
Interaction of TLK1 and AKTIP as a Potential Regulator of AKT Activation in Castration-Resistant Prostate Cancer Progression

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids and Antibodies
2.2. Cell Culture
2.3. Cell Treatment
2.4. Cell Transfection
2.5. His Pull-Down Assay
2.6. Co-Immunoprecipitation (Co-IP)
2.7. Site-Directed Mutagenesis
2.8. In Vitro Kinase (IVK) Assay
2.9. Identification of AKTIP Phospho-Peptides
2.10. Cell Proliferation Assay
2.11. Western Blotting
3. Results
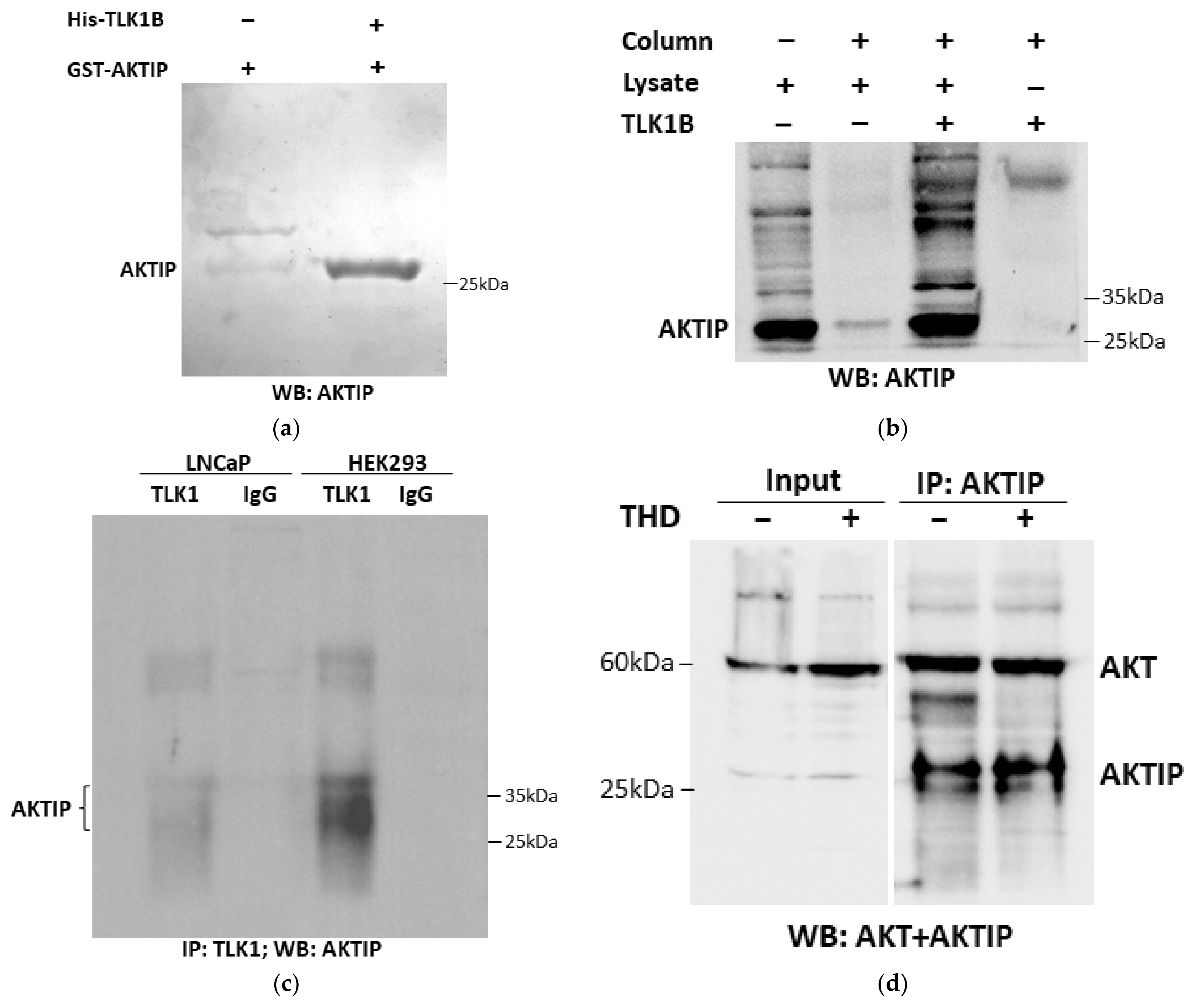
3.1. TLK1 Interactions with AKTIP Both In Vitro and In Vivo
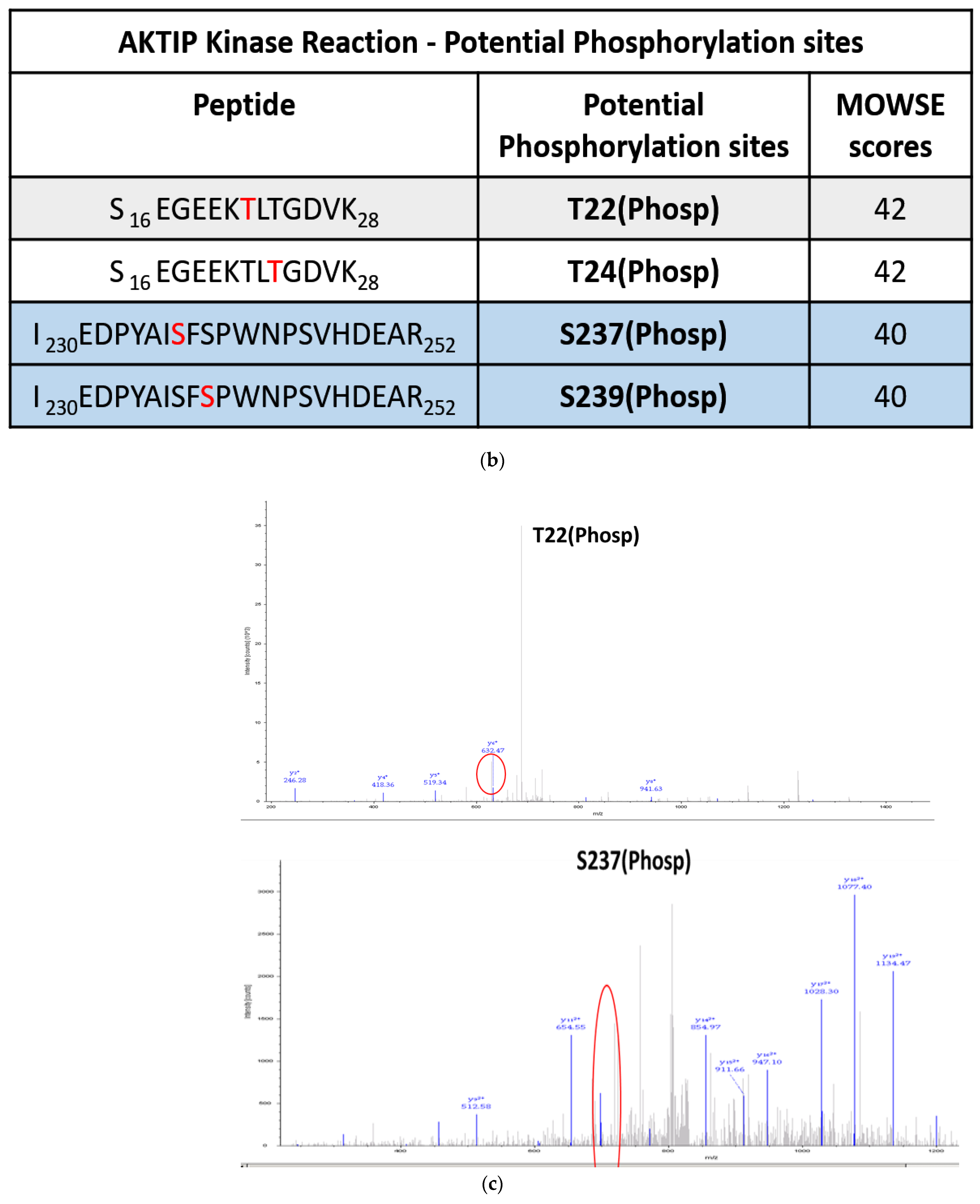
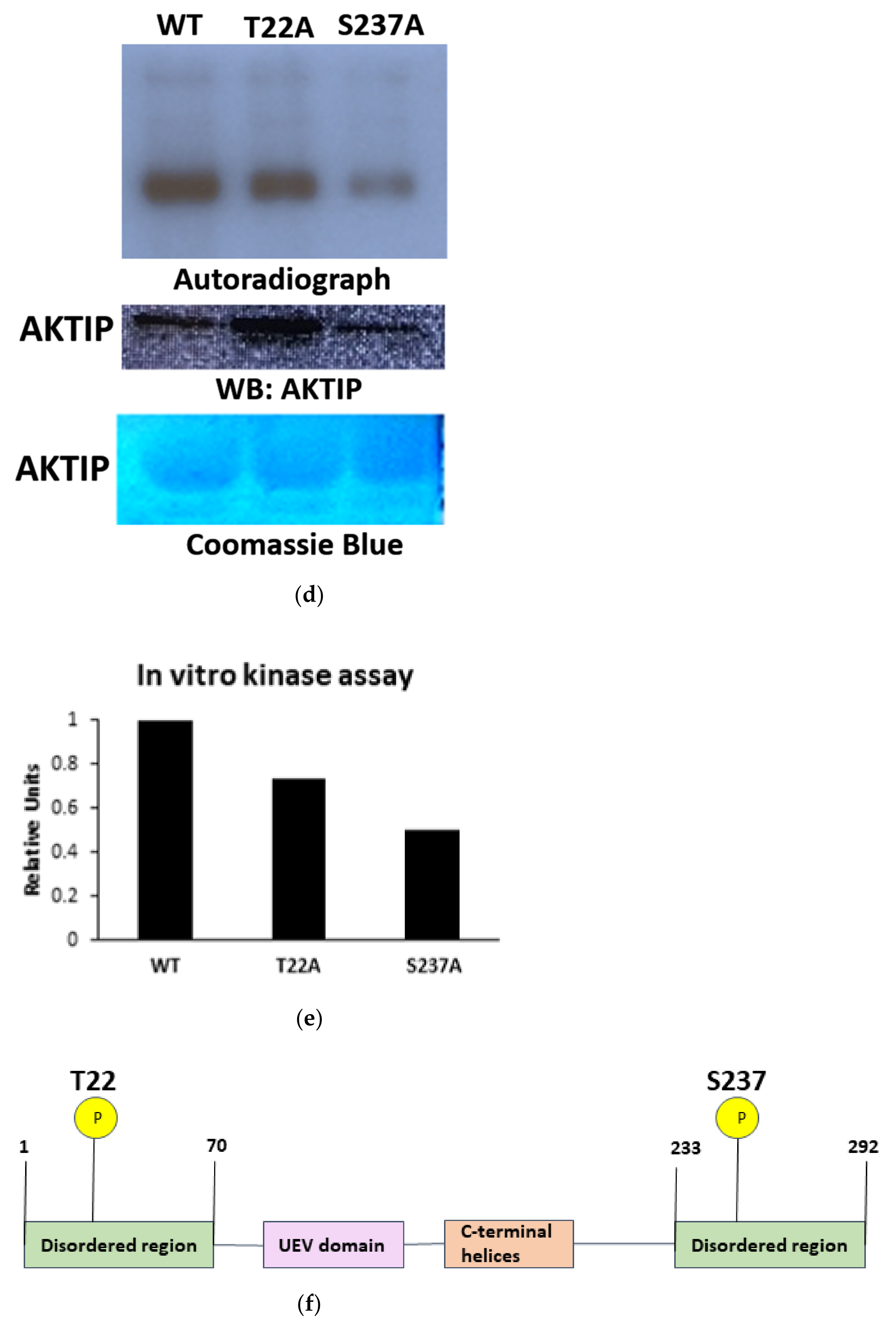
3.2. TLK1 Phosphorylates AKTIP In Two Unique Residues, Which Lie in the Intrinsically Disordered Region
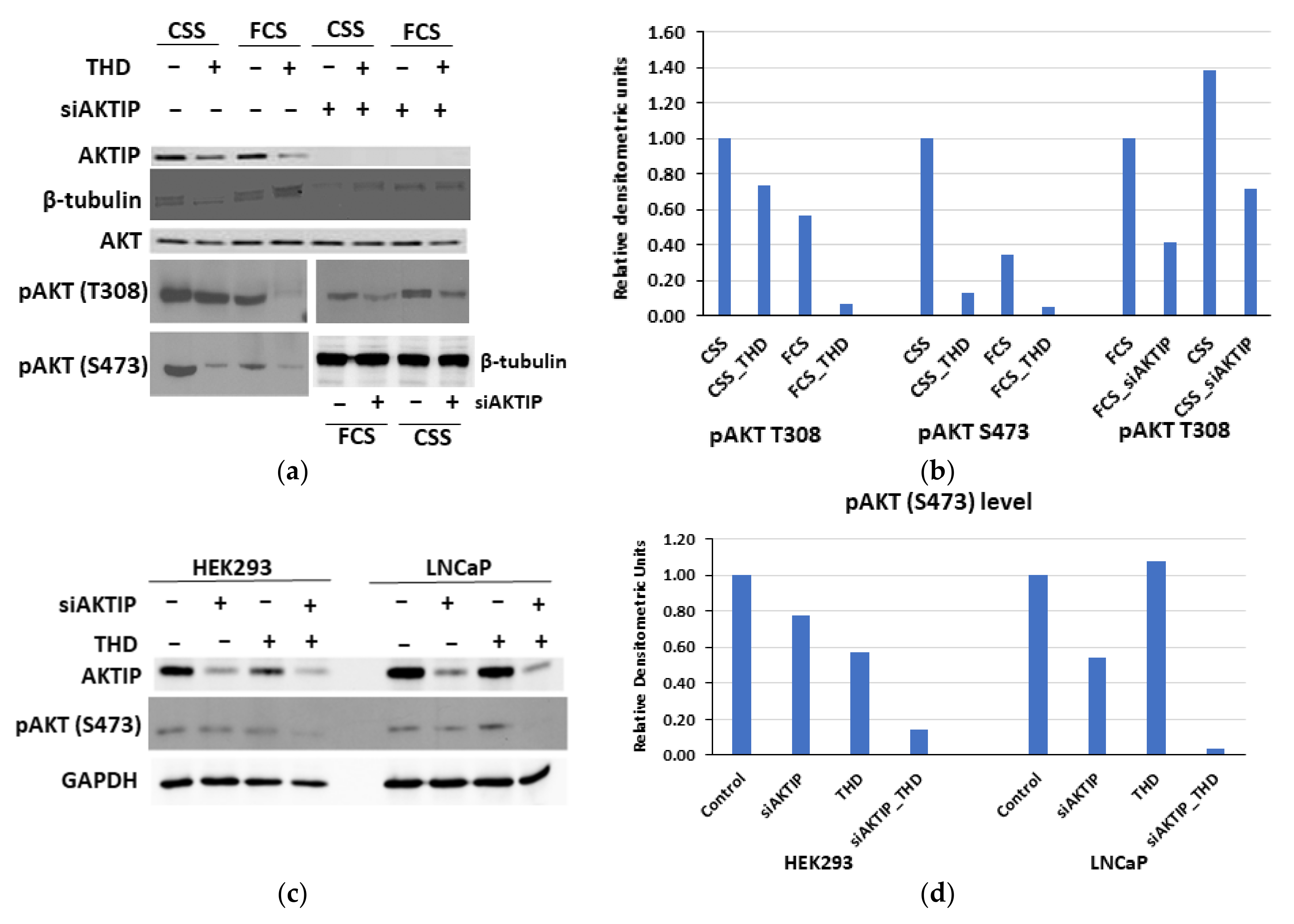
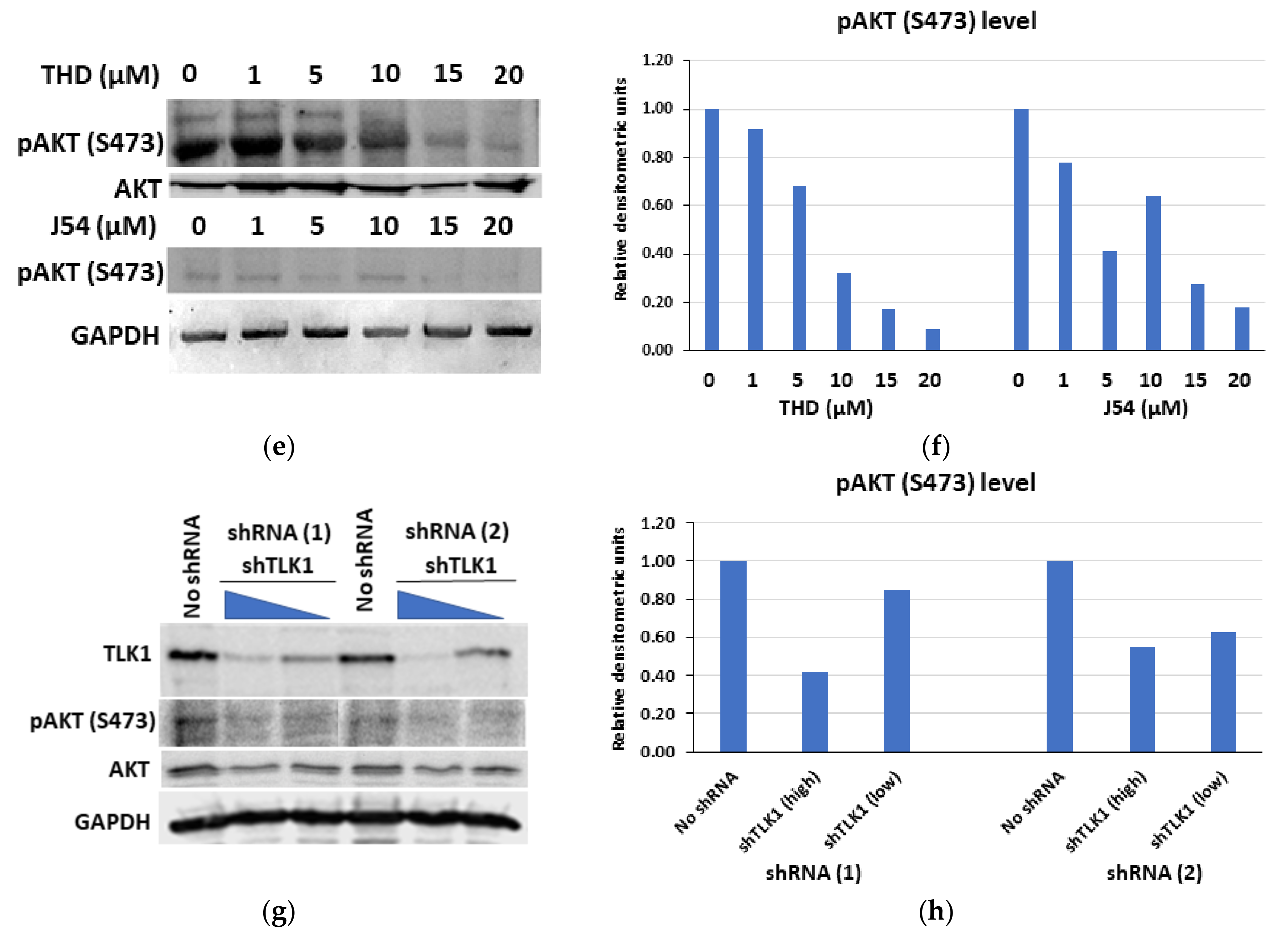
3.3. The Genetic Depletion and Pharmacologic Inhibition of Both TLK1 and AKTIP Results in Reduced Activating Phosphorylation of AKT
3.4. TLK1 Inhibition Reduces Cellular Proliferation Rate, Probably by Downregulating AKT Activation, in Addition to Other Pathways
