
Plasma-Assisted Synthesis of Surfactant-Free and D-Fructose-Coated Gold Nanoparticles for Multiple Applications
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials and Solution Preparations
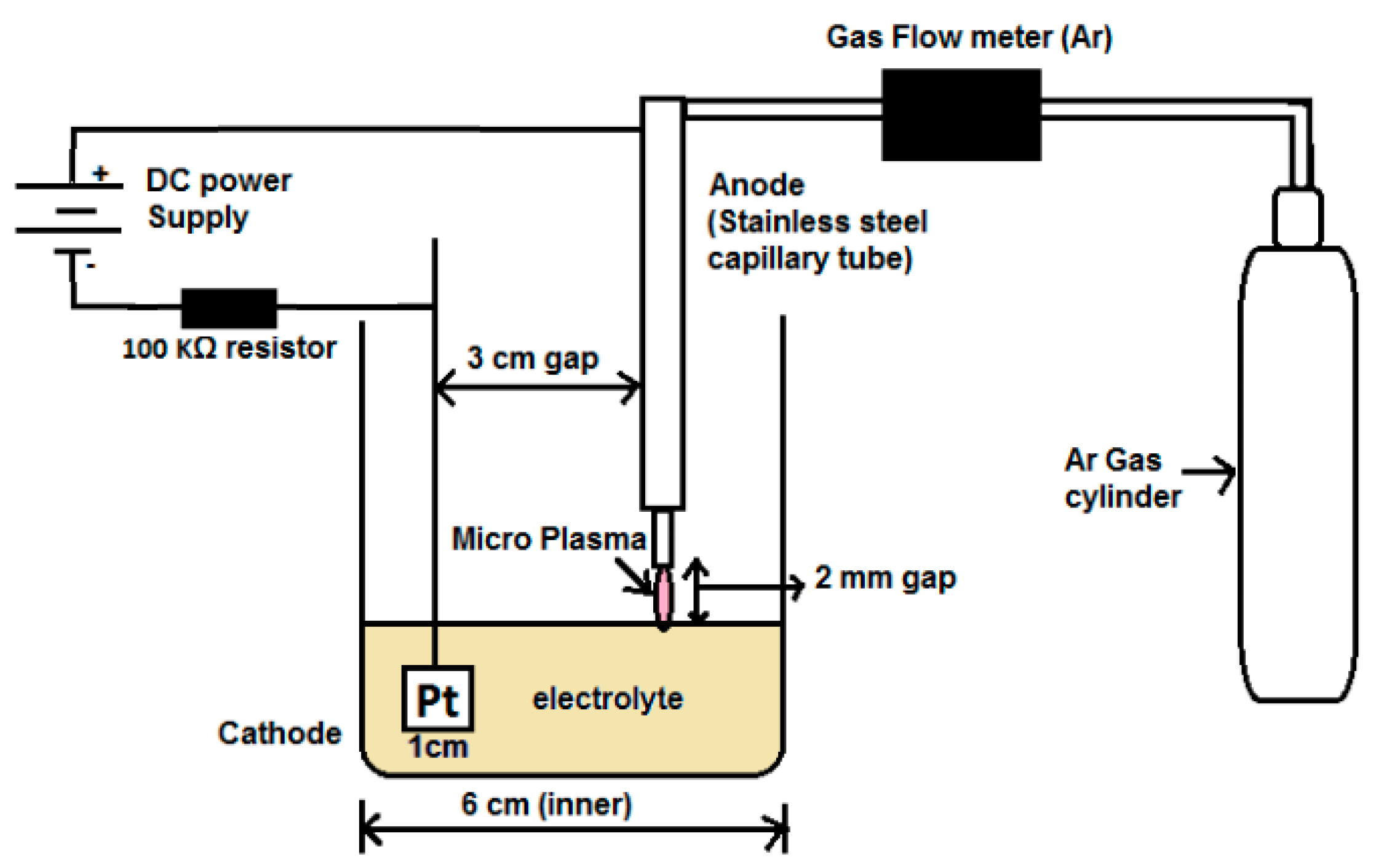
2.2. Experimental Setup
2.3. Synthesis and Characterization
2.4. SERS Analysis
2.5. Catalysis Experiments
3. Results and Discussion
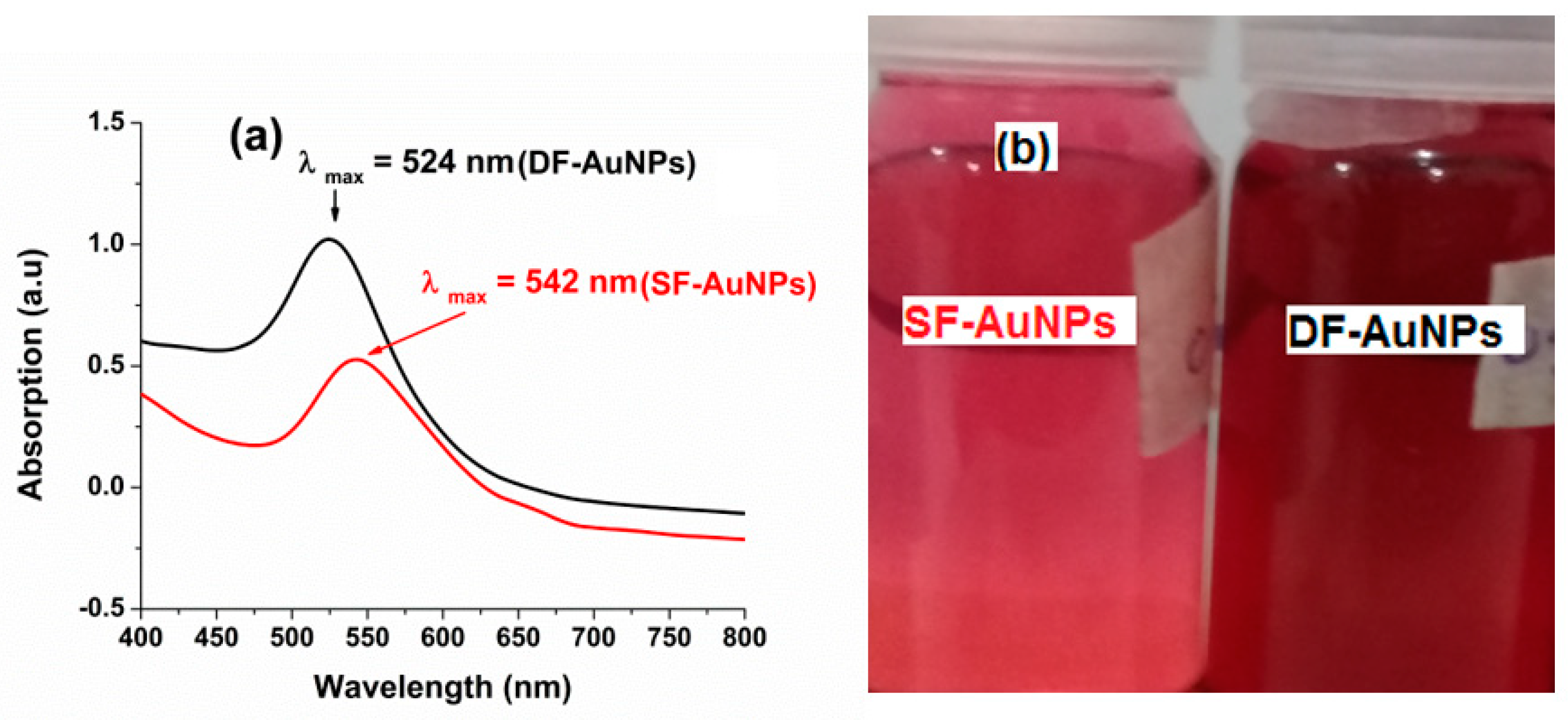
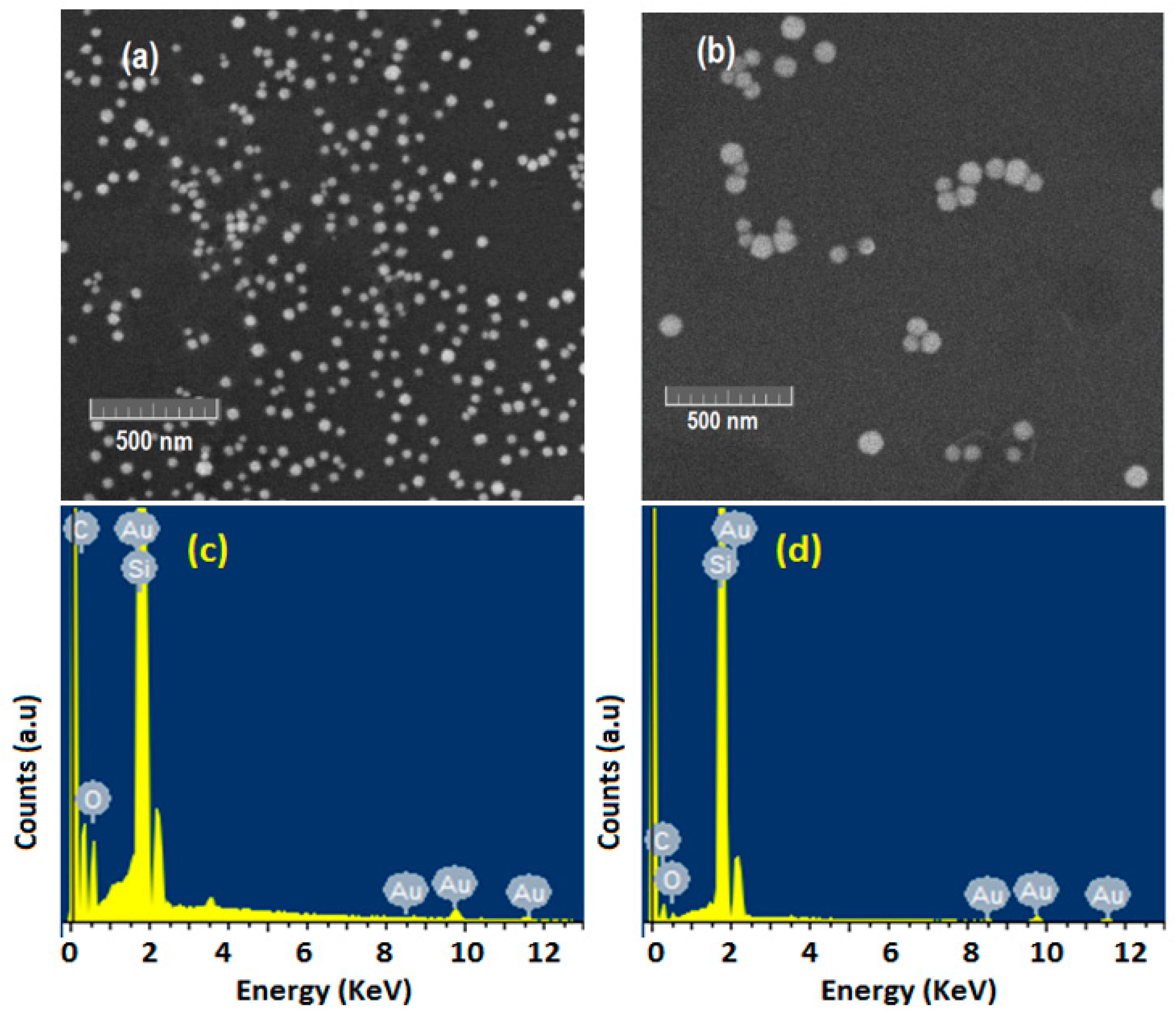
3.1. Synthesis and Characterization of and
3.2. SERSSF–AuNPsDF–AuNPs
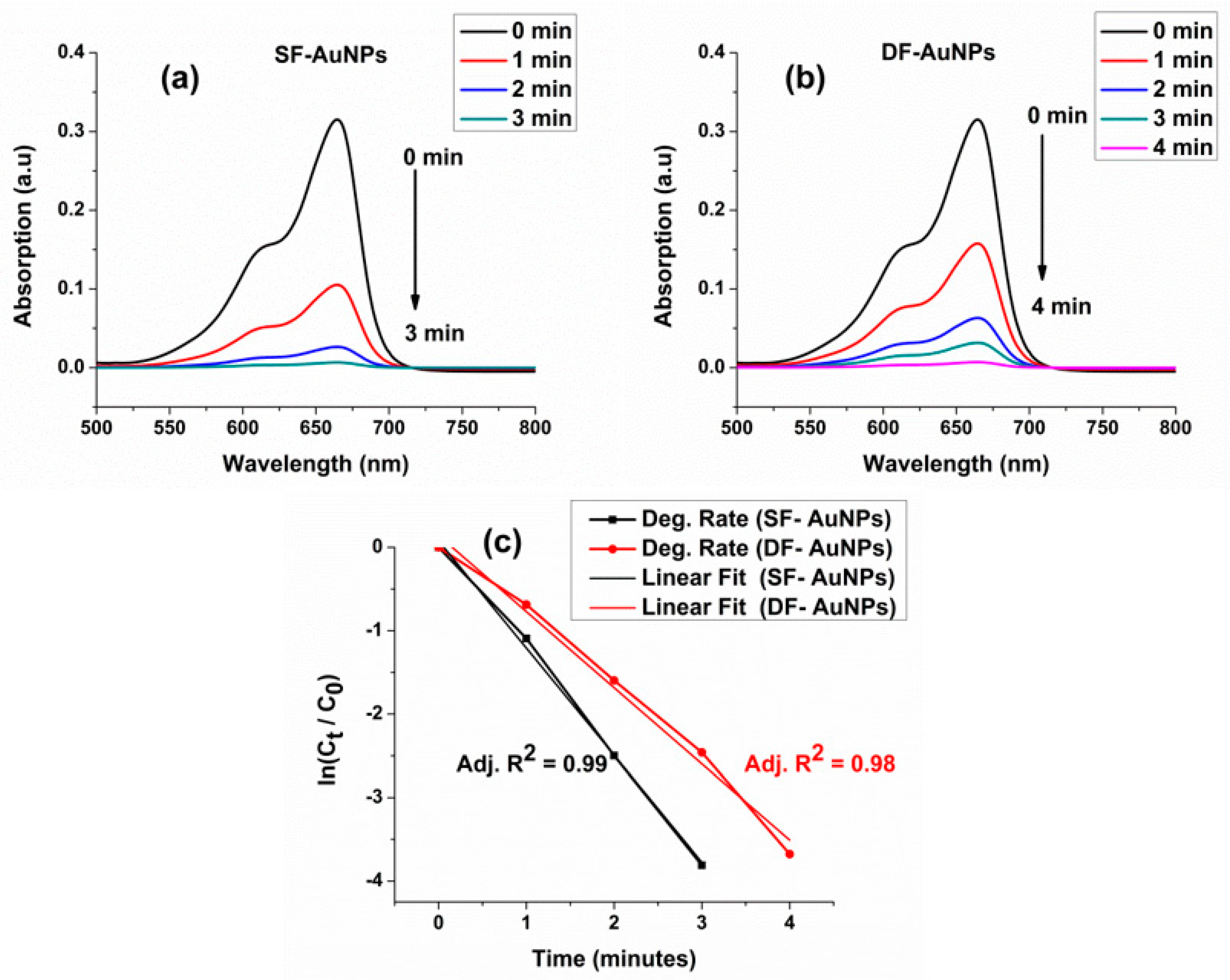
3.3. Catalytic Properties of and
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bailly, A.-L.; Correard, F.; Popov, A.; Tselikov, G.; Chaspoul, F.; Appay, R.; Al-Kattan, A.; Kabashin, A.V.; Braguer, D.; Esteve, M.-A. In vivo evaluation of safety, biodistribution and pharmacokinetics of laser-synthesized gold nanoparticles. Sci. Rep. 2019, 9, 12890. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Ng, H.P.; Xu, Y.; Li, Y.; Zheng, Y.; Yu, J.; Han, F.; Peng, F.; Fu, L. Gold nanoparticles: Synthesis, stability test, and application for the rice growth. J. Nanomater. 2014, 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Khan, H.I.; Khalid, M.U.; Abdullah, A.; Ali, A.; Bhatti, A.S. Facile synthesis of gold nanostars over a wide size range and their excellent surface enhanced Raman scattering and fluorescence quenching properties. J. Vac. Sci. Technol. B Nanotechnol. Microelectron. Mater. Process. Meas. Phenom. 2018, 36, 03E101. [Google Scholar] [CrossRef]
