
Molecular Functions of Hydrogen Sulfide in Cancer

Abstract
:1. Introduction
2. H2S Chemistry, Synthesis, and Molecular Mechanisms of Action
2.1. H2S Chemistry
2.2. H2S Synthesis
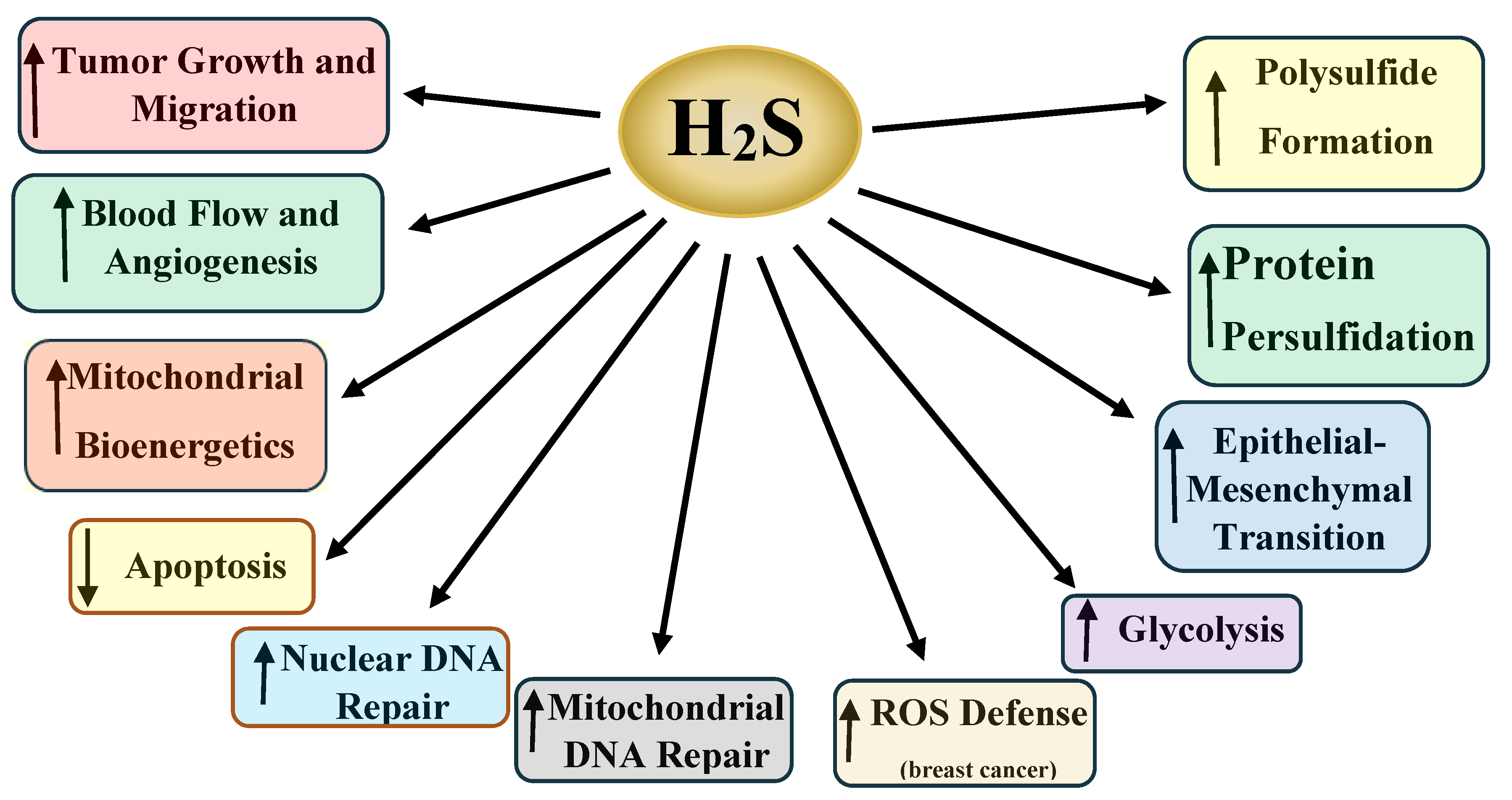
2.3. Molecular Mechanisms of H2S Activity
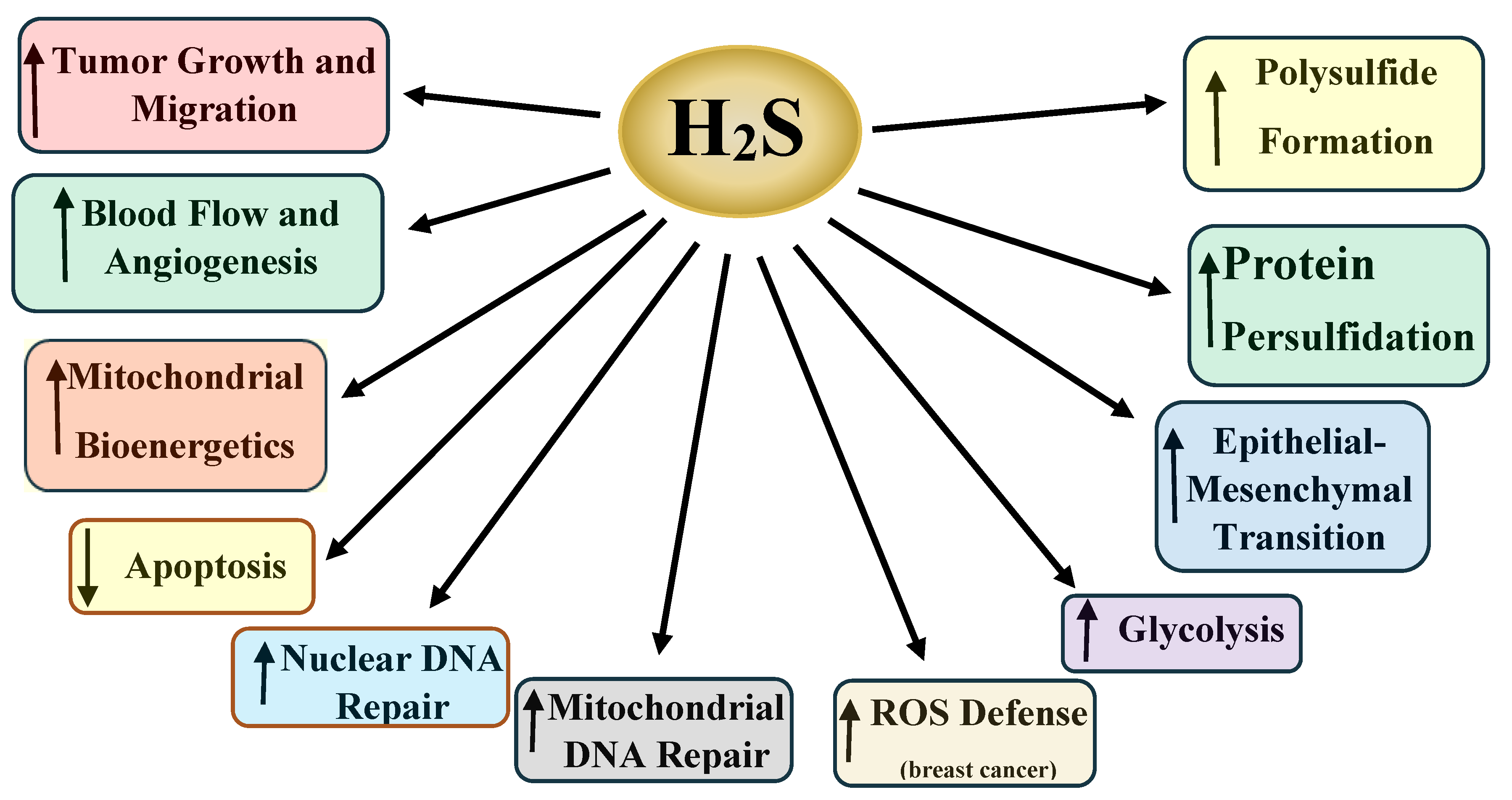
3. H2S and Cancer
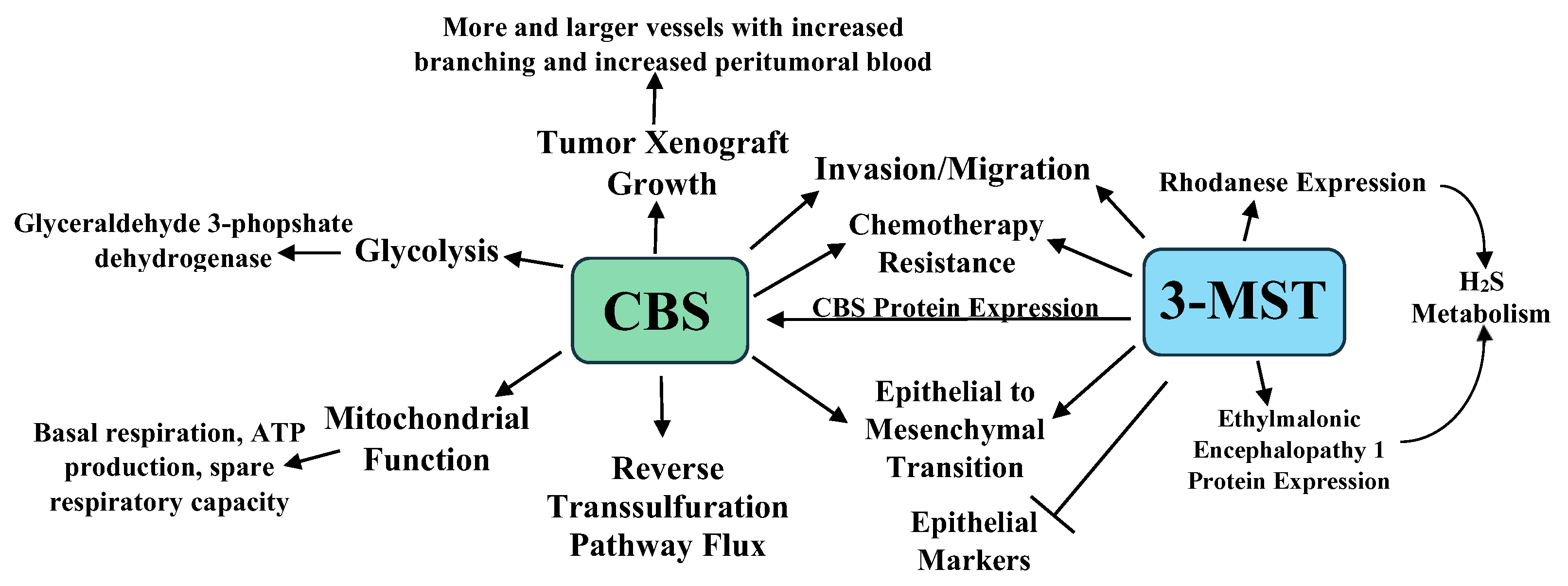
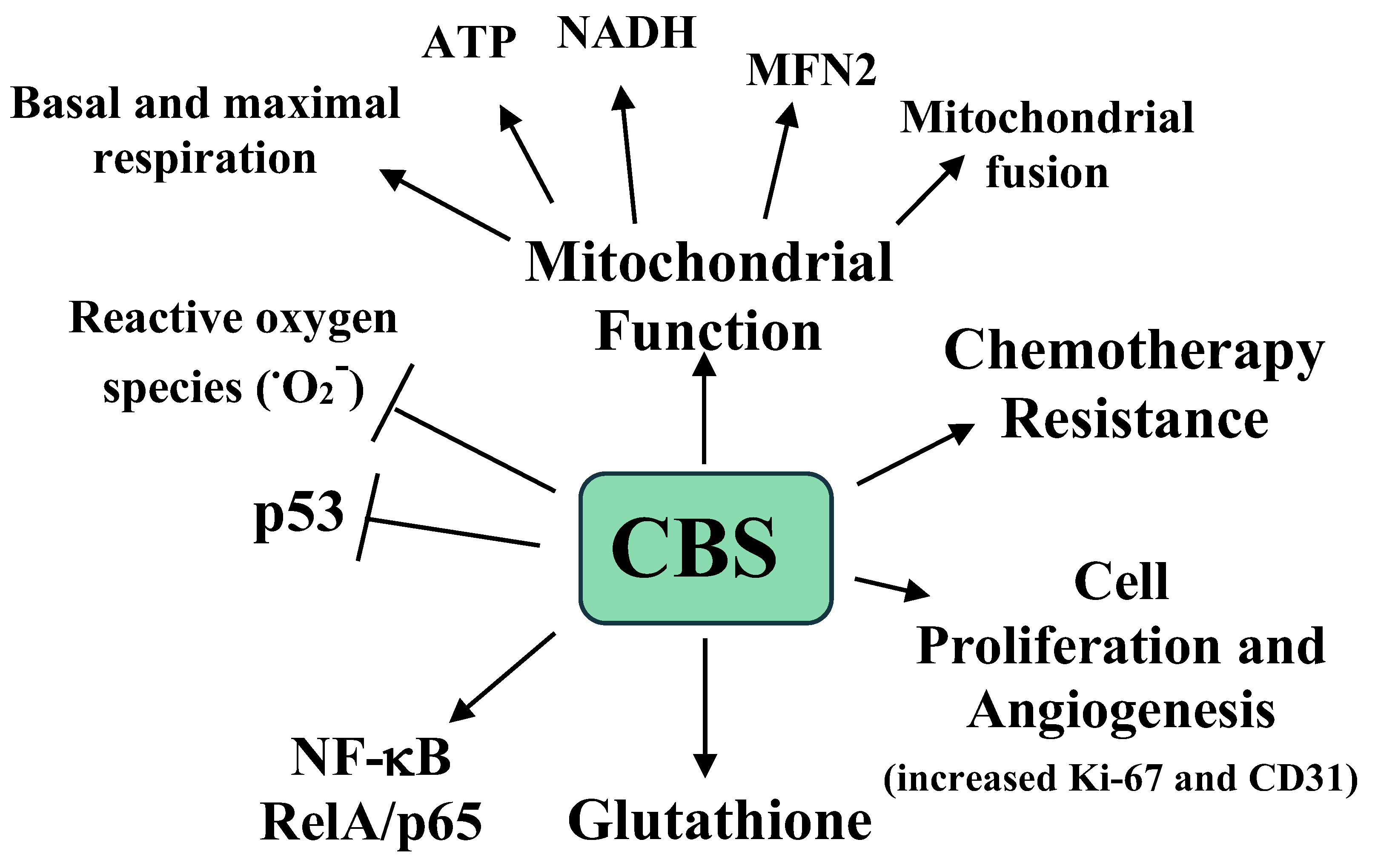
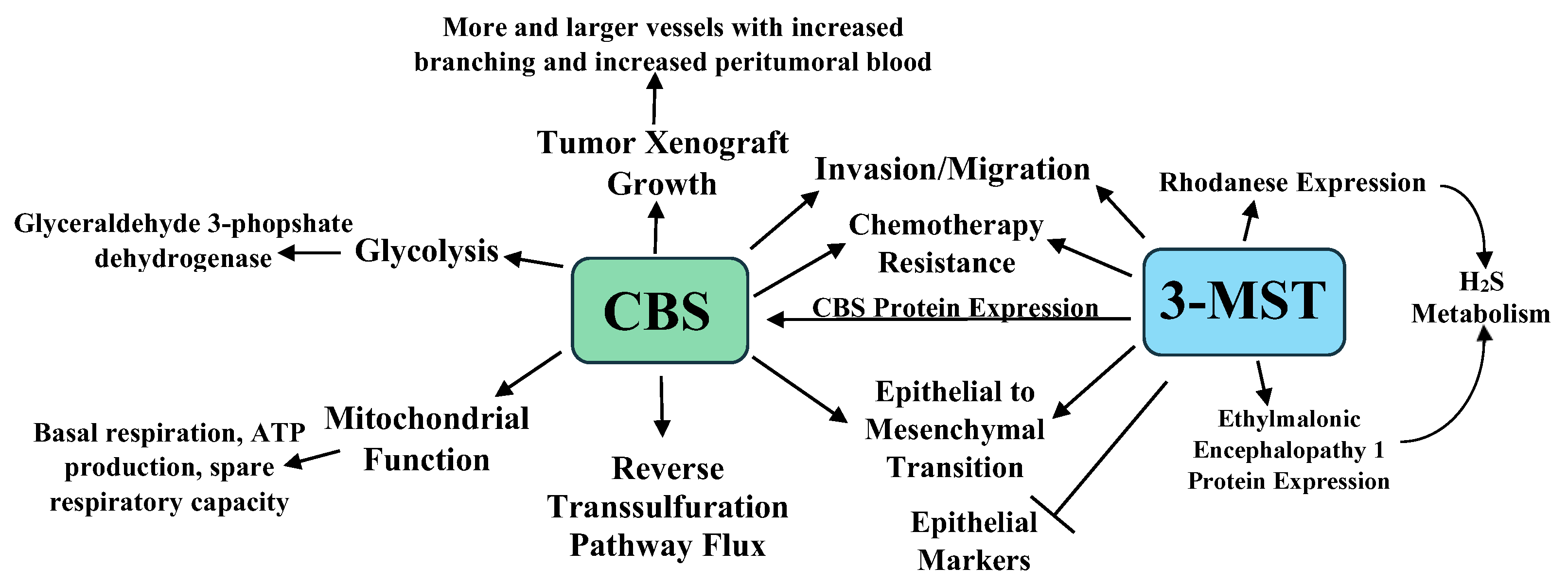
3.1. H2S and Colorectal Cancer
3.2. H2S and Ovarian Cancer
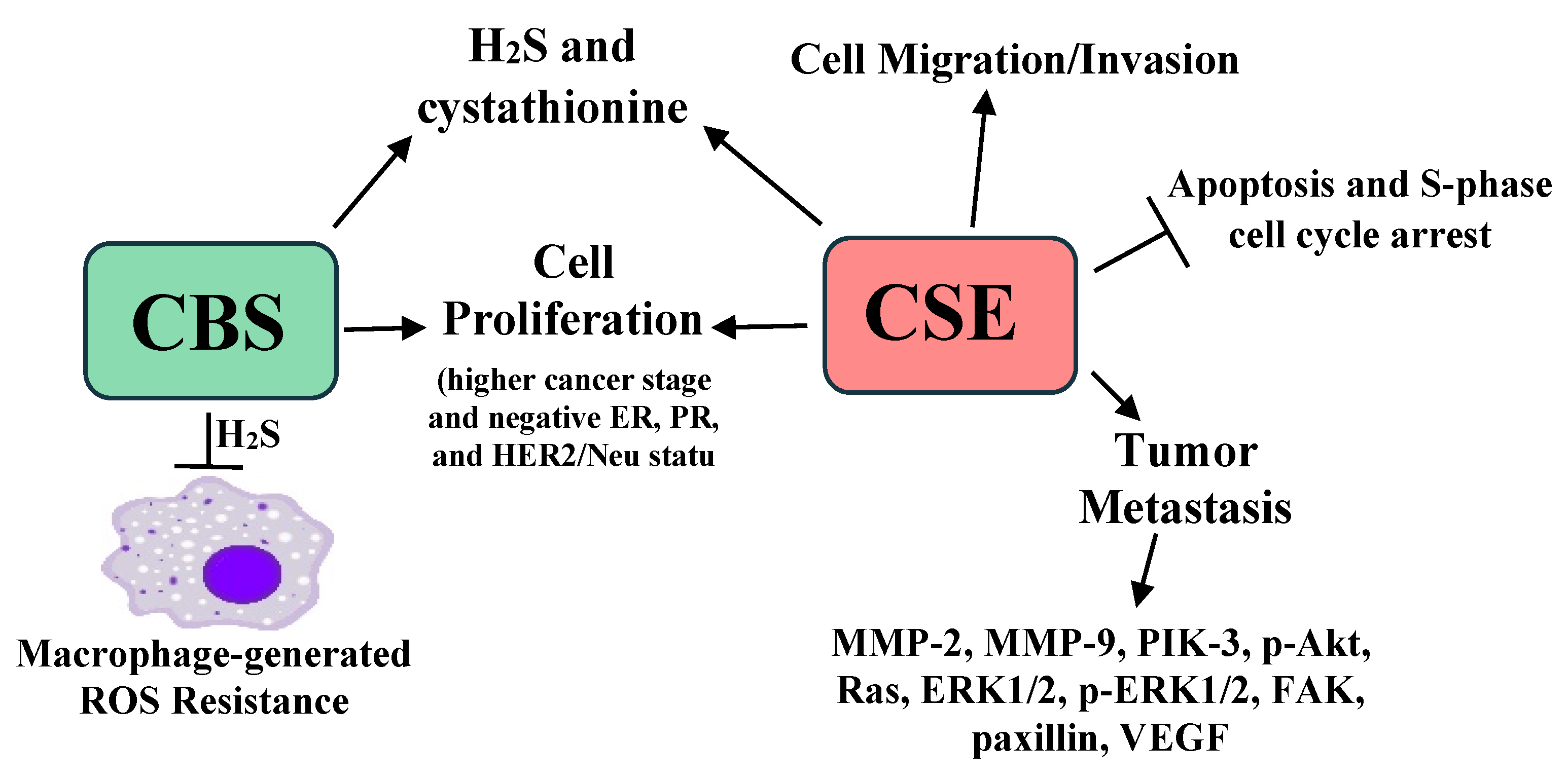
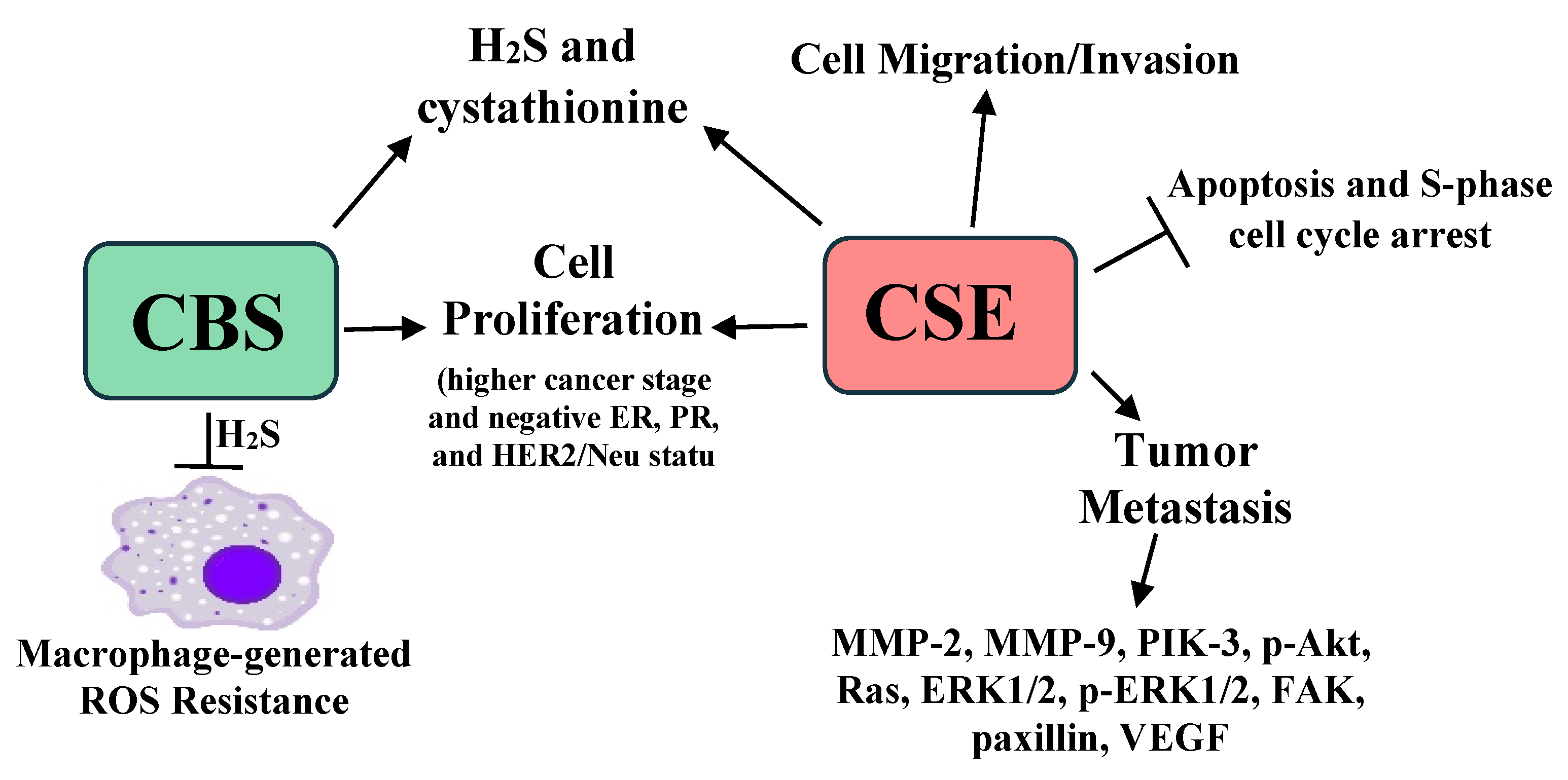
3.3. Breast Cancer
3.4. Bladder Cancer
3.5. Renal Cancer
3.6. Prostate Cancer
3.7. Thyroid
3.8. Pulmonary Adenocarcinoma
3.9. Melanoma
3.10. Oral Squamous Cell Carcinoma
4. Protein Sulfhydration and Cancer
4.1. NF-κB
4.2. GAPDH
4.3. Lactate Dehydrogenase (LDHA)
5. Polysulfides and Cancer
6. H2S-Associated Tumor Markers in Bodily Gas and Fluids
7. H2S and Nuclear DNA Repair
8. The Immunomodulatory Role of H2S in Cancer
9. Conclusions, Issues, and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reiffenstein, R.J.; Hulbert, W.C.; Roth, S.H. Toxicology of hydrogen sulfide. Annu. Rev. Pharmacol. Toxocol. 1992, 32, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.A.; Roth, S.H.; Zhang, A.; Brookes, J.; Skrajny, B.; Bennington, R. Inhibition of respiratory and bioenergetic mechanisms by hydrogen sulfide in mammalian brain. J. Toxicol. Environ. Health A 1998, 54, 491–507. [Google Scholar]
- Warenycia, M.W.; Smith, K.A.; Blashko, C.S.; Kombian, S.B.; Reiffenstein, R.J. Monoamine oxidase inhibition as a sequel of hydrogen sulfide intoxication: Increases in brain catecholamine and 5-hydroxytryptamine levels. Arch. Toxicol. 1989, 63, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shen, X.; Pardue, S.; Meram, A.T.; Rajendran, S.; Ghali, G.; Kevil, C.G.; Shackelford, R.E. The Ataxia telangiectasia-mutated and Rad3-related protein kinase regulates cellular hydrogen sulfide concentrations. DNA Repair 2019, 73, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [Green Version]
- Nagy, P. Mechanistic chemical perspective of hydrogen sulfide signaling. Methods Enzymol. 2015, 554, 3–29. [Google Scholar]
- Geng, B.; Yang, J.; Qi, Y.; Zhao, J.; Pang, Y.; Du, J.; Tang, C. H2S generated by heart in rat and its effects on cardiac function. Biochem. Biophys. Res. Commun. 2004, 313, 362–368. [Google Scholar] [CrossRef]
- Fu, Z.; Liu, X.; Geng, B.; Fang, L.; Tang, C. Hydrogen sulfide protects rat lung from ischemia-reperfusion injury. Life Sci. 2008, 82, 1196–1202. [Google Scholar] [CrossRef]
- Distrutti, E.; Sediari, L.; Mencarelli, A.; Renga, B.; Otlandi, S.; Antonelli, E.; Roviezzo, F.; Morelli, A.; Cirino, G.; Wallace, J.L.; et al. Evidence that hydrogen sulfide exerts antinociceptive effects on the gastrointestinal tract by activating KATP channels. J. Pharmacol. Exp. Ther. 2006, 316, 325–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhao, X.; Jin, H.; Wei, H.; Li, W.; Bu, D.; Tang, X.; Ren, Y.; Tang, C.; Du, J. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Desai, K.M.; Chang, T.; Untereiner, A.; Wu, L. Hydrogen sulfide and the metabolic syndrome. Expert Rev. Clin. Pharmacol. 2011, 4, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Panagaki, T.; Randi, E.B.; Augsburger, F.; Szabo, C. Overproduction of H2S, generated by CBS, inhibits mitochondrial Complex IV and suppresses oxidative phosphorylation in Down syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 18769–18771. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Snyder, S.H. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 2018, 149, 101–109. [Google Scholar] [CrossRef]
