipihi, B.; Purnama, B.; Delimiyanti, M.K.; Kubo, M.; Satou, K. Combined use of k-mer numerical features and position-specific categorical features in fixed-length DNA sequence classification. J. Biomed. Sci. Eng. 2017, 10, 390–401. [Google Scholar] [CrossRef]
- Zhang, X.; Beinke, B.; Kindhi, B.A.; Wiering, M. Comparing machine learning algorithms with or without feature extraction for DNA classification. arXiv 2020, arXiv:2011.00485. [Google Scholar]
- Do, D.T.; Le, N.Q.K. Using extreme gradient boosting to identify origin of replication in Saccharomyces cerevisiae via hybrid features. Genomics 2020, 112, 2445–2451. [Google Scholar] [CrossRef]
- Xu, H.; Jia, P.; Zhao, Z. Deep4mC: Systematic assessment and computational prediction for DNA N4-methylcytosine sites by deep learning. Brief. Bioinform. 2021, 22, bbaa099. [Google Scholar] [CrossRef]
- Remita, A.M.; Diallo, A.B. Statistical linear models in virus genomic alignment-free classification: Application to hepatitis C viruses. In Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 18–21 November 2019; pp. 474–481. [Google Scholar]
- Lopez-Rincon, A.; Tonda, A.; Mendoza-Maldonado, L.; Mulders, D.G.; Molenkamp, R.; Perez-Romero, C.A.; Claassen, E.; Garssen, J.; Kraneveld, A.D. Classification and specific primer design for accurate detection of SARS-CoV-2 using deep learning. Sci. Rep. 2021, 11, 947. [Google Scholar] [CrossRef] [PubMed]
- Ullah, W.; Hussain, T.; Khan, Z.A.; Haroon, U.; Baik, S.W. Intelligent dual stream CNN and echo state network for anomaly detection. Knowl. -Based Syst. 2022, 253, 109456. [Google Scholar] [CrossRef]
- Ullah, W.; Ullah, A.; Hussain, T.; Muhammad, K.; Heidari, A.A.; Del Ser, J.; Baik, S.W.; De Albuquerque, V.H.C. Artificial Intelligence of Things-assisted two-stream neural network for anomaly detection in surveillance Big Video Data. Future Gener. Comput. Syst. 2022, 129, 286–297. [Google Scholar] [CrossRef]
- Feng, X.; Tustison, N.J.; Patel, S.H.; Meyer, C.H. Brain tumor segmentation using an ensemble of 3d u-nets and overall survival prediction using radiomic features. Front. Comput. Neurosci. 2020, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.A.; Ullah, W.; Ullah, A.; Rho, S.; Lee, M.Y.; Baik, S.W. An Adaptive Filtering Technique for Segmentation of Tuberculosis in Microscopic Images. In Proceedings of the 4th International Conference on Natural Language Processing and Information Retrieval, Seoul, Korea, 20 December 2020; pp. 184–187. [Google Scholar]
- Van Den Oord, A.; Dieleman, S.; Zen, H.; Simonyan, K.; Vinyals, O.; Graves, A.; Kalchbrenner, N.; Senior, A.W.; Kavukcuoglu, K. WaveNet: A generative model for raw audio. SSW 2016, 125, 2. [Google Scholar]
- Wei, S.-E.; Ramakrishna, V.; Kanade, T.; Sheikh, Y. Convolutional pose machines. In Proceedings of the IEEE conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 4724–4732. [Google Scholar]
- Newell, A.; Yang, K.; Deng, J. Stacked hourglass networks for human pose estimation. In Proceedings of the European Conference on Computer Vision, Amsterdam, The Netherlands, 11–14 October 2016; Springer: Berlin/Heidelberg, Germany, 2016; pp. 483–499. [Google Scholar]
- Dantone, M.; Gall, J.; Leistner, C.; Van Gool, L. Body parts dependent joint regressors for human pose estimation in still images. IEEE Trans. Pattern Anal. Mach. Intell. 2014, 36, 2131–2143. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Dollár, P.; Girshick, R.; He, K.; Hariharan, B.; Belongie, S. Feature pyramid networks for object detection. In Proceedings of the IEEE conference on computer vision and pattern recognition, Honolulu, HI, USA, 21–26 July 2017; pp. 2117–2125. [Google Scholar]
- Ding, L.; Xu, C. Weakly-supervised action segmentation with iterative soft boundary assignment. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Salt Lake City, UT, USA, 18–23 June 2018; pp. 6508–6516. [Google Scholar]
- Ullah, W.; Muhammad, K.; Haq, I.U.; Ullah, A.; Ullah Khattak, S.; Sajjad, M. Splicing sites prediction of human genome using machine learning techniques. Multimed. Tools Appl. 2021, 80, 30439–30460. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Backbone | Overall Accuracy (%) |
|---|---|---|
| DNC * + SVM [34] | Statistical patterns | 52.0 |
| TNC * + SVM [34] | Statistical patterns | 54.3 |
| TetraNC * + SVM [34] | Statistical patterns | 52.2 |
| CompositeNC * + SVM [34] | Statistical patterns | 57.3 |
| DeepBind [26] | Deep model | 61.3 |
| DeeperBind [26] | Deep model | 67.8 |
| Attention-CNN-LSTM | Deep model | 79.5 |
| Our CNN-LSTM | Deep model | 71.4 |
| Our CNN-GRU | Deep model | 71.1 |
| Our CNN-BDLSTM | Deep model | 74.8 |
| Our CNN-BDGRU | Deep model | 75.3 |
| Our proposed TCN | Deep model | 88.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, W.; Ullah, A.; Malik, K.M.; Saudagar, A.K.J.; Khan, M.B.; Hasanat, M.H.A.; AlTameem, A.; AlKhathami, M. Multi-Stage Temporal Convolution Network for COVID-19 Variant Classification. Diagnostics 2022, 12, 2736. https://doi.org/10.3390/diagnostics12112736
Ullah W, Ullah A, Malik KM, Saudagar AKJ, Khan MB, Hasanat MHA, AlTameem A, AlKhathami M. Multi-Stage Temporal Convolution Network for COVID-19 Variant Classification. Diagnostics. 2022; 12(11):2736. https://doi.org/10.3390/diagnostics12112736
Chicago/Turabian StyleUllah, Waseem, Amin Ullah, Khalid Mahmood Malik, Abdul Khader Jilani Saudagar, Muhammad Badruddin Khan, Mozaherul Hoque Abul Hasanat, Abdullah AlTameem, and Mohammed AlKhathami. 2022. "Multi-Stage Temporal Convolution Network for COVID-19 Variant Classification" Diagnostics 12, no. 11: 2736. https://doi.org/10.3390/diagnostics12112736
APA StyleUllah, W., Ullah, A., Malik, K. M., Saudagar, A. K. J., Khan, M. B., Hasanat, M. H. A., AlTameem, A., & AlKhathami, M. (2022). Multi-Stage Temporal Convolution Network for COVID-19 Variant Classification. Diagnostics, 12(11), 2736. https://doi.org/10.3390/diagnostics12112736