
Developing ROR1 Targeting CAR-T Cells against Solid Tumors in Preclinical Studies

 ,
,
Abstract
:Simple Summary
Abstract
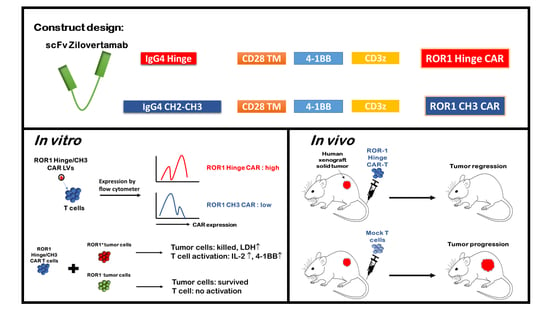
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. CAR Constructs and Lentivirus Preparation
2.3. Generation of ROR-1 CAR-T
2.4. LDH Assay
2.5. Flow Cytometry
2.6. Animal Protocol
2.7. Statistical Analyses
3. Results
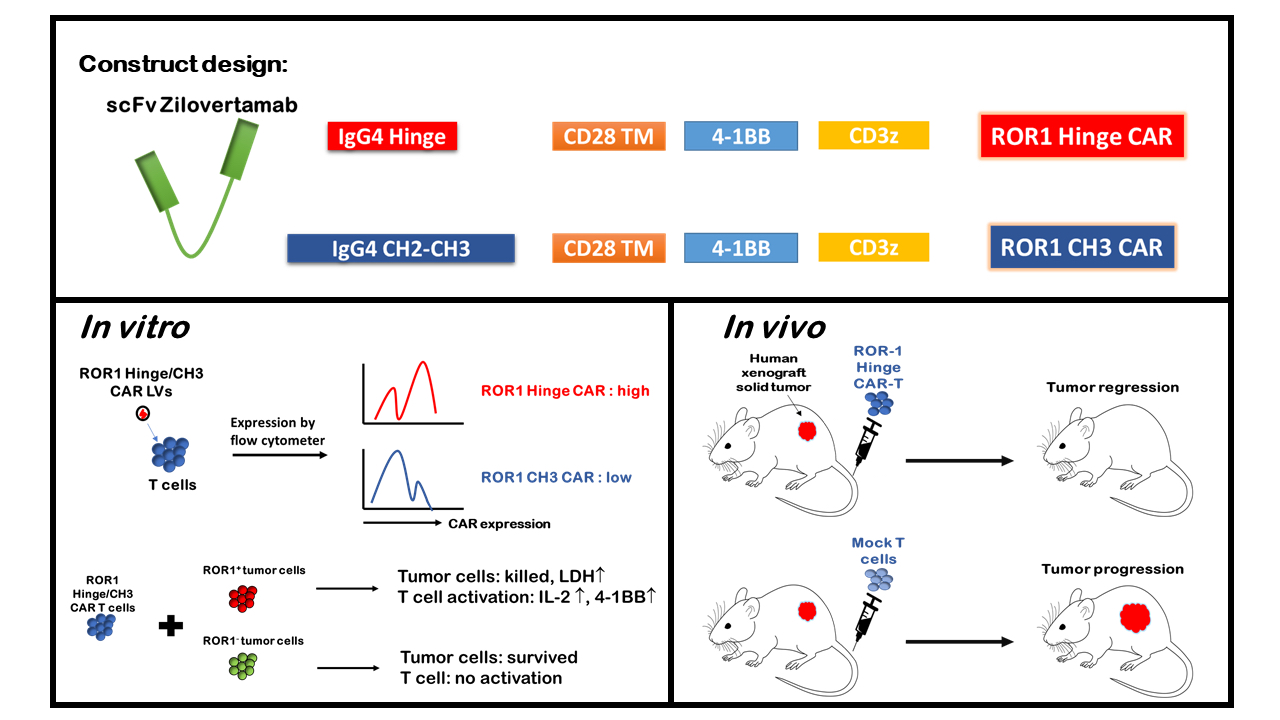
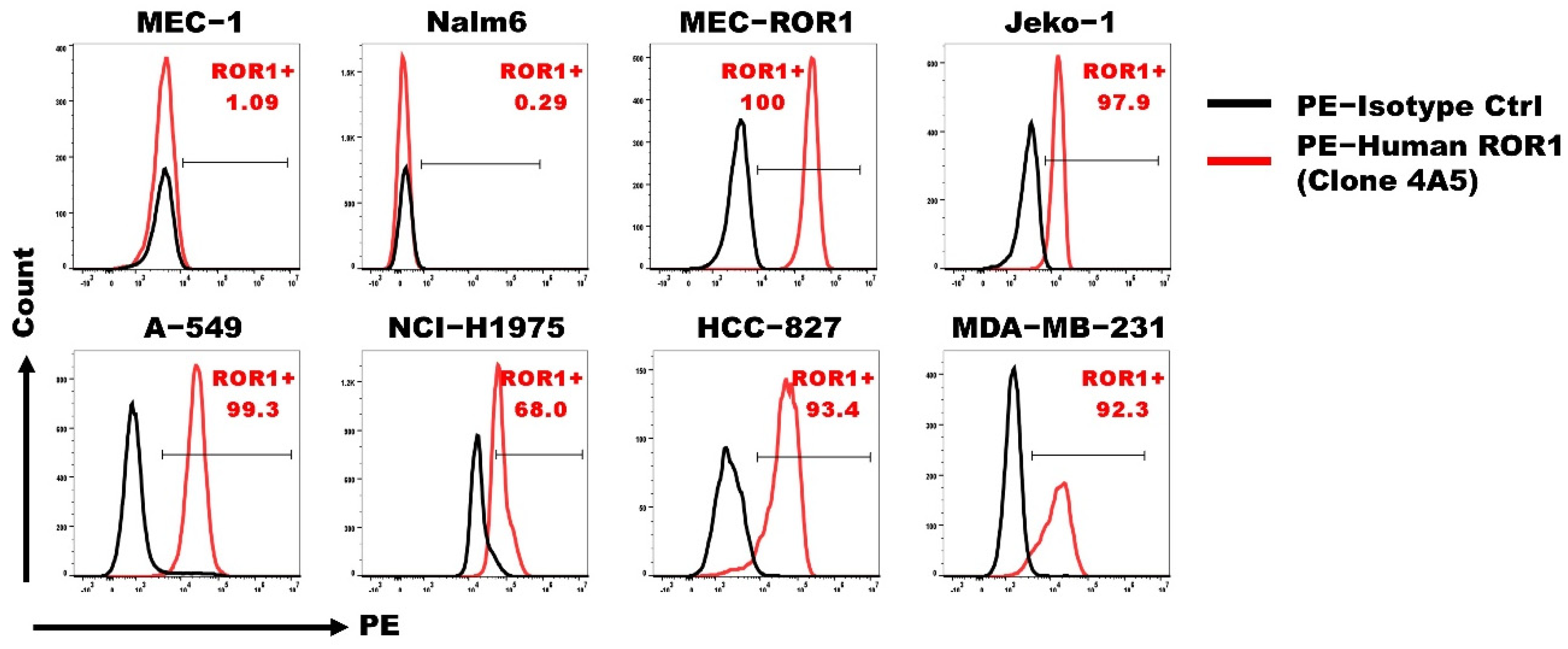
3.1. ROR1 Is Expressed on Various