- Wu, D.; Si, W.; Wang, M.; Lv, S.; Ji, A.; Li, Y. Hydrogen sulfide in cancer: Friend or foe? Nitric Oxide 2015, 50, 38–45. [Google Scholar] [CrossRef]
- Meram, A.T.; Chen, J.; Patel, S.; Kim, D.D.; Shirley, B.; Covello, P.; Coppola, D.; Wei, E.X.; Ghali, G.; Kevil, C.G. Hydrogen Sulfide Is Increased in Oral Squamous Cell Carcinoma Compared to Adjacent Benign Oral Mucosae. Anticancer Res. 2018, 38, 3843–3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabibzadeh, S. Nature creates, adapts, protects and sustains life using hydrogen sulfide. Front. Biosci. 2016, 21, 528–560. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Shen, X.; Bir, S.C.; Kevil, C.G. Hydrogen sulfide chemical biology: Pathophysiological roles and detection. Nitric Oxide 2013, 35, 5–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuevasanta, E.; Denicola, A.; Alvarez, B.; Moller, M.N. Solubility and permeation of hydrogen sulfide in lipid membranes. PLoS ONE 2012, 7, e34562. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef]
- Kimura, Y.; Kimura, H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef]
- Whiteman, M.; Armstrong, J.S.; Chu, S.H.; Jia-Ling, S.; Wong, B.S.; Cheung, N.S.; Halliwell, B.; Moore, P.K. The novel neuromodulator hydrogen sulfide: An endogenous peroxynitrite ‘scavenger’? J. Neurochem. 2004, 90, 765–768. [Google Scholar] [CrossRef]
- Whiteman, M.; Cheung, N.S.; Zhu, Y.-Z.; Chu, S.H.; Siau, J.L.; Wong, B.S.; Armstrong, J.S.; Moore, P.K. Hydrogen sulphide: A novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem. Biophys. Res. Commun. 2005, 326, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Li, L.; Kostetski, I.; Chu, S.H.; Siau, J.L.; Bhatia, M.; Moore, P.K. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem. Biophys. Res. Commun. 2006, 343, 303–310. [Google Scholar] [CrossRef]
- Olson, K.; DeLeon, E.R.; Liu, F. Controversies in hydrogen sulfide biology. Nitric Oxide 2014, 41, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Carballal, S.; Trujillo, M.; Cuevasanta, E.; Bartesaghi, S.; Möller, M.N.; Folkes, L.K.; García-Bereguiaín, M.A.; Gutiérrez-Merino, C.; Wardman, P.; Denicola, A.; et al. Reactivity of hydrogen sulfide with peroxynitrite and other oxidants of biological interest. Free Radic. Biol. Med. 2011, 50, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Kabil, O.; Banerjee, R. High turnover rates for hydrogen sulfide allow for rapid regulation of its tissue concentrations. Antioxid. Redox. Signal. 2012, 17, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Gutscher, M.; Pauleau, A.-L.; Marty, L.; Brach, T.; Wabnitz, G.H.; Samstag, Y.; Meyer, A.J.; Dick, T.P. Real-time imaging of the intracellular glutathione redox potential. Nat. Methods 2008, 5, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Peter, E.A.; Bir, S.; Wang, R.; Kevil, C.G. Analytical measurement of discrete hydrogen sulfide pools in biological specimens. Free Radic. Biol. Med. 2012, 52, 2276–2283. [Google Scholar] [CrossRef] [Green Version]
- Łowicka, E.; Bełtowski, J. Hydrogen sulfide (H2S)—The third gas of interest for pharmacologists. Pharmacol. Rep. 2007, 59, 4–24. [Google Scholar]