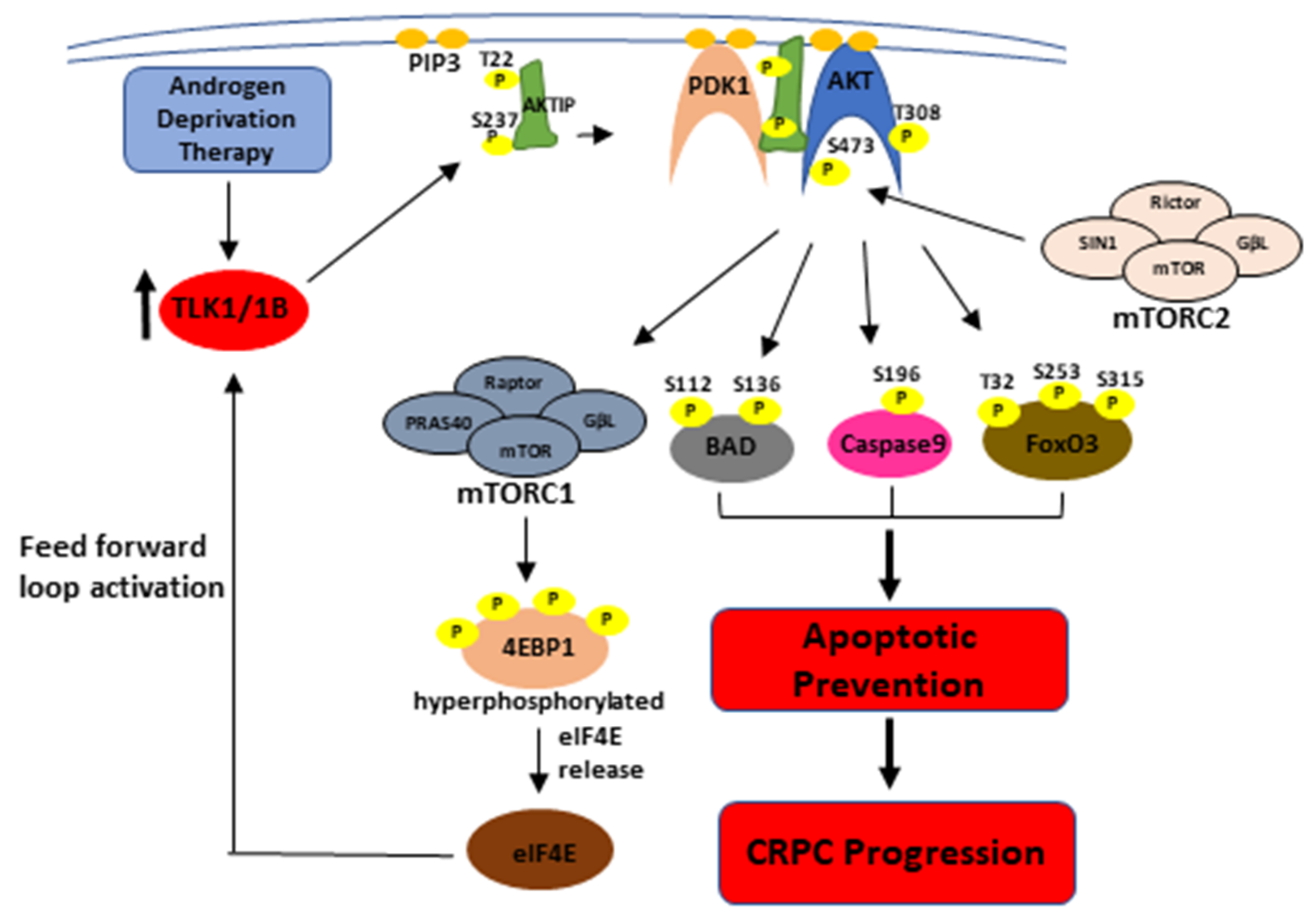
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Denmeade, S.R.; Isaacs, J.T. Activation of programmed (apoptotic) cell death for the treatment of prostate cancer. Adv. Pharmacol. 1996, 35, 281–306. [Google Scholar]
- Gurumurthy, S.; Vasudevan, K.M.; Rangnekar, V.M. Regulation of Apoptosis in Prostate Cancer. Cancer Metastasis Rev. 2001, 20, 225–243. [Google Scholar] [CrossRef]
- Silljé, H.; Takahashi, K.; Tanaka, K.; Van Houwe, G.; Nigg, E. Mammalian homologues of the plant Tousled gene code for cell-cycle-regulated kinases with maximal activities linked to ongoing DNA replication. EMBO J. 1999, 18, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- Silljé, H.H.; Nigg, E.A. Identification of human Asf1 chromatin assembly factors as substrates of Tousled-like kinases. Curr. Biol. 2001, 11, 1068–1073. [Google Scholar] [CrossRef]
- Sunavala-Dossabhoy, G.; Li, Y.; Williams, B.; De Benedetti, A. A dominant negative mutant of TLK1 causes chromosome missegregation and aneuploidy in normal breast epithelial cells. BMC Cell Biol. 2003, 4, 16. [Google Scholar] [CrossRef]