- Baba, T.; Lee, Y.; Ueno, A.; Kometani, R.; Maeda, E. Triple-walled gold surfaces with small-gaps for nonresonance surface enhanced Raman scattering of rhodamine 6G molecules. J. Vac. Sci. Technol. B Nanotechnol. Microelectron. Mater. Process. Meas. Phenom. 2016, 34, 011802. [Google Scholar] [CrossRef]
- León, E.R.; Rodríguez, E.L.; Beas, C.R.; Plascencia-Villa, G.; Palomares, R.A.I. Study of methylene blue degradation by gold nanoparticles synthesized within natural zeolites. J. Nanomater. 2016, 2016, 9541683. [Google Scholar] [CrossRef] [Green Version]
- Suvith, V.; Philip, D. Catalytic degradation of methylene blue using biosynthesized gold and silver nanoparticles. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 118, 526–532. [Google Scholar] [CrossRef]
- Tan, E.; Yin, P.; Lang, X.; Wang, X.; You, T.; Guo, L. Functionalized gold nanoparticles as nanosensor for sensitive and selective detection of silver ions and silver nanoparticles by surface-enhanced Raman scattering. Analyst 2012, 137, 3925–3928. [Google Scholar] [CrossRef]
- Hwang, J.S.; Yang, M. Sensitive and reproducible gold SERS sensor based on interference lithography and electrophoretic deposition. Sensors 2018, 18, 4076. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Lim, D.-J.; Vines, J.B.; Yoon, J.-H.; Ryu, N.-E. Gold nanoparticles for photothermal cancer therapy. Front. Chem. 2019, 7, 167. [Google Scholar]
- Calavia, P.G.; Bruce, G.; Pérez-García, L.; Russell, D.A. Photosensitiser-gold nanoparticle conjugates for photodynamic therapy of cancer. Photochem. Photobiol. Sci. 2018, 17, 1534–1552. [Google Scholar] [CrossRef] [Green Version]
- Kong, F.-Y.; Zhang, J.-W.; Li, R.-F.; Wang, Z.-X.; Wang, W.-J.; Wang, W. Unique roles of gold nanoparticles in drug delivery, targeting and imaging applications. Molecules 2017, 22, 1445. [Google Scholar] [CrossRef]