- Rajendran, S.; Shen, X.; Glawe, J.; Kolluru, G.K.; Kevil, C.G. Nitric Oxide and Hydrogen Sulfide Regulation of Ischemic Vascular Growth and Remodeling. Compr. Physiol. 2019, 9, 1213–1247. [Google Scholar] [PubMed]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox. Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef]
- Kimura, Y.; Toyofuku, Y.; Koike, S.; Shibuya, N.; Nagahara, N.; Lefer, D.; Ogasawara, Y.; Kimura, H. Identification of H2S3 and H2S produced by 3-mercaptopyruvate sulfurtransferase in the brain. Sci. Rep. 2015, 5, 14774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuto, J.M.; Ignarro, L.J.; Nagy, P.; Wink, D.A.; Kevil, C.G.; Feelisch, M.; Cortese-Krott, M.M.; Bianco, C.L.; Kumagai, Y.; Hobbs, A.J.; et al. Biological hydropersulfides and related polysulfides—A new concept and perspective in redox biology. FEBS Lett. 2018, 592, 2140–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, K.; Hishiki, T.; Yamamoto, S.; Yamamoto, T.; Miura, N.; Kubo, A.; Itoh, M.; Chen, W.Y.; Takano, M.; Yoshikawa, T.; et al. On-tissue polysulfide visualization by surface-enhanced Raman spectroscopy benefits patients with ovarian cancer to predict post-operative chemosensitivity. Redox Biol. 2021, 41, 101926. [Google Scholar] [CrossRef]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Minkler, P.; Grove, D.; Wang, R.; Willard, B.; Dweik, R.; Hine, C. Non-enzymatic hydrogen sulfide production from cysteine in blood is catalyzed by iron and vitamin B6. Commun. Biol. 2019, 2, 194. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Carlström, M.; Borniquel, S.; Jädert, C.; Kevil, C.G.; Lundberg, J.O. Microbial regulation of host hydrogen sulfide bioavailability and metabolism. Free Radic. Biol. Med. 2013, 60, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Vasas, A.; Dóka, E.; Fábián, I.; Nagy, P. Kinetic and thermodynamic studies on the disulfide-bond reducing potential of hydrogen sulfide. Nitric Oxide 2015, 46, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Szabo, C.; Coletta, C.; Chao, C.; Módis, K.; Szczesny, B.; Papapetropoulos, A.; Hellmich, M.R. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12474–12479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Módis, K.; Coletta, C.; Asimakopoulou, A.; Szczesny, B.; Chao, C.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Effect of S-adenosyl-L-methionine (SAM), an allosteric activator of cystathionine-beta-synthase (CBS) on colorectal cancer cell proliferation and bioenergetics in vitro. Nitric Oxide 2014, 41, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, C.M.; Zatarain, J.R.; Nicholls, M.E.; Porter, C.; Widen, S.G.; Thanki, K.; Johnson, P.; Jawad, M.U.; Moyer, M.P.; Randall, J.W.; et al. Upregulation of Cystathionine-β-Synthase in Colonic Epithelia Reprograms Metabolism and Promotes Carcinogenesis. Cancer Res. 2017, 77, 5741–5754. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Min, X.; Shen, M.; Hua, Q.; Han, Y.; Zhao, L.; Liu, L.; Huang, G.; Liu, J.; Zhao, X. ACLY facilitates colon cancer cell metastasis by CTNNB1. J. Exp. Clin. Cancer Res. 2019, 38, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascenção, K.; Dilek, N.; Augsburger, F.; Panagaki, T.; Zuhra, K.; Szabo, C. Pharmacological induction of mesenchymal-epithelial transition via inhibition of H2S biosynthesis and consequent suppression of ACLY activity in colon cancer cells. Pharmacol. Res. 2021, 165, 105393. [Google Scholar] [CrossRef]
- Chen, S.; Yue, T.; Huang, Z.; Zhu, J.; Bu, D.; Wang, X.; Pan, Y.; Liu, Y.; Wang, P. Inhibition of hydrogen sulfide synthesis reverses acquired resistance to 5-FU through miR-215-5p-EREG/TYMS axis in colon cancer cells. Cancer Lett. 2019, 466, 49–60. [Google Scholar] [CrossRef]