- Han, Z.; Saam, J.R.; Adams, H.P.; Mango, S.E.; Schumacher, J.M. The C. elegans Tousled-like Kinase (TLK-1) Has an Essential Role in Transcription. Curr. Biol. 2003, 13, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, A. The Tousled-Like-Kinases as guardians of genome integrity. ISRN Mol. Biol. 2012, 2012. [Google Scholar] [CrossRef]
- Segura-Bayona, S.; Stracker, T.H. The Tousled-like kinases regulate genome and epigenome stability: Implications in development and disease. Cell. Mol. Life Sci. 2019, 76, 3827–3841. [Google Scholar] [CrossRef]
- Sunavala-Dossabhoy, G. Preserving salivary gland physiology against genotoxic damage - the Tousled way. Oral Dis. 2018, 24, 1390–1398. [Google Scholar] [CrossRef]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of androgen independent prostate tumors. Int. J. Cancer 2019, 145, 1055–1067. [Google Scholar] [CrossRef]
- Singh, V.; Khalil, I.; De Benedetti, A. The TLK1/Nek1 axis contributes to mitochondrial integrity and apoptosis prevention via phosphorylation of VDAC1. Cell Cycle 2020, 19, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Khalil, I.; Ghosh, I.; Singh, V.; Chen, J.; Zhu, H.; De Benedetti, A. NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output. Cancers 2020, 12, 3666. [Google Scholar] [CrossRef]
- Singh, V.; Connelly, Z.M.; Shen, X.; De Benedetti, A. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle 2017, 16, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Lesche, R.; Peetz, A.; Van Der Hoeven, F.; Ruther, U. Ftl, a novel gene related to ubiquitin-conjugating enzymes, is deleted in the Fused toes mouse mutation. Mamm. Genome 1997, 8, 879–883. [Google Scholar] [CrossRef]
- Van Der Hoeven, F.; Schimmang, T.; Volkmann, A.; Mattei, M.-G.; Kyewski, B.; Ruther, U. Programmed cell death is affected in the novel mouse mutant Fused toes (Ft). Development 1994, 120, 2601–2607. [Google Scholar] [CrossRef] [PubMed]
- Remy, I.; Michnick, S.W. Regulation of Apoptosis by the Ft1 Protein, a New Modulator of Protein Kinase B/Akt. Mol. Cell. Biol. 2004, 24, 1493–1504. [Google Scholar] [CrossRef]