Human Tumor Cell Lines
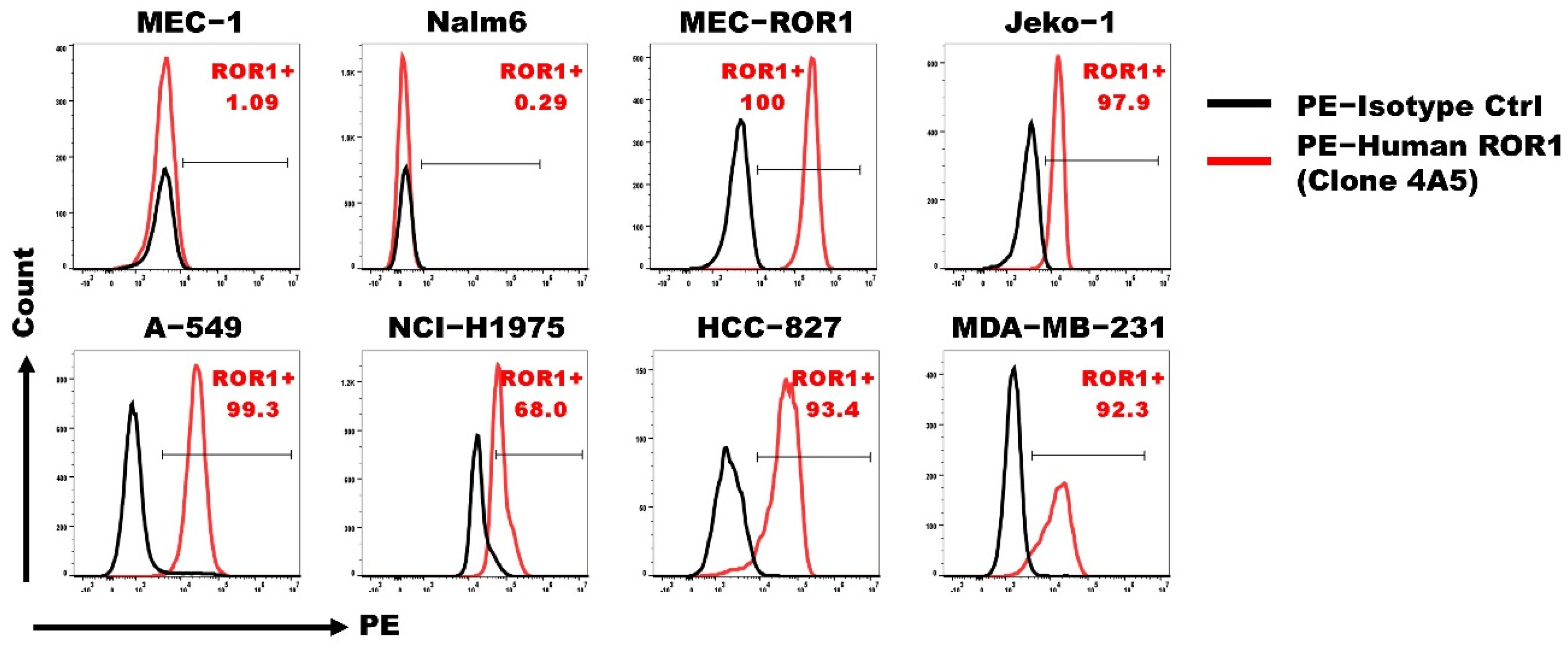
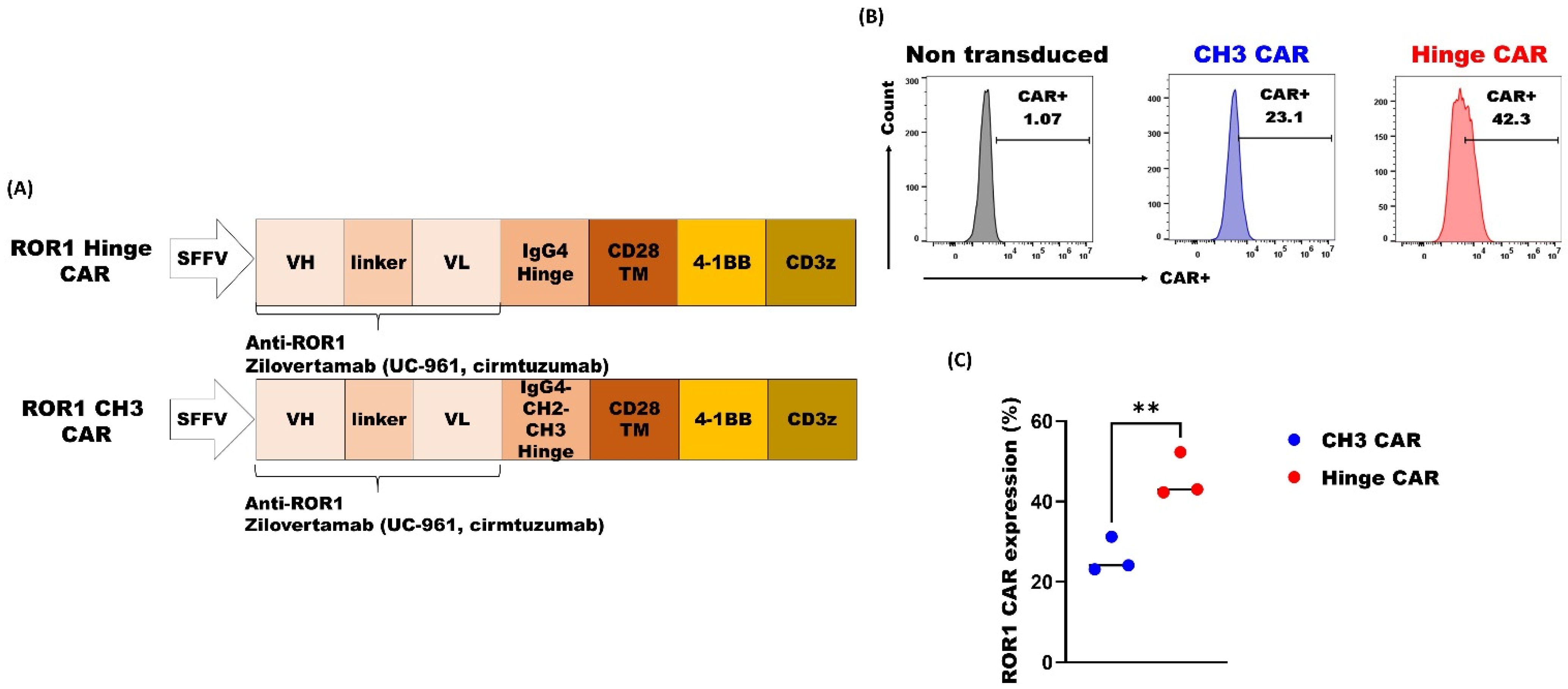
3.2. ROR1 CAR with Shorter Spacer Region Exhibited Superior Expression