- Yue, T.; Zuo, S.; Bu, D.; Zhu, J.; Chen, S.; Ma, Y.; Ma, J.; Guo, S.; Wen, L.; Zhang, X.; et al. Aminooxyacetic acid (AOAA) sensitizes colon cancer cells to oxaliplatin via exaggerating apoptosis induced by ROS. J. Cancer 2020, 11, 1828–1838. [Google Scholar] [CrossRef]
- Untereiner, A.A.; Pavlidou, A.; Druzhyna, N.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Drug resistance induces the upregulation of H2S-producing enzymes in HCT116 colon cancer cells. Biochem. Pharmacol. 2018, 149, 174–185. [Google Scholar] [CrossRef]
- Jelovac, D.; Armstrong, D.K. Recent progress in the diagnosis and treatment of ovarian cancer. CA Cancer J. Clin. 2011, 61, 183–203. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Saha, S.; Giri, K.; Lanza, I.R.; Nair, K.S.; Jennings, N.B.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Basal, E.; Weaver, A.L.; et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS ONE 2013, 8, e79167. [Google Scholar] [CrossRef]
- Chakraborty, P.K.; Murphy, B.; Mustafi, S.B.; Dey, A.; Xiong, X.; Rao, G.; Naz, S.; Zhang, M.; Yang, D.; Dhanasekaran, D.N.; et al. Cystathionine beta-synthase regulates mitochondrial morphogenesis in ovarian cancer. FASEB J. 2018, 32, 4145–4157. [Google Scholar] [CrossRef] [Green Version]
- Benson, J.R.; Jatoi, I. The global breast cancer burden. Future Oncol. 2012, 8, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Kawahara, B.; Mahata, S.K.; Tsai, R.; Yoon, A.; Hwang, L.; Hu-Moore, K.; Villanueva, C.; Vajihuddin, A.; Parameshwar, P.; et al. Cystathionine: A novel oncometabolite in human breast cancer. Arch. Biochem. Biophys. 2016, 604, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Kawahara, B.; Gupta, D.; Tsai, R.; Khachatryan, M.; Roy-Chowdhuri, S.; Bose, S.; Yoon, A.; Faull, K.; Farias-Eisner, R.; et al. Role of cystathionine beta-synthase in human breast cancer. Free Radic. Biol. Med. 2015, 86, 228–238. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Shi, X.; Liang, H.; Ye, J.; Wang, L.; Han, H.; Fang, H.; Kang, W.; Wang, T. Cystathionine-gamma-lyase promotes process of breast cancer in association with STAT3 signaling pathway. Oncotarget 2017, 8, 65677–65686. [Google Scholar] [CrossRef] [Green Version]
- da Silva, J.L.; Nunes, N.C.C.; Izetti, P.; de Mesquita, G.G.; de Melo, A.C. Triple negative breast cancer: A thorough review of biomarkers. Crit. Rev. Oncol. Hematol. 2020, 145, 102855. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, H.; Liu, Y.; Zhang, W.; Duan, X.; Li, M.; Shi, X.; Wang, T. Cystathionine-gamma-lyase promotes the metastasis of breast cancer via the VEGF signaling pathway. Int. J. Oncol. 2019, 55, 473–487. [Google Scholar] [PubMed] [Green Version]
- Liu, Y.; Wang, L.; Zhang, X.; Deng, Y.; Pan, L.; Li, H.; Shi, X.; Wang, T. A novel cystathionine gamma-lyase inhibitor, I194496, inhibits the growth and metastasis of human TNBC via downregulating multiple signaling pathways. Sci. Rep. 2021, 11, 8963. [Google Scholar] [CrossRef]
- Letašiová, S.; Medvedová, A.; Šovčíková, A.; Dušinská, M.; Volkovová, K.; Mosoiu, C.; Bartonová, A. Bladder cancer, a review of the environmental risk factors. Environ. Health 2012, 1, S11. [Google Scholar] [CrossRef] [Green Version]
- Gai, J.-W.; Qin, W.; Liu, M.; Wang, H.-F.; Zhang, M.; Li, M.; Zou, W.-H.; Ma, G.-M.; Liu, G.-M.; Song, W.-H.; et al. Expression profile of hydrogen sulfide and its synthases correlates with tumor stage and grade in urothelial cell carcinoma of bladder. Urol. Oncol. 2016, 34, 166.e15–166.e20. [Google Scholar] [CrossRef]