- Farooq, M.U.; Novosad, V.; Rozhkova, E.A.; Wali, H.; Ali, A.; Fateh, A.A.; Neogi, P.B.; Neogi, A.; Wang, Z. Gold nanoparticles-enabled efficient dual delivery of anticancer therapeutics to HeLa cells. Sci. Rep. 2018, 8, 2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Wang, L.; Zibart, A.; Koch, C. Corrosion protection of electrically conductive surfaces. Metals 2012, 2, 450–477. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, M.; Miyagi, N.; Mita, M.; Yamasaki, K.; Maekawa, K. On-demand gold laser-plating onto stainless steel for electrical contacts. In Proceedings of the 2016 International Conference on Electronics Packaging (ICEP), Hokkaido, Japan, 20–22 April 2016; pp. 323–328. [Google Scholar]
- Kim, M.; Lee, J.H.; Nam, J.M. Plasmonic Photothermal Nanoparticles for Biomedical Applications. Adv. Sci. 2019, 6, 1900471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hainfeld, J.F.; O’Connor, M.J.; Lin, P.; Qian, L.; Slatkin, D.N.; Smilowitz, H.M. Infrared-transparent gold nanoparticles converted by tumors to infrared absorbers cure tumors in mice by photothermal therapy. PLoS ONE 2014, 9, e88414. [Google Scholar] [CrossRef] [Green Version]
- Norman, T.J.; Grant, C.D.; Magana, D.; Zhang, J.Z.; Liu, J.; Cao, D.; Bridges, F.; Van Buuren, A. Near infrared optical absorption of gold nanoparticle aggregates. J. Phys. Chem. B 2002, 106, 7005–7012. [Google Scholar] [CrossRef]
- Elbert, K.C.; Lee, J.D.; Wu, Y.; Murray, C.B. Improved chemical and colloidal stability of gold nanoparticles through dendron capping. Langmuir 2018, 34, 13333–13338. [Google Scholar] [CrossRef]