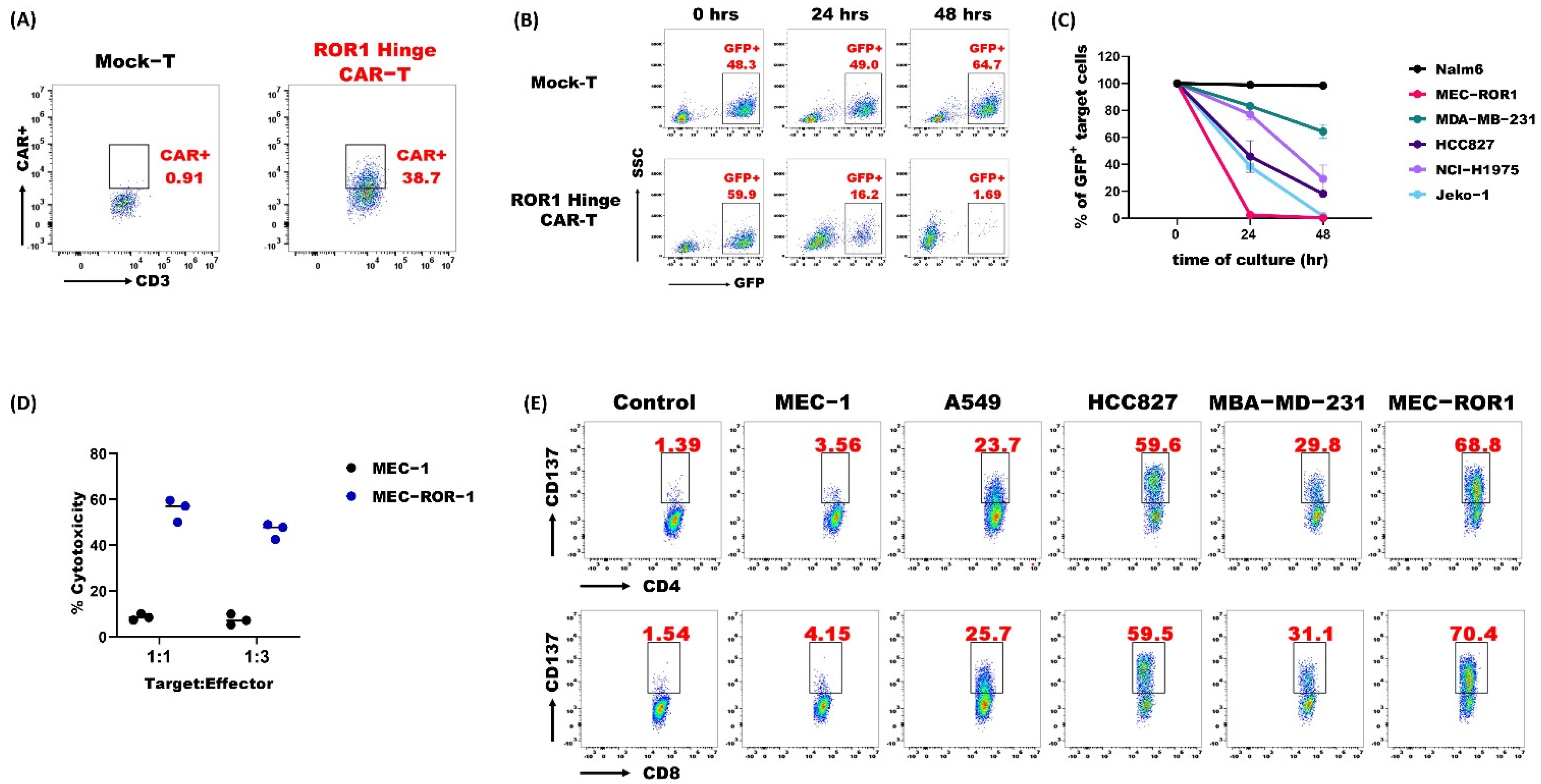
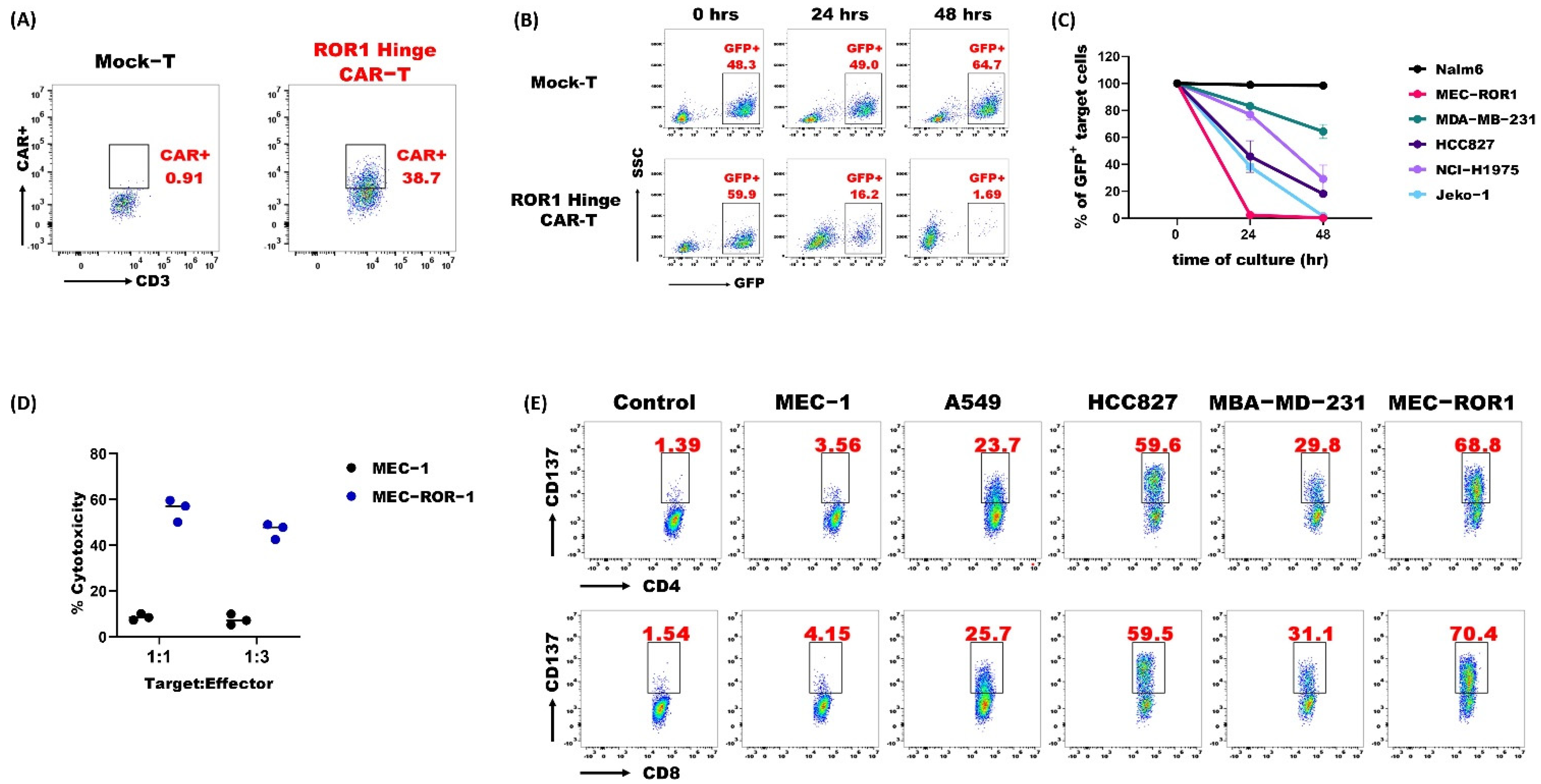
3.3. ROR1 Hinge CAR-T-Cells Demonstrated Potent Anti-Tumor Activities