- Liu, H.; Chang, J.; Zhao, Z.; Li, Y.; Hou, J. Effects of exogenous hydrogen sulfide on the proliferation and invasion of human bladder cancer cells. J. Cancer Res. Ther. 2017, 13, 829–832. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN. Int. J. Cancer 2012, 136, E359–E386. [Google Scholar] [CrossRef]
- Sonke, E.; Verrydt, M.; Postenka, C.O.; Pardhan, S.; Willie, C.J.; Mazzola, C.R.; Hammers, M.D.; Pluth, M.D.; Lobb, I.; Power, N.E.; et al. Inhibition of endogenous hydrogen sulfide production in clear-cell renal cell carcinoma cell lines and xenografts restricts their growth, survival and angiogenic potential. Nitric Oxide 2015, 49, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Shackelford, R.E.; Abdulsattar, J.; Wei, E.X.; Cotelingam, J.; Coppola, D.; Herrera, G.A. Increased Nicotinamide Phosphoribosyltransferase and Cystathionine-β-Synthase in Renal Oncocytomas, Renal Urothelial Carcinoma, and Renal Clear Cell Carcinoma. Anticancer Res. 2017, 37, 3423–3427. [Google Scholar]
- Breza, J.; Soltysova, A.; Hudecova, S.; Penesova, A.; Szadvari, I.; Babula, P.; Chovancova, B.; Lencesova, L.; Pos, O.; Breza, J. Endogenous H2S producing enzymes are involved in apoptosis induction in clear cell renal cell carcinoma. BMC Cancer 2018, 18, 541. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.H.; Huang, J.T.; Chen, W.L.; Wang, R.H.; Kao, M.C.; Pan, Y.R.; Chan, S.H.; Tsai, K.W.; Kung, H.J.; Lin, K.T. Dysregulation of cystathionine gamma-lyase promotes prostate cancer progression and metastasis. EMBO Rep. 2019, 20, e45986. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Li, S.; Wu, L.; Lai, C.; Yang, G. Hydrogen sulfide represses androgen receptor transactivation by targeting at the second zinc finger module. J. Biol. Chem. 2014, 289, 20824–20835. [Google Scholar] [CrossRef] [Green Version]
- Schlumberger, M.J. Papillary and follicular thyroid carcinoma. N. Engl. J. Med. 1998, 338, 297–306. [Google Scholar] [CrossRef]
- Turbat-Herrera, E.A.; Kilpatrick, M.J.; Chen, J.; Meram, A.T.; Cotelingam, J.; Ghali, G.; Kevil, C.G.; Coppola, D.; Shackelford, R.E. Cystathione β-Synthase Is Increased in Thyroid Malignancies. Anticancer Res. 2018, 38, 6085–6090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Ma, N.; Wei, P.; Zeng, Z.; Meng, J. Expression of hydrogen sulfide synthases and Hh signaling pathway components correlate with the clinicopathological characteristics of papillary thyroid cancer patients. Int. J. Clin. Exp. Pathol. 2018, 11, 1818–1824. [Google Scholar] [PubMed]
- Wu, D.; Li, J.; Zhang, Q.; Tian, W.; Zhong, P.; Liu, Z.; Wang, H.; Wang, H.; Ji, A.; Li, Y. Exogenous Hydrogen Sulfide Regulates the Growth of Human Thyroid Carcinoma Cells. Oxid. Med. Cell. Longev. 2019, 16, 6927298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczesny, B.; Marcatti, M.; Zatarain, J.R.; Druzhyna, N.; Wiktorowicz, J.E.; Nagy, P.; Hellmich, M.R.; Szabo, C. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA Repair and suppressing cellular bioenergetics. Sci. Rep. 2016, 6, 36125. [Google Scholar] [CrossRef] [Green Version]
- Ali, Z.; Yousaf, N.; Larkin, J. Melanoma epidemiology, biology and prognosis. EJC Suppl. 2013, 11, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Panza, E.; De Cicco, P.; Armogida, C.; Scognamiglio, G.; Gigantino, V.; Botti, G.; Germano, D.; Napolitano, M.; Papapetropoulos, A.; Bucci, M.; et al. Role of the cystathionine gamma lyase/hydrogen sulfide pathway in human melanoma progression. Pigment Cell Melanoma Res. 2015, 28, 61–72. [Google Scholar] [CrossRef]