- Burla, R.; Carcuro, M.; Raffa, G.D.; Galati, A.; Raimondo, D.; Rizzo, A.; La Torre, M.; Micheli, E.; Ciapponi, L.; Cenci, G.; et al. AKTIP/Ft1, a New Shelterin-Interacting Factor Required for Telomere Maintenance. PLoS Genet. 2015, 11, e1005167. [Google Scholar] [CrossRef]
- Burla, R.; Carcuro, M.; La Torre, M.; Fratini, F.; Crescenzi, M.; D’Apice, M.R.; Spitalieri, P.; Raffa, G.D.; Astrologo, L.; Lattanzi, G.; et al. The telomeric protein AKTIP interacts with A- and B-type lamins and is involved in regulation of cellular senescence. Open Biol. 2016, 6. [Google Scholar] [CrossRef]
- Prabakaran, D.; Muthusami, S.; Sivaraman, T.; Yu, J.-R.; Park, W.-Y. Silencing of FTS increases radiosensitivity by blocking radiation-induced Notch1 activation and spheroid formation in cervical cancer cells. Int. J. Biol. Macromol. 2019, 126, 1318–1325. [Google Scholar] [CrossRef]
- Muthusami, S.; Prabakaran, D.S.; Yu, J.-R.; Park, W.-Y. FTS is responsible for radiation-induced nuclear phosphorylation of EGFR and repair of DNA damage in cervical cancer cells. J. Cancer Res. Clin. Oncol. 2014, 141, 203–210. [Google Scholar] [CrossRef]
- Brazil, D.; Hemmings, B.A. Ten years of protein kinase B signalling: A hard Akt to follow. Trends Biochem. Sci. 2001, 26, 657–664. [Google Scholar] [CrossRef]
- Brazil, D.P.; Park, J.; Hemmings, B.A. PKB Binding Proteins: Getting in on the Akt. Cell 2002, 111, 293–303. [Google Scholar] [CrossRef]
- A Lawlor, M.; Alessi, D. PKB/Akt: A key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 2001, 114, 2903–2910. [Google Scholar] [CrossRef] [PubMed]
- Belham, C.; Wu, S.; Avruch, J. Intracellular signalling: PDK1–a kinase at the hub of things. Curr. Biol. 1999, 9, R93–R96. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Alessi, D.R. The PI3K–PDK1 connection: More than just a road to PKB. Biochem. J. 2000, 346, 561–576. [Google Scholar] [CrossRef]
- Zinda, M.J.; Johnson, A.M.; Paul, J.D.; Horn, C.; Konicek, B.W.; Lu, Z.H.; Sandusky, G.; E Thomas, J.; Neubauer, B.L.; Lai, M.T.; et al. AKT-1, -2, and -3 are expressed in both normal and tumor tissues of the lung, breast, prostate, and colon. Clin. Cancer Res. 2001, 7, 2475–2479. [Google Scholar]
- Lin, H.-K.; Yeh, S.; Kang, H.-Y.; Chang, C. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 7200–7205. [Google Scholar] [CrossRef] [PubMed]
- Bhoir, S.; Shaik, A.; Thiruvenkatam, V.; Kirubakaran, S. High yield bacterial expression, purification and characterisation of bioactive Human Tousled-like Kinase 1B involved in cancer. Sci. Rep. 2018, 8, 4796. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Bhoir, S.; Chikhale, R.; Hussain, J.; Dwyer, D.; Bryce, R.A.; Kirubakaran, S.; De Benedetti, A. Generation of Phenothiazine with Potent Anti-TLK1 Activity for Prostate Cancer Therapy. iScience 2020, 23. [Google Scholar] [CrossRef]
- Khalil, M.I.; Singh, V.; King, J.; De Benedetti, A. TLK1-MK5 Axis Drives Prostate Cancer Cell Motility and Pathologic Features of Aggressiveness. 2021. Available online: https://assets.researchsquare.com/files/rs-434116/v1/508d23e1-6d71-4f47-b966-5830cf7306e2.pdf?c=1619546029 (accessed on 1 May 2021).