- Shah, M.; Badwaik, V.; Kherde, Y.; Waghwani, H.K.; Modi, T.; Aguilar, Z.P.; Rodgers, H.; Hamilton, W.; Marutharaj, T.; Webb, C. Gold nanoparticles: Various methods of synthesis and antibacterial applications. Front. Biosci. 2014, 19, 1320–1344. [Google Scholar] [CrossRef] [Green Version]
- Song, J.Y.; Kim, B.S. Rapid biological synthesis of silver nanoparticles using plant leaf extracts. Bioprocess Biosyst. Eng. 2009, 32, 79. [Google Scholar] [CrossRef]
- Verma, H.N.; Singh, P.; Chavan, R. Gold nanoparticle: Synthesis and characterization. Vet. World 2014, 7, 72. [Google Scholar] [CrossRef] [Green Version]
- Jeevanandam, J.; Chan, Y.S.; Danquah, M.K. Biosynthesis of metal and metal oxide nanoparticles. ChemBioEng Rev. 2016, 3, 55–67. [Google Scholar] [CrossRef]
- Jamkhande, P.G.; Ghule, N.W.; Bamer, A.H.; Kalaskar, M.G. Metal nanoparticles synthesis: An overview on methods of preparation, advantages and disadvantages, and applications. J. Drug Deliv. Sci. Technol. 2019, 53, 101174. [Google Scholar] [CrossRef]
- Parveen, K.; Banse, V.; Ledwani, L. Green synthesis of nanoparticles: Their advantages and disadvantages. AIP Conf. Proc. 2016, 1724, 020048. [Google Scholar]
- Vigderman, L.; Zubarev, E.R. High-yield synthesis of gold nanorods with longitudinal SPR peak greater than 1200 nm using hydroquinone as a reducing agent. Chem. Mater. 2013, 25, 1450–1457. [Google Scholar] [CrossRef]
- Patel, J.; Němcová, L.; Maguire, P.; Graham, W.; Mariotti, D. Synthesis of surfactant-free electrostatically stabilized gold nanoparticles by plasma-induced liquid chemistry. Nanotechnology 2013, 24, 245604. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, T.; Thirunavukkarasu, A.; Rathnavelu, S.M.; Kasivelu, G. In Vivo Non-toxicity of Gold Nanoparticles on Wistar Rats. J. Clust. Sci. 2019, 30, 513–519. [Google Scholar] [CrossRef]
- Pastoriza-Santos, I.; Pérez-Juste, J.; Liz-Marzán, L.M. Silica-coating and hydrophobation of CTAB-stabilized gold nanorods. Chem. Mater. 2006, 18, 2465–2467. [Google Scholar] [CrossRef]
- Morais, T.; Soares, M.E.; Duarte, J.A.; Soares, L.; Maia, S.; Gomes, P.; Pereira, E.; Fraga, S.; Carmo, H.; de Lourdes Bastos, M. Effect of surface coating on the biodistribution profile of gold nanoparticles in the rat. Eur. J. Pharm. Biopharm. 2012, 80, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Khatoon, N.; Yasin, H.; Younus, M.; Ahmed, W.; Rehman, N.; Zakaullah, M.; Iqbal, M.Z. Synthesis and spectroscopic characterization of gold nanoparticles via plasma-liquid interaction technique. AIP Adv. 2018, 8, 015130. [Google Scholar] [CrossRef]
- Seoudi, R.; Said, D.A. Studies on the effect of the capping materials on the spherical gold nanoparticles catalytic activity. World J. Nano Sci. Eng. 2011, 1, 51. [Google Scholar] [CrossRef] [Green Version]
- Soliman, M.G.; Pelaz, B.; Parak, W.J.; Del Pino, P. Phase transfer and polymer coating methods toward improving the stability of metallic nanoparticles for biological applications. Chem. Mater. 2015, 27, 990–997. [Google Scholar] [CrossRef] [Green Version]
- Qi, G.; Jia, K.; Fu, C.; Xu, S.; Xu, W. A highly sensitive SERS sensor for quantitative analysis of glucose based on the chemical etching of silver nanoparticles. J. Opt. 2015, 17, 114020. [Google Scholar] [CrossRef]
- Bruggeman, P.; Ribežl, E.; Maslani, A.; Degroote, J.; Malesevic, A.; Rego, R.; Vierendeels, J.; Leys, C. Characteristics of atmospheric pressure air discharges with a liquid cathode and a metal anode. Plasma Sources Sci. Technol. 2008, 17, 025012. [Google Scholar] [CrossRef]
- Scholl, J.A.; Koh, A.L.; Dionne, J.A. Quantum plasmon resonances of individual metallic nanoparticles. Nature 2012, 483, 421. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zuo, S.; Wu, D.; Zhang, J.; Zhu, W.; Becker, K.H.; Fang, J. Microplasma-assisted synthesis of colloidal gold nanoparticles and their use in the detection of cardiac troponin I (cTn-I). Plasma Process. Polym. 2015, 12, 380–391. [Google Scholar] [CrossRef]
- Li, X.; Lin, L.; Chiang, W.-H.; Chang, K.; Xu, H. Microplasma synthesized gold nanoparticles for surface enhanced Raman spectroscopic detection of methylene blue. React. Chem. Eng. 2022, 7, 346–353. [Google Scholar] [CrossRef]
- Yang, C.; Yu, Y.; Xie, Y.; Zhang, D.; Zeng, P.; Dong, Y.; Yang, B.; Liang, R.; Ou, Q.; Zhang, S. One-step synthesis of size-tunable gold nanoparticles/reduced graphene oxide nanocomposites using argon plasma and their applications in sensing and catalysis. Appl. Surf. Sci. 2019, 473, 83–90. [Google Scholar] [CrossRef]
- Liu, T.; Yang, F.; Li, Y.; Ren, L.; Zhang, L.; Xu, K.; Wang, X.; Xu, C.; Gao, J. Plasma synthesis of carbon nanotube-gold nanohybrids: Efficient catalysts for green oxidation of silanes in water. J. Mater. Chem. A 2014, 2, 245–250. [Google Scholar] [CrossRef]
- Nguyen, L.N.; Kaushik, N.; Lamichhane, P.; Mumtaz, S.; Paneru, R.; Bhartiya, P.; Kwon, J.S.; Mishra, Y.K.; Nguyen, L.Q.; Kaushik, N.K. In situ plasma-assisted synthesis of polydopamine-functionalized gold nanoparticles for biomedical applications. Green Chem. 2020, 22, 6588–6599. [Google Scholar] [CrossRef]
- Yasin, H.; Ahmed, W.; Ali, A.; Bhatti, A.; Rehman, N. Micro-plasma assisted synthesis of multifunctional D-fructose coated silver nanoparticles. Mater. Res. Express 2019, 6, 1050a2. [Google Scholar] [CrossRef]
- Hieda, J.; Saito, N.; Takai, O. Exotic shapes of gold nanoparticles synthesized using plasma in aqueous solution. J. Vac. Sci. Technol. A Vac. Surf. Film. 2008, 26, 854–856. [Google Scholar] [CrossRef] [Green Version]
- Mironov, I.; Makotchenko, E. The Hydrolysis of AuCl 4− and the Stability of Aquachlorohydroxocomplexes of Gold (III) in Aqueous Solution. J. Solut. Chem. 2009, 38, 725–737. [Google Scholar] [CrossRef]
- Akolkar, R.; Sankaran, R.M. Charge transfer processes at the interface between plasmas and liquids. J. Vac. Sci. Technol. A Vac. Surf. Film. 2013, 31, 050811. [Google Scholar] [CrossRef]
- Chen, Q.; Li, J.; Li, Y. A review of plasma–liquid interactions for nanomaterial synthesis. J. Phys. D Appl. Phys. 2015, 48, 424005. [Google Scholar] [CrossRef] [Green Version]
- Kooij, E.S.; Ahmed, W.; Zandvliet, H.J.; Poelsema, B. Localized plasmons in noble metal nanospheroids. J. Phys. Chem. C 2011, 115, 10321–10332. [Google Scholar] [CrossRef]
- Kelly, K.L.; Coronado, E.; Zhao, L.L.; Schatz, G.C. The optical properties of metal nanoparticles: The influence of size, shape, and dielectric environment. J. Phys. Chem. B 2003, 107, 668–677. [Google Scholar] [CrossRef]
- Pastoriza-Santos, I.; Liz-Marzán, L.M. Colloidal silver nanoplates. State of the art and future challenges. J. Mater. Chem. 2008, 18, 1724–1737. [Google Scholar] [CrossRef]
- McFarland, A.D.; Van Duyne, R.P. Single silver nanoparticles as real-time optical sensors with zeptomole sensitivity. Nano Lett. 2003, 3, 1057–1062. [Google Scholar] [CrossRef] [Green Version]
- Fenger, R.; Fertitta, E.; Kirmse, H.; Thünemann, A.F.; Rademann, K. Size dependent catalysis with CTAB-stabilized gold nanoparticles. Phys. Chem. Chem. Phys. 2012, 14, 9343–9349. [Google Scholar] [CrossRef]
- Silva, J.A.d.; Dias, R.P.; da Hora, G.C.; Soares, T.A.; Meneghetti, M.R. Molecular dynamics simulations of cetyltrimethylammonium bromide (CTAB) micelles and their interactions with a gold surface in aqueous solution. J. Braz. Chem. Soc. 2018, 29, 191–199. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasin, H.M.; Ahmed, W.; Rehman, N.U.; Majd, A.; Alkhedher, M.; Tag El Din, E.M. Plasma-Assisted Synthesis of Surfactant-Free and D-Fructose-Coated Gold Nanoparticles for Multiple Applications. Materials 2022, 15, 7579. https://doi.org/10.3390/ma15217579
Yasin HM, Ahmed W, Rehman NU, Majd A, Alkhedher M, Tag El Din EM. Plasma-Assisted Synthesis of Surfactant-Free and D-Fructose-Coated Gold Nanoparticles for Multiple Applications. Materials. 2022; 15(21):7579. https://doi.org/10.3390/ma15217579
Chicago/Turabian StyleYasin, Hafiz M., W. Ahmed, N. U. Rehman, Abdul Majd, Mohammad Alkhedher, and ElSayed M. Tag El Din. 2022. "Plasma-Assisted Synthesis of Surfactant-Free and D-Fructose-Coated Gold Nanoparticles for Multiple Applications" Materials 15, no. 21: 7579. https://doi.org/10.3390/ma15217579