- Gu, X.; Zhu, Y.Z. Therapeutic applications of organosulfur compounds as novel hydrogen sulfide donors and/or mediators. Expert. Rev. Clin. Pharmacol. 2011, 4, 123–133. [Google Scholar] [CrossRef]
- Mondal, A.; Banerjee, A.; Bose, S.; Mazumder, S.; Haber, R.A.; Bishayee, A. Garlic constituents for cancer prevention and therapy: From phytochemistry to novel formulations. Pharmacol. Res. 2021. [Google Scholar] [CrossRef]
- Szabo, C.; Papapetropoulos, A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharmacol. Rev. 2017, 69, 497–564. [Google Scholar] [CrossRef] [Green Version]
- Ueda, Y.; Richmond, A. NF-κB activation in melanoma. Pigment Cell Res. 2006, 19, 112–124. [Google Scholar] [CrossRef] [Green Version]
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Synder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 10, ra72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untereiner, A.A.; Oláh, G.; Módis, K.; Hellmich, M.R.; Szabo, C. H2S-induced S-sulfhydration of lactate dehydrogenase a (LDHA) stimulates cellular bioenergetics in HCT116 colon cancer cells. Biochem. Pharmacol. 2017, 136, 86–98. [Google Scholar] [CrossRef]
- Greiner, R.; Pálinkás, Z.; Bäsell, K.; Becher, D.; Antelmann, H.; Nagy, P.; Dick, T.P. Polysulfides link H2S to protein thiol oxidation. Antioxid. Redox. Signal. 2013, 19, 1749–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Huang, J.; Cushnir, J.R.; Španěl, P.; Smith, D.; Hanna, G.B. Selected ion flow tube-MS analysis of headspace vapor from gastric content for the diagnosis of gastro-esophageal cancer. Anal. Chem. 2012, 84, 9550–9557. [Google Scholar] [CrossRef]
- Bhatt, A.; Parsi, M.A.; Stevens, T.; Gabbard, S.; Kumaravel, A.; Jang, S. Volatile organic compounds in plasma for the diagnosis of esophageal adenocarcinoma: A pilot study. Gastrointest. Endosc. 2016, 84, 597–603. [Google Scholar] [CrossRef]
- Yamagishi, K.; Onuma, K.; Chiba, Y.; Yagi, S.; Aoki, S.; Sato, T.; Sugawara, Y.; Hosoya, N.; Saeki, Y.; Takahashi, M.; et al. Generation of gaseous sulfur-containing compounds in tumour tissue and suppression of gas diffusion as an antitumour treatment. Gut 2012, 61, 554–561. [Google Scholar] [CrossRef]
- Chwatko, G.; Forma, E.; Wilkosz, J.; Głowacki, R.; Jóźwiak, P.; Różański, W.; Bryś, M.; Krześlak, A. Thiosulfate in urine as a facilitator in the diagnosis of prostate cancer for patients with prostate-specific antigen less or equal 10 ng/mL. Clin. Chem. Lab. Med. 2013, 51, 1825–1831. [Google Scholar] [CrossRef]
- Zhang, W.; Braun, A.; Bauman, Z.; Olteanu, H.; Madzelan, P.; Banerjee, R. Expression profiling of homocysteine junction enzymes in the NCI60 panel of human cancer cell lines. Cancer Res. 2005, 65, 1554–1560. [Google Scholar] [CrossRef] [Green Version]
- Stabler, S.; Koyama, T.; Zhao, Z.; Martinez-Ferrer, M.; Allen, R.H.; Luka, Z.; Loukachevitch, L.V.; Clark, P.E.; Wagner, C.; Bhowmick, N.A. Serum methionine metabolites are risk factors for metastatic prostate cancer progression. PLoS ONE 2011, 6, e22486. [Google Scholar] [CrossRef] [Green Version]
- Stephan, C.; Jung, K.; Miller, K.; Ralla, B. New biomarkers in serum and urine for detection of prostate cancer. Aktuelle Urol. 2015, 46, 129–143. [Google Scholar]