- Lu, K.P.; Hunter, T. Evidence for a NIMA-like mitotic pathway in vertebrate cells. Cell 1995, 81, 413–424. [Google Scholar] [CrossRef][Green Version]
- Gao, N.; Flynn, D.C.; Zhang, Z.; Zhong, X.-S.; Walker, V.; Liu, K.J.; Shi, X.; Jiang, B.-H. G1cell cycle progression and the expression of G1cyclins are regulated by PI3K/AKT/mTOR/p70S6K1 signaling in human ovarian cancer cells. Am. J. Physiol. Physiol. 2004, 287, C281–C291. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, L.; Wu, X.; Li, R.; Wen, J.; Sha, J.; Wen, X. The PI3K/AKT pathway in the pathogenesis of prostate cancer. Front. Biosci. Landmark Ed. 2016, 21, 1084–1091. [Google Scholar]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.; et al. Cell Autonomous Role of PTEN in Regulating Castration-Resistant Prostate Cancer Growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arija, J.A.A.; Shih, K.C.; Radavoi, G.D.; Xu, N. Randomized phase II study evaluating Akt blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Sunavala-Dossabhoy, G.; Fowler, M.; De Benedetti, A. Translation of the radioresistance kinase TLK1B is induced by gamma-irradiation through activation of mTOR and phosphorylation of 4E-BP1. BMC Mol. Biol. 2004, 5, 1. [Google Scholar] [CrossRef]
- Norton, K.S.; McClusky, D.; Sen, S.; Yu, H.; Meschonat, C.; Debenedetti, A.; Li, B.D. TLK1B is elevated with eIF4E overexpression in breast cancer. J. Surg. Res. 2004, 116, 98–103. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of Cell Death Protease Caspase-9 by Phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta BBA Bioenerg. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Oudard, S.; Fizazi, K.; Sengeløv, L.; Daugaard, G.; Saad, F.; Hansen, S.; Hjälm-Eriksson, M.; Jassem, J.; Thiery-Vuillemin, A.; Caffo, O.; et al. Cabazitaxel Versus Docetaxel As First-Line Therapy for Patients With Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase III Trial—FIRSTANA. J. Clin. Oncol. 2017, 35, 3189–3197. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, M.I.; Madere, C.; Ghosh, I.; Adam, R.M.; De Benedetti, A. Interaction of TLK1 and AKTIP as a Potential Regulator of AKT Activation in Castration-Resistant Prostate Cancer Progression. Pathophysiology 2021, 28, 339-354. https://doi.org/10.3390/pathophysiology28030023
Khalil MI, Madere C, Ghosh I, Adam RM, De Benedetti A. Interaction of TLK1 and AKTIP as a Potential Regulator of AKT Activation in Castration-Resistant Prostate Cancer Progression. Pathophysiology. 2021; 28(3):339-354. https://doi.org/10.3390/pathophysiology28030023
Chicago/Turabian StyleKhalil, Md Imtiaz, Christopher Madere, Ishita Ghosh, Rosalyn M. Adam, and Arrigo De Benedetti. 2021. "Interaction of TLK1 and AKTIP as a Potential Regulator of AKT Activation in Castration-Resistant Prostate Cancer Progression" Pathophysiology 28, no. 3: 339-354. https://doi.org/10.3390/pathophysiology28030023
APA StyleKhalil, M. I., Madere, C., Ghosh, I., Adam, R. M., & De Benedetti, A. (2021). Interaction of TLK1 and AKTIP as a Potential Regulator of AKT Activation in Castration-Resistant Prostate Cancer Progression. Pathophysiology, 28(3), 339-354. https://doi.org/10.3390/pathophysiology28030023