

Selenoprotein F Knockout Caused Glucose Metabolism Disorder in Young Mice by Disrupting Redox Homeostasis



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Fasting Blood Glucose Level Test, GTT and ITT
2.4. Measurements of Serum and Hepatic Biochemical Parameters
2.5. Liver and Muscle Glycogen Determination
2.6. Western Blot Analysis
2.7. Histopathology of the Liver and Pancreas
2.8. Statistical Analysis
3. Results
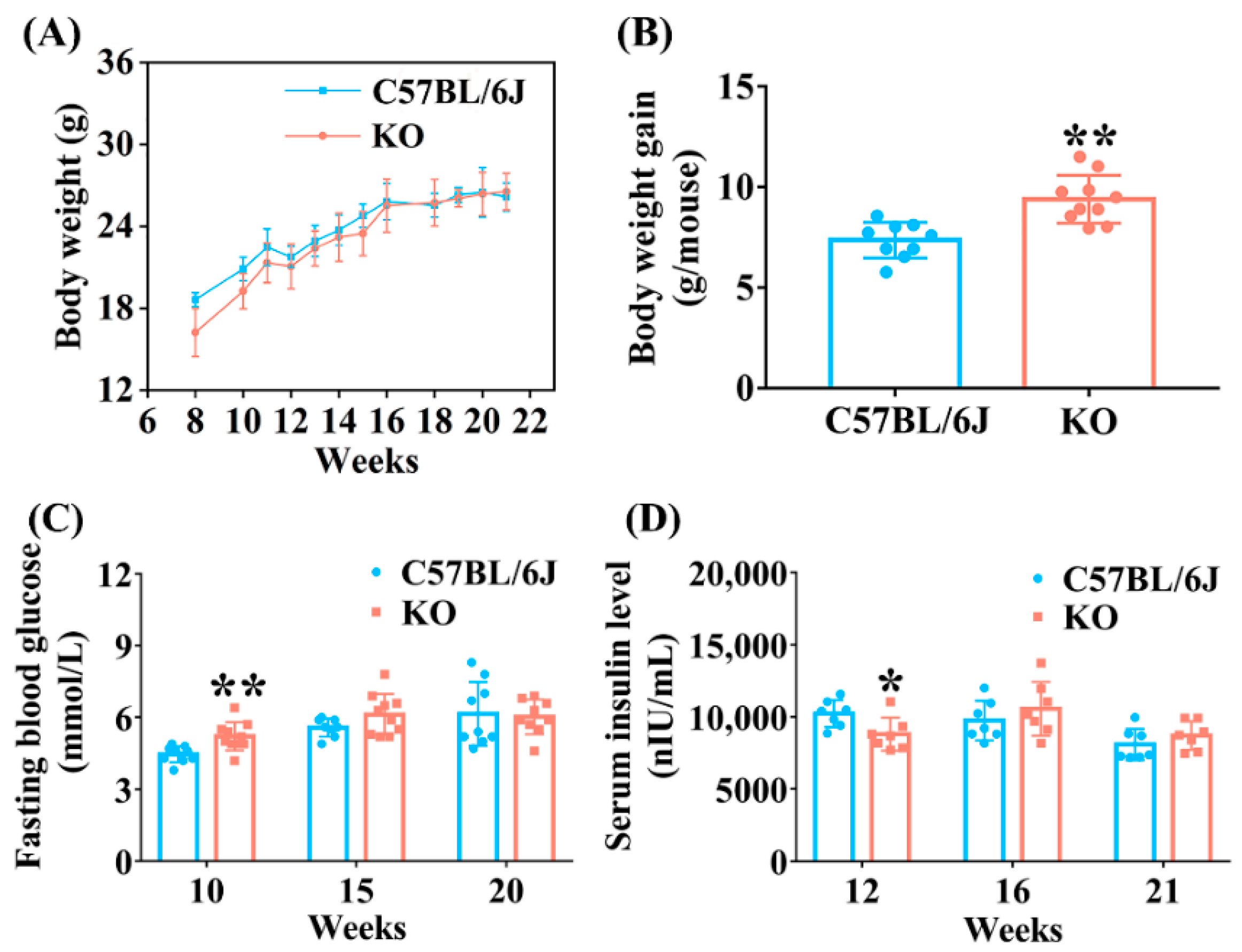
3.1. Metabolic Phenotype Analysis of SELENOF KO Mice
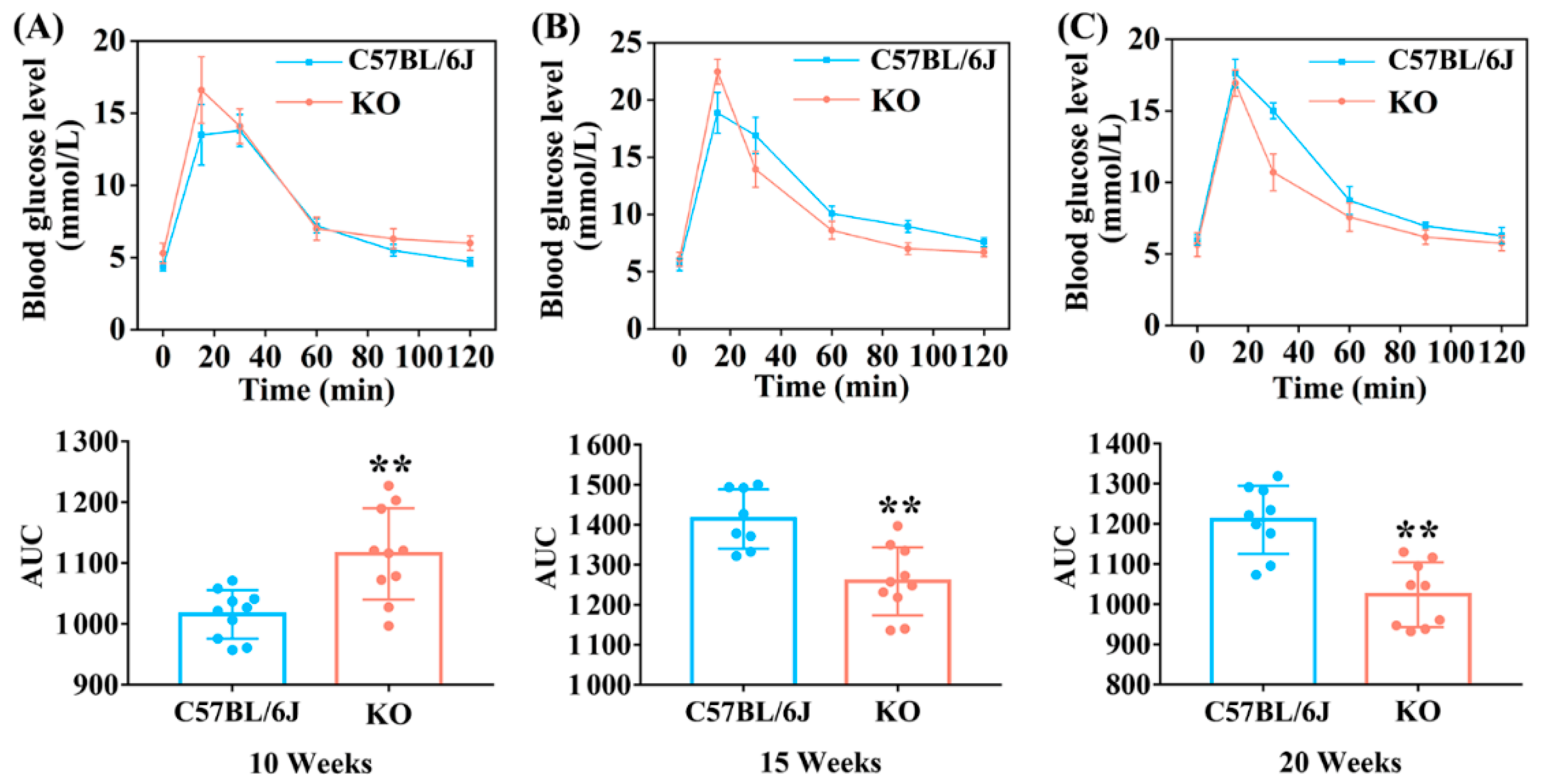
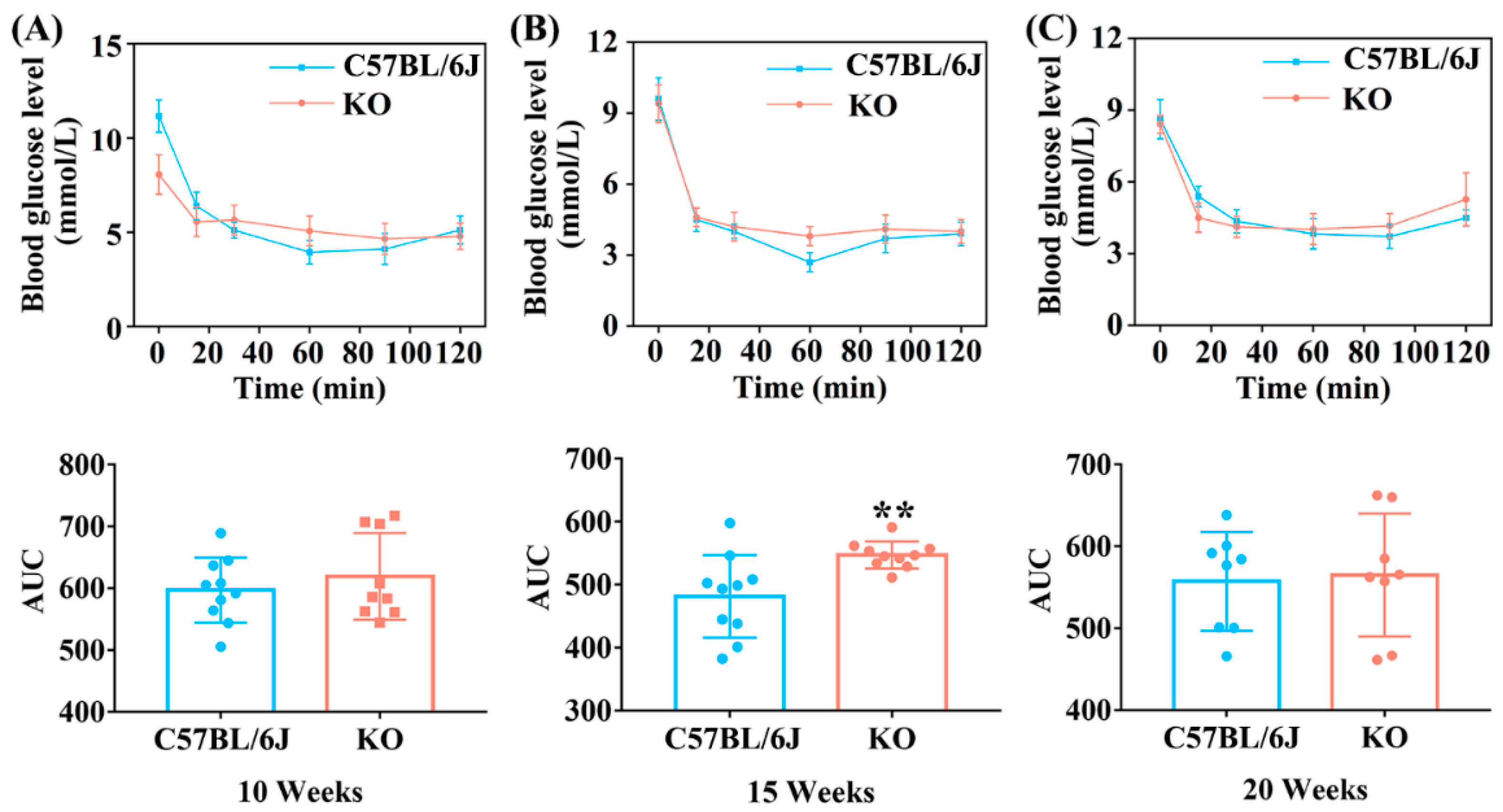
3.2. SELENOF KO Led to Impaired Glucose Tolerance and Decreased Insulin Sensitivity in Young Mice
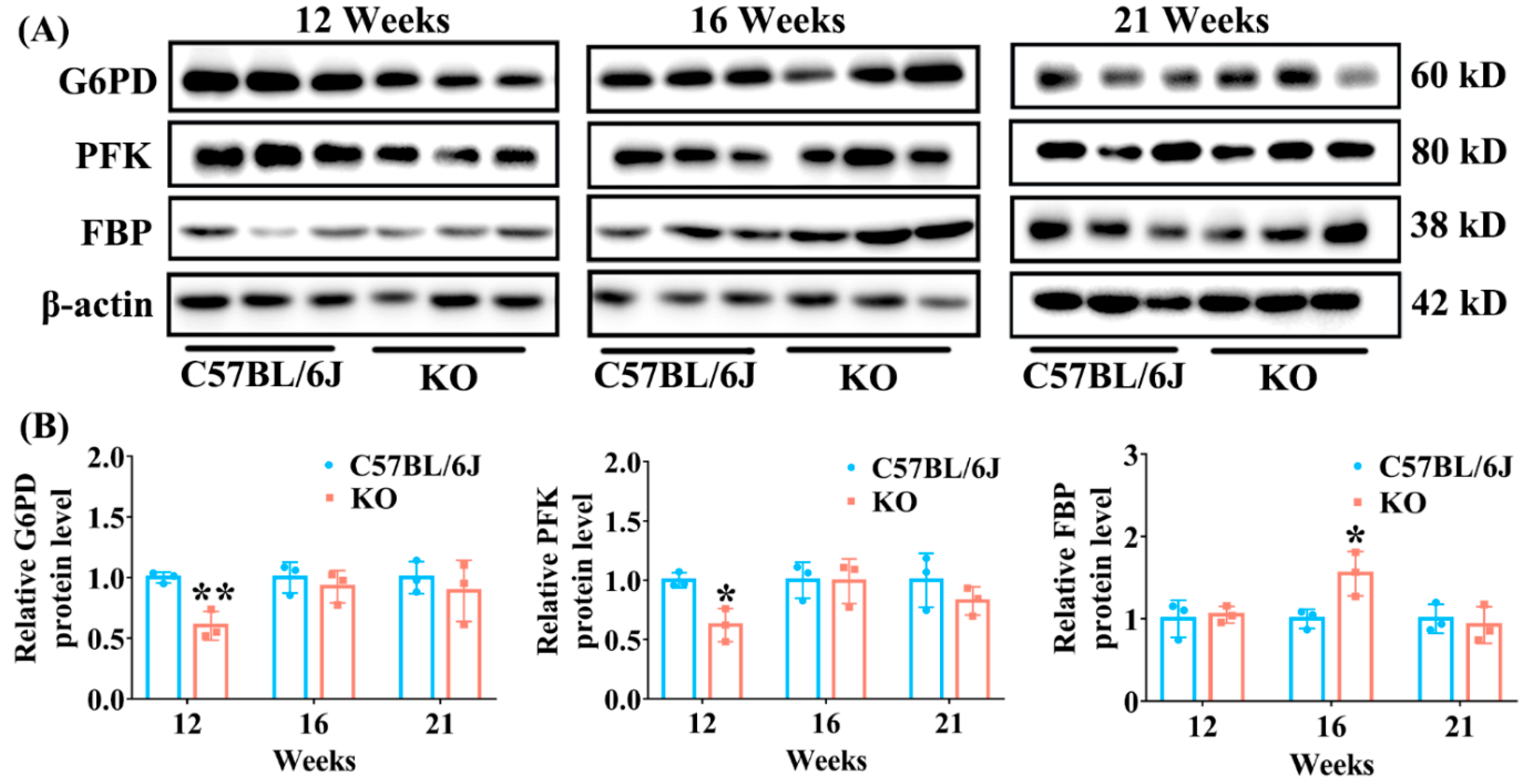
3.3. Effect of SELENOF KO on the Expression of Key Enzymes Involved in Glucose Metabolism Pathways
3.4. SELENOF KO Reduced Glycogen Accumulation in the Liver and Muscle of Mice at an Earlier Age
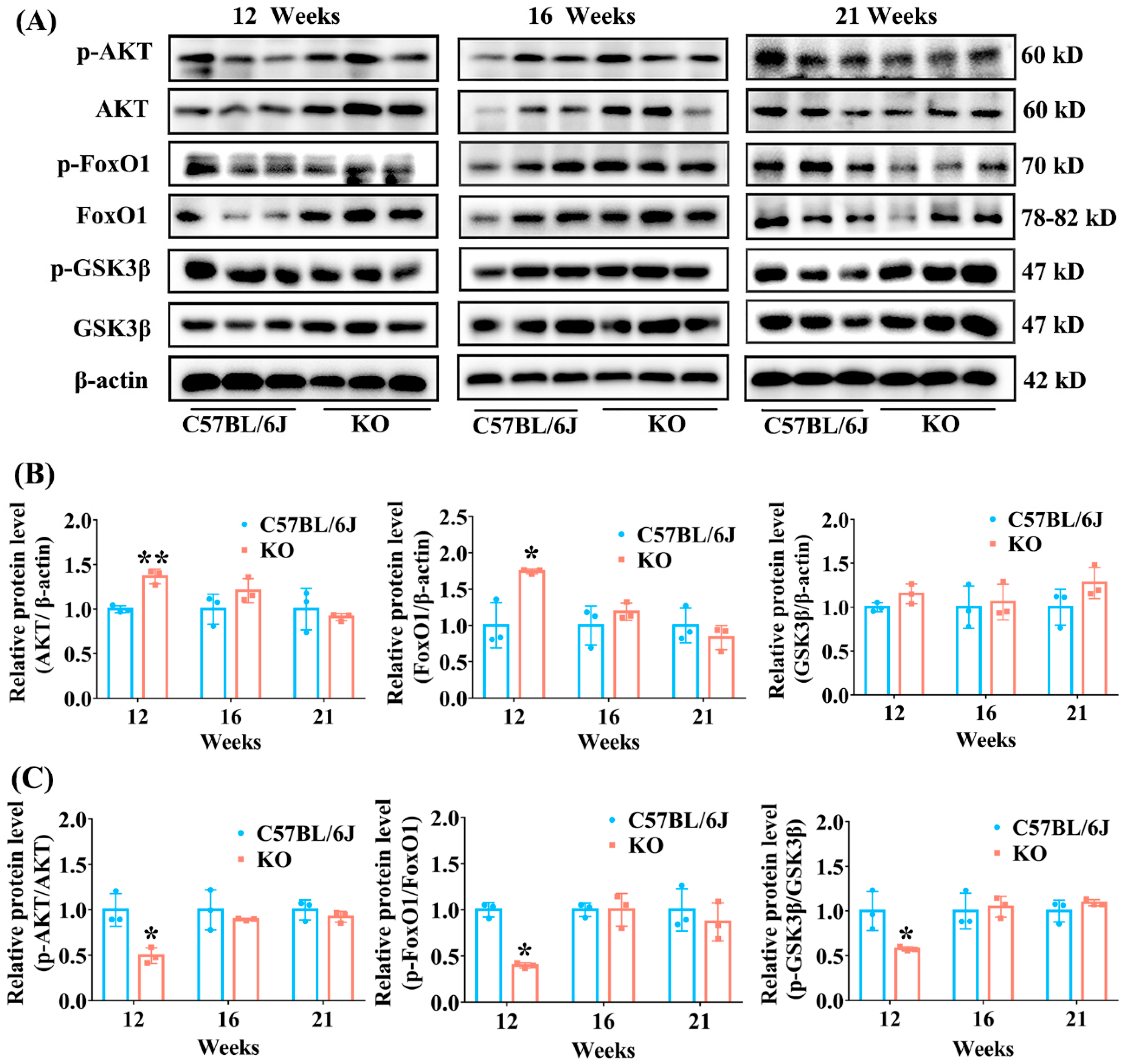
3.5. SELENOF KO Resulted in Inhibition of AKT Signaling Pathway in Young Mice
3.6. SELENOF Knockout Resulted in the Disruption of Redox Homeostasis in Young Mice
3.7. SELENOF KO Increased the Expression of Hepatic SELENOS in Young Mice
3.8. SELENOF KO Might Not Cause ER Stress in the Liver and Pancreas of Mice with Different Ages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aguilar, M.; Bhuket, T.; Torres, S.; Liu, B.; Wong, R. Prevalence of the metabolic syndrome in the United States, 2003–2012. JAMA 2015, 313, 1973–1974. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Zhao, Y.; Yin, Z.; Wang, D.W.; Chen, C. The role of miR-320 in glucose and lipid metabolism disorder-associated diseases. Int. J. Biol. Sci. 2021, 17, 402–416. [Google Scholar] [CrossRef] [PubMed]
- Kuryιéowicz, A.; Cakaιa-Jakimowicz, M.; Puzianowska-Kuznicka, M. Targeting abdominal obesity and its complications with dietary phytoestrogens. Nutrients 2020, 12, 582. [Google Scholar] [CrossRef]
- Luo, Y.; Yu, J.; Yang, M.; Wang, P.; Yang, C.; Xu, J. Identification of nonylphenol and glucolipid metabolism-related proteins in the serum of type 2 diabetes patients. Iran. J. Public Health 2019, 48, 2210–2215. [Google Scholar]
- Fairweather-Tait, S.J.; Bao, Y.; Broadley, M.R.; Collings, R.; Ford, D.; Hesketh, J.E.; Hurst, R. Selenium in human health and disease. Antioxid Redox Sign. 2011, 14, 1337–1383. [Google Scholar] [CrossRef] [PubMed]
- Gorini, F.; Vassalle, C. Selenium and selenoproteins at the intersection of type 2 diabetes and thyroid pathophysiology. Antioxidants 2022, 11, 1188. [Google Scholar] [CrossRef]
- Rayman, M. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Duntas, L.H.; Rayman, M.P. The role of selenium in type-2 diabetes mellitus and its metabolic comorbidities. Redox Biol. 2022, 50, 102236. [Google Scholar] [CrossRef]
- Akbaraly, T.N.; Arnaud, J.; Rayman, M.P.; Hininger-Favier, I.; Roussel, A.-M.; Berr, C.; Fontbonne, A. Plasma selenium and risk of dysglycemia in an elderly French population: Results from the prospective epidemiology of vascular ageing study. Nutr. Metab. 2010, 7, 21. [Google Scholar] [CrossRef]
- Kljai, K.; Runje, R. Selenium and glycogen levels in diabetic patients. Biol. Trace Elem. Res. 2001, 83, 223–229. [Google Scholar] [CrossRef]
- Navarro-Alarcón, M.; López-G de la Serrana, H.; Pérez-Valero, V.; López-Martínez, C. Serum and urine selenium concentrations as indicators of body status in patients with diabetes mellitus. Sci. Total Environ. 1999, 228, 79–85. [Google Scholar] [CrossRef]
- Ryan, T.; Wu, L.; Cao, E.; Mattson, K.; Witwer, W. Opposing impacts on healthspan and longevity by limiting dietary selenium in telomere dysfunctional mice. Aging Cell 2016, 16, 125–135. [Google Scholar]
- Huang, Y.C.; Wu, T.L.; Zeng, H.; Cheng, W.H. Dietary selenium requirement for the prevention of glucose intolerance and insulin resistance in middle-aged mice. J. Nutr. 2021, 151, 1894–1900. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed]
- Sunde, R.A.; Raines, A.M.; Barnes, K.M.; Evenson, J.K. Selenium status highly regulates selenoprotein mRNA levels for only a subset of the selenoproteins in the selenoproteome. Biosci. Rep. 2009, 29, 329–338. [Google Scholar] [CrossRef]
- Wang, X.; Vatamaniuk, M.Z.; Roneker, C.A.; Pepper, M.P.; Hu, L.G.; Simmons, R.A.; Lei, X.G.; Signaling, R. Knockouts of SOD1 and GPX1 exert different impacts on murine islet function and pancreatic integrity. Antioxid. Redox Sign. 2011, 14, 391–401. [Google Scholar] [CrossRef]
- Ren, B.Y.; Liu, M.; Ni, J.Z.; Tian, J. Role of selenoprotein F in protein folding and secretion: Potential involvement in human disease. Nutrients 2018, 10, 1619. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Ferguson, A.D.; Fomenko, D.E.; Chelliah, Y.; Hatfield, D.L.; Gladyshev, V.N. A novel cysteine-rich domain of Sep15 mediates the interaction with UDP-glucose:glycoprotein glucosyltransferase. J. Biol. Chem. 2005, 280, 37839–37845. [Google Scholar] [CrossRef]
- Vyacheslav, M.; Labunskyy, M.H.Y.; Hatfield, D.L.; Gladyshe, V.N. Sep15, a thioredoxin-like selenoprotein, is involved in the unfolded protein response and differentially regulated by adaptive and acute ER stresses. Biochemistry 2009, 48, 8458–8465. [Google Scholar]
- Yim, S.H.; Everley, R.A.; Schil Db Erg, F.A.; Lee, S.G.; Orsi, A.; Barbati, Z.R.; Karatepe, K.; Fomenko, D.E.; Tsuji, P.A.; Luo, H.R. Role of Selenof as a gatekeeper of secreted disulfide-rich glycoproteins. Cell Rep. 2018, 23, 1387–1398. [Google Scholar] [CrossRef]
- Kumaraswamy, E.; Malykh, A.; Korotkov, K.V.; Kozyavkin, S.; Gladyshev, V.N. Structure-expression relationships of the 15-kDa selenoprotein gene. Possible role of the protein in cancer etiology. J. Biol. Chem. 2000, 275, 35540–35547. [Google Scholar] [CrossRef]
- Zheng, X.X.; Ren, B.Y.; Li, X.M.; Yan, H.H.; Xie, Q.G.; Liu, H.M.; Zhou, J.; Tian, J.; Huang, K.X. Selenoprotein F knockout leads to glucose and lipid metabolism disorders in mice. J. Biol. Inorg. Chem. 2020, 25, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.X.; Ren, B.Y.; Wang, H.; Huang, R.K.; Zhou, J.; Liu, H.M.; Tian, J.; Huang, K.X. Hepatic proteomic analysis of selenoprotein F knockout mice by iTRAQ: An implication for the roles of selenoprotein F in metabolism and diseases. J. Proteom. 2020, 215, 103653. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.M.; Heatlie, P.L. Lowry method of protein quantification: Evidence for photosensitivity. Anal. Biochem. 1984, 140, 391–393. [Google Scholar] [CrossRef]
- Nogales, F.; Ojeda, M.L.; Fenutria, M.; Murillo, M.L.; Carreras, O. Role of selenium and glutathione peroxidase on development, growth, and oxidative balance in rat offspring. Reproduction 2013, 146, 659–667. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.Z.; Wu, L.; Liu, Q.; Zhang, D.F.; Yin, J.J. Expression of selenoprotein genes in muscle is crucial for the growth of rainbow trout (oncorhynchus mykiss) fed diets supplemented with selenium yeast. Aquaculture 2018, 492, 82–90. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, X.; Zhou, R.; Gu, Y.; Zhu, X.; Tang, Z.; Yuan, X.; Chen, W.; Zhang, R.; Qian, C.; et al. Delay in glucose peak time during the oral glucose tolerance test as an indicator of insulin resistance and insulin secretion in type 2 diabetes patients. J. Diabetes Investig. 2018, 9, 1288–1295. [Google Scholar] [CrossRef]
- González-Ortiz, L.J.; Martínez-Abundis, E.; González-Ortiz, M. A new model to fit glucose concentration during the insulin tolerance test improving the predictive capability to estimate insulin sensitivity. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 78–79. [Google Scholar] [CrossRef]
- Wang, K.P.; Wang, H.X.; Liu, Y.G.; Shui, W.Z.; Wang, J.F.; Cao, P.; Wang, H.J.; You, R.X.; Zhang, Y. Dendrobium officinale polysaccharide attenuates type 2 diabetes mellitus via the regulation of PI3K/Akt-mediated glycogen synthesis and glucose metabolism. J. Funct. Foods 2018, 40, 261–271. [Google Scholar] [CrossRef]
- Benchoula, K. FoxO1 signaling as a therapeutic target for type 2 diabetes and obesity. Eur. J. Pharmacol. 2021, 891, 173758. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Asensi, M.; Sastre, J.; Pallardo, F.V.; Lloret, A.; Via, J. Ratio of reduced to oxidized glutathione as indicator of oxidative stress status and DNA damage. Method Enzymol. 1999, 299, 267–276. [Google Scholar]
- Ghosh, R.; Colon Negron, K.; Papa, F.R. Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol. Metab. 2019, 27, S60–S68. [Google Scholar] [CrossRef] [PubMed]
- Villalobos Labra, R.; Subiabre, M.; Toledo, F.; Pardo, F.; Sobrevia, L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol. Aspects Med. 2019, 66, 49–61. [Google Scholar] [CrossRef]
- Kasaikina, M.V.; Fomenko, D.E.; Labunskyy, V.M.; Lachke, S.A.; Qiu, W.; Moncaster, J.A.; Zhang, J.; Wojnarowicz, M.W., Jr.; Natarajan, S.K.; Malinouski, M.; et al. Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract development revealed by the analysis of Sep 15 knockout mice. J. Biol. Chem. 2011, 286, 33203–33212. [Google Scholar] [CrossRef]
- Ferguson, A.D.; Labunskyy, V.M.; Fomenko, D.E.; Araç, D.; Chelliah, Y.; Amezcua, C.A.; Rizo, J.; Gladyshev, V.N.; Deisenhofer, J. NMR structures of the selenoproteins Sep15 and SelM reveal redox activity of a new thioredoxin-like family. J. Biol. Chem. 2006, 281, 3536–3543. [Google Scholar] [CrossRef]
- Liao, P.S.; Liu, H.M.; He, C.M. Chemical synthesis of human selenoprotein F and elucidation of its thiol-disulfide oxidoreductase activity. Chem. Sci. 2022, 13, 6322–6327. [Google Scholar] [CrossRef]
- Coskun, O.; Kanter, M.; Korkmaz, A.; Oter, S. Quercetin, a flavonoid antioxidant, prevents and protects streptozotocin-induced oxidative stress and β-cell damage in rat pancreas. Pharmacol. Res. 2005, 51, 117–123. [Google Scholar] [CrossRef]
- Gu, C.; Xu, H. Effect of oxidative damage due to excessive protein ingestion on pancreas function in mice. Int. J. Mol. Sci. 2010, 11, 4591–4600. [Google Scholar] [CrossRef]
- Lenzen, S. Oxidative stress: The vulnerable β-cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Zhang, Y.L.; Sun, X.Y.; Shi, X.; Xu, S.W. Microplastics and di (2-ethylhexyl) phthalate synergistically induce apoptosis in mouse pancreas through the GRP78/CHOP/Bcl-2 pathway activated by oxidative stress. Food Chem. Toxicol. 2022, 167, 113315. [Google Scholar] [CrossRef]
- Noguchi, R.; Kubota, H.; Yugi, K.; Toyoshima, Y.; Komori, Y.; Soga, T.; Kuroda, S. The selective control of glycolysis, gluconeogenesis and glycogenesis by temporal insulin patterns. Mol. Syst. Biol. 2013, 9, 664. [Google Scholar] [CrossRef]
- Rines, A.K.; Sharabi, K.; Tavares, C.; Puigserver, P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat. Rev. Drug Discov. 2016, 15, 786–804. [Google Scholar] [CrossRef] [PubMed]
- Fasciolo, G.; Napolitano, G.; Aprile, M.; Cataldi, S.; Costa, V.; Ciccodicola, A.; Di Meo, S.; Venditti, P. Hepatic insulin resistance in hyperthyroid rat liver: Vitamin E supplementation highlights a possible role of ROS. Antioxidants 2022, 11, 1295. [Google Scholar] [CrossRef] [PubMed]
- Ke, W.X.; Wang, P.; Wang, X.H.; Zhou, X.L.; Hu, X.S.; Chen, F. Dietary platycodon grandiflorus attenuates hepatic insulin resistance and oxidative stress in high-fat-diet induced non-alcoholic fatty liver disease. Nutrients 2020, 12, 480. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.M.; Zhao, J.H.; Xie, X.Q.; Chen, T.; Yin, Y.; Zhai, R.H.; Wang, X.L.; An, W.; Li, J.X. Anthocyanins from the fruits of lycium ruthenicum murray improve high-fat diet-induced insulin resistance by ameliorating inflammation and oxidative stress in mice. Food Funct. 2021, 12, 3855–3871. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.H.; Liu, C.; Feng, X.H.; Xie, Y.T.; Yue, H.T.; Dong, J.; Zhao, Z.K.; Chen, G.L.; Yang, J. Camel whey protein (CWP) ameliorates liver injury in type 2 diabetes mellitus rats and insulin resistance (IR) in HepG2 cells via activation of the PI3K/Akt signaling pathway. Food Funct. 2022, 13, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Miao, L.; Xiao, J.; Cheang, W.S. 3,3’,4,5’-tetramethoxy-trans-stilbene improves insulin resistance by activating the IRS/PI3K/Akt pathway and inhibiting oxidative stress. Curr. Issues Mol. Biol. 2022, 44, 147. [Google Scholar] [CrossRef]
- Jiting, Y.; Changyuan, W.; Yue, J.; Qiang, M.; Qi, L. Catalpol ameliorates hepatic insulin resistance in type 2 diabetes through acting on AMPK/NOX4/PI3K/AKT pathway. Pharmacol. Res. 2018, 130, 466–480. [Google Scholar]
- Wu, J.; Chen, Y.B.; Li, X.; Ran, L.Y.; Liu, X.D.; Wang, X.H.; Zhen, M.M.; Shao, S.S.; Zeng, L.; Wang, C.R. Functionalized gadofullerene ameliorates impaired glycolipid metabolism in type 2 diabetic mice. J. Genet Genom. 2022, 49, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Walder, K.; Kantham, L.; McMillan, J.S.; Trevaskis, J.; Kerr, L.; De Silva, A.; Sunderland, T.; Godde, N.; Gao, Y.; Bishara, N. Tanis: A link between type 2 diabetes and inflammation? Diabetes 2002, 51, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Walder, K.; Sunderland, T.; Kantham, L.; Feng, H.C.; Quick, M.; Bishara, N.; De Silva, A.; Augert, G.; Tenne-Brown, J. Elevation in Tanis expression alters glucose metabolism and insulin sensitivity in H4IIE cells. Diabetes 2003, 52, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, J.; Arnér, E. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Li, K.; Feng, T.J.; Liu, L.Y.; Liu, H.M.; Huang, K.X.; Zhou, J. Hepatic Proteomic Analysis of Selenoprotein T Knockout Mice by TMT: Implications for the Role of Selenoprotein T in Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2021, 22, 8515. [Google Scholar] [CrossRef] [PubMed]
- Prevost, G.; Arabo, A.; Jian, L.; Quelennec, E.; Cartier, D.; Hassan, S.; Falluel-Morel, A.; Tanguy, Y.; Gargani, S.; Lihrmann, I. The PACAP-regulated gene selenoprotein T is abundantly expressed in mouse and human β-cells and its targeted inactivation impairs glucose tolerance. Endocrinology 2013, 154, 3796–3806. [Google Scholar] [CrossRef] [PubMed]
- Kalyani, R.R.; Egan, J.M. Diabetes and altered glucose metabolism with aging. Endocrinol. Metab. Clin. 2013, 42, 333–347. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (Weeks) | Mice Types | MDA (nmol/mL) | GSH (nmol/mL) | GSSG (nmol/mL) | GSH/ GSSG | GPX (U/mL) | T-SOD (U/mL) | CAT (U/mL) |
|---|---|---|---|---|---|---|---|---|
| 12 | C57BL/6J | 11.4 ± 1.0 | 9.9 ± 1.6 | 4.3 ± 0.3 | 2.3 ± 0.3 | 535.6 ± 90.7 | 149.3 ± 9.4 | 16.9 ± 2.2 |
| KO | 12.0 ± 0.7 | 7.5 ± 1.1 ** | 4.1 ± 0.5 | 1.8 ± 0.3 * | 350.6 ± 33.8 ** | 138.4 ± 5.6 | 15.6 ± 2.5 | |
| 16 | C57BL/6J | 10.5 ± 1.0 | 10.3 ± 2.1 | 5.2 ± 0.7 | 2.0 ± 0.3 | 422.0 ± 85.1 | 141.8 ± 5.4 | 14.2 ± 2.1 |
| KO | 12.9 ± 1.1 * | 8.6 ± 1.7 | 5.0 ± 1.0 | 1.7 ± 0.3 | 382.5 ± 35.6 | 127.6 ± 7.1 * | 14.5 ± 2.3 | |
| 21 | C57BL/6J | 11.5 ± 1.3 | 10.0 ± 1.8 | 5.5 ± 0.5 | 1.8 ± 0.4 | 438.5 ± 69.0 | 148.1 ± 6.2 | 13.4 ± 2.1 |
| KO | 11.8 ± 1.4 | 8.3 ± 1.8 | 5.1 ± 0.5 | 1.6 ± 0.3 | 395.6 ± 69.7 | 125.6 ± 8.4 * | 13.1 ± 1.9 |
| Age (Weeks) | Mice Types | MDA (nmol/ mg Protein) | GSH (nmol/mg Protein) | GSSG (nmol/mg Protein) | GSH/ GSSG | GPX (U/mg Protein) | CAT (U/mg Protein) | T-SOD (U/mg Protein) |
|---|---|---|---|---|---|---|---|---|
| 12 | C57BL/6J | 1.8 ± 0.3 | 2.27 ± 0.25 | 0.26 ± 0.04 | 8.8 ± 1.5 | 396.8 ± 36.2 | 165.8 ± 39.1 | 311.3 ± 32.3 |
| KO | 2.2 ± 0.3 * | 1.93 ± 0.29 * | 0.35 ± 0.05 * | 5.6 ± 0.7 ** | 336.4 ± 40.8 ** | 175.6 ± 36.9 | 284.5 ± 23.9 | |
| 16 | C57BL/6J | 1.6 ± 0.3 | 2.19 ± 0.45 | 0.38 ± 0.06 | 5.8 ± 0.9 | 410.3 ± 43.8 | 169.3 ± 38.8 | 284.1 ± 22.8 |
| KO | 1.9 ± 0.4 | 2.02 ± 0.37 | 0.39 ± 0.07 | 5.3 ± 1.1 | 374.8 ± 41.1 | 182.0 ± 37.8 | 309.7 ± 31.2 | |
| 21 | C57BL/6J | 1.8 ± 0.3 | 2.60 ± 0.38 | 0.50 ± 0.06 | 5.2 ± 0.9 | 430.3 ± 90.3 | 186.9 ± 43.6 | 254.8 ± 34.6 |
| KO | 2.0 ± 0.4 | 2.30 ± 0.21 | 0.45 ± 0.05 | 5.1 ± 0.6 | 382.9 ± 41.4 | 183.6 ± 41.6 | 278.5 ±30.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Zhang, Y.; Zhou, J.; Liu, H. Selenoprotein F Knockout Caused Glucose Metabolism Disorder in Young Mice by Disrupting Redox Homeostasis. Antioxidants 2022, 11, 2105. https://doi.org/10.3390/antiox11112105
Li M, Zhang Y, Zhou J, Liu H. Selenoprotein F Knockout Caused Glucose Metabolism Disorder in Young Mice by Disrupting Redox Homeostasis. Antioxidants. 2022; 11(11):2105. https://doi.org/10.3390/antiox11112105
Chicago/Turabian StyleLi, Min, Yun Zhang, Jun Zhou, and Hongmei Liu. 2022. "Selenoprotein F Knockout Caused Glucose Metabolism Disorder in Young Mice by Disrupting Redox Homeostasis" Antioxidants 11, no. 11: 2105. https://doi.org/10.3390/antiox11112105
APA StyleLi, M., Zhang, Y., Zhou, J., & Liu, H. (2022). Selenoprotein F Knockout Caused Glucose Metabolism Disorder in Young Mice by Disrupting Redox Homeostasis. Antioxidants, 11(11), 2105. https://doi.org/10.3390/antiox11112105