- Hasan, T.; Arora, R.; Bansal, A.K.; Bhattacharya, R.; Sharma, G.S.; Singh, R.L. Disturbed homocysteine metabolism is associated with cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Q.-Z.; Zhang, K.; Li, L.-Y.; Pluth, M.D.; Yi, L.; Xi, Z. Dual-biomarker-triggered fluorescence probes for differentiating cancer cells and revealing synergistic antioxidant effects under oxidative stress. Chem. Sci. 2019, 10, 1945–1952. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Zhang, J.; Xi, Z.; Li, L.-Y.; Gu, X.; Zhang, Q.-Z.; Yi, L. A new H2S-specific near-infrared fluorescence-enhanced probe that can visualize the H2S level in colorectal cancer cells in mice. Chem. Sci. 2017, 8, 2776–2781. [Google Scholar] [CrossRef] [Green Version]
- Shackelford, R.; Ozluk, E.; Islam, M.Z.; Hopper, B.; Meram, A.; Ghali, G.; Kevil, C.G. Hydrogen sulfide and DNA repair. Redox Biol. 2021, 38, 101675. [Google Scholar] [CrossRef]
- Parikh, R.A.; Appleman, L.J.; Bauman, J.E.; Sankunny, M.; Lewis, D.W.; Vlad, A.; Gollin, S.M. Upregulation of the ATR-CHEK1 pathway in oral squamous cell carcinomas. Genes Chromosomes Cancer 2014, 53, 25–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Fatah, T.M.A.; Middleton, F.K.; Arora, A.; Agarwal, D.; Chen, T.; Moseley, P.M.; Perry, C.; Doherty, R.; Chan, S.; Green, A.R.; et al. Untangling the ATR-CHEK1 network for prognostication, prediction and therapeutic target validation in breast cancer. Mol. Oncol. 2015, 9, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Dean, D.C.; Hornicek, F.J.; Wang, J.; Jia, Y.; Duan, Z.; Shi, H. ATR and p-ATR are emerging prognostic biomarkers and DNA damage response targets in ovarian cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920982853. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-C.; Yang, J.-C.; Lu, M.-C.; Lee, C.-L.; Peng, C.-Y.; Hsu, W.-Y.; Dai, Y.-H.; Chang, F.-R.; Zhang, D.-Y.; Wu, W.-J. ATR-Chk1 signaling inhibition as a therapeutic strategy to enhance cisplatin chemosensitivity in urothelial bladder cancer. Oncotarget 2016, 7, 1947–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundar, R.; Brown, J.; Russo, A.I.; Yap, T.A. Targeting ATR in cancer medicine. Curr. Probl. Cancer 2017, 41, 302–315. [Google Scholar] [CrossRef]
- Dilek, N.; Papapetropoulos, A.; Toliver-Kinsky, T.; Csaba Szabo, C. Hydrogen sulfide: An endogenous regulator of the immune system. Pharmacol. Res. 2020, 161, 105119. [Google Scholar] [CrossRef]
- Cicco, P.D.; Ercolano, G.; Rubino, V.; Terrazzano, G.; Ruggiero, G.; Cirino, G.; Ianaro, A. Modulation of the functions of myeloid-derived suppressor cells: A new strategy of hydrogen sulfide anti-cancer effects. Br. J. Pharmacol. 2020, 177, 884–897. [Google Scholar] [CrossRef] [PubMed]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-β-Synthase: Molecular regulation and pharmacological inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Panopoulos, P.; Chasapis, C.T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G.A.; Papapetropoulos, A. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br. J. Pharmacol. 2013, 169, 922–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2008, 14, e1007362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Ding, L.; Xie, Z.Z.; Yang, Y.; Whiteman, M.; Moore, P.K.; Bian, J.-S. A Review of Hydrogen Sulfide Synthesis, Metabolism, and Measurement: Is Modulation of Hydrogen Sulfide a Novel Therapeutic for Cancer? Antioxid. Redox Signal. 2019, 31, 1–38. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type | H2S Synthesizing Enzyme Expression | Comments | References |
|---|---|---|---|
| Colon Cancer | CBS increased | Higher colon cancer CBS promotes cancer growth, mitochondrial bioenergetic activity, and increased glycolysis, migration, invasion, and chemotherapy resistance. While not increased, 3-MST expression promotes colon cancer EMT. | [42,43,48,49] |
| Ovarian Cancer | CBS and CSE increased | Increased CBS and CSE promote events including cancer growth, more active mitochondrial bioenergetics and morphologic integrity, migration, invasion, chemotherapy resistance, and a poor prognosis. Data also indicates a role for polysulfides in ovarian cancer. | [36,51,52] |
| Breast Cancer | CBS and CSE increased | Increased CBS and CSE promote cell growth, migration, and chemotherapy resistance. Membranous CBS protects cells from macrophage-derived ROS and confers a worse prognosis. CSE promotes breast cancer metastasis. | [39,56,58] |
| Bladder Cancer | CBS, CSE, and 3-MST increased | H2S likely promotes bladder cancer cell proliferation and invasion, and MMP-2 and MMP-9 protein expression. H2S synthesis in tumor lysates positively correlates with tumor stage and grade. | [61,62] |
| Renal Cancer | Most studies show suppressed or unchanged expression | H2S appears to promote tumor growth. The present studies show contradictory results, possibly based on analysis methods employed. | [64,65,66] |
| Prostate Cancer | Increased or decreased CSE in different studies | CSE promotes cell proliferation, migration, invasion, and poor patient survival. CSE promotes cell migration by an enzymatic activity-independent mechanism. Another study shows that CSE suppression promotes prostatic cancer. | [68,69] |
| Thyroid Cancer | CBS and CSE increased | Different studies show increased CBS or CSE. NaHS promotes thyroid cancer proliferation, migration, and cell viability. | [71,72,73] |
| Pulmonary adenocarcinoma | CBS, CSE, and 3-MST increased | H2S promotes cell proliferation, mitochondrial bioenergetics, and mitochondrial DNA repair. | [74] |
| Melanoma | Mildly increased CSE expression | CSE expression inhibited melanoma cell growth and H2S donors increased apoptosis by NF-κB inhibition. | [76] |
| Oral squamous cell carcinoma | CBS, CSE, and 3-MST increased | Direct measurements of tumor H2S concentrations revealed 13% higher H2S than in adjacent benign oral squamous epithelium. | [16] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shackelford, R.E.; Mohammad, I.Z.; Meram, A.T.; Kim, D.; Alotaibi, F.; Patel, S.; Ghali, G.E.; Kevil, C.G. Molecular Functions of Hydrogen Sulfide in Cancer. Pathophysiology 2021, 28, 437-456. https://doi.org/10.3390/pathophysiology28030028
Shackelford RE, Mohammad IZ, Meram AT, Kim D, Alotaibi F, Patel S, Ghali GE, Kevil CG. Molecular Functions of Hydrogen Sulfide in Cancer. Pathophysiology. 2021; 28(3):437-456. https://doi.org/10.3390/pathophysiology28030028
Chicago/Turabian StyleShackelford, Rodney E., Islam Z. Mohammad, Andrew T. Meram, David Kim, Fawaz Alotaibi, Stavan Patel, Ghali E. Ghali, and Christopher G. Kevil. 2021. "Molecular Functions of Hydrogen Sulfide in Cancer" Pathophysiology 28, no. 3: 437-456. https://doi.org/10.3390/pathophysiology28030028
APA StyleShackelford, R. E., Mohammad, I. Z., Meram, A. T., Kim, D., Alotaibi, F., Patel, S., Ghali, G. E., & Kevil, C. G. (2021). Molecular Functions of Hydrogen Sulfide in Cancer. Pathophysiology, 28(3), 437-456. https://doi.org/10.3390/pathophysiology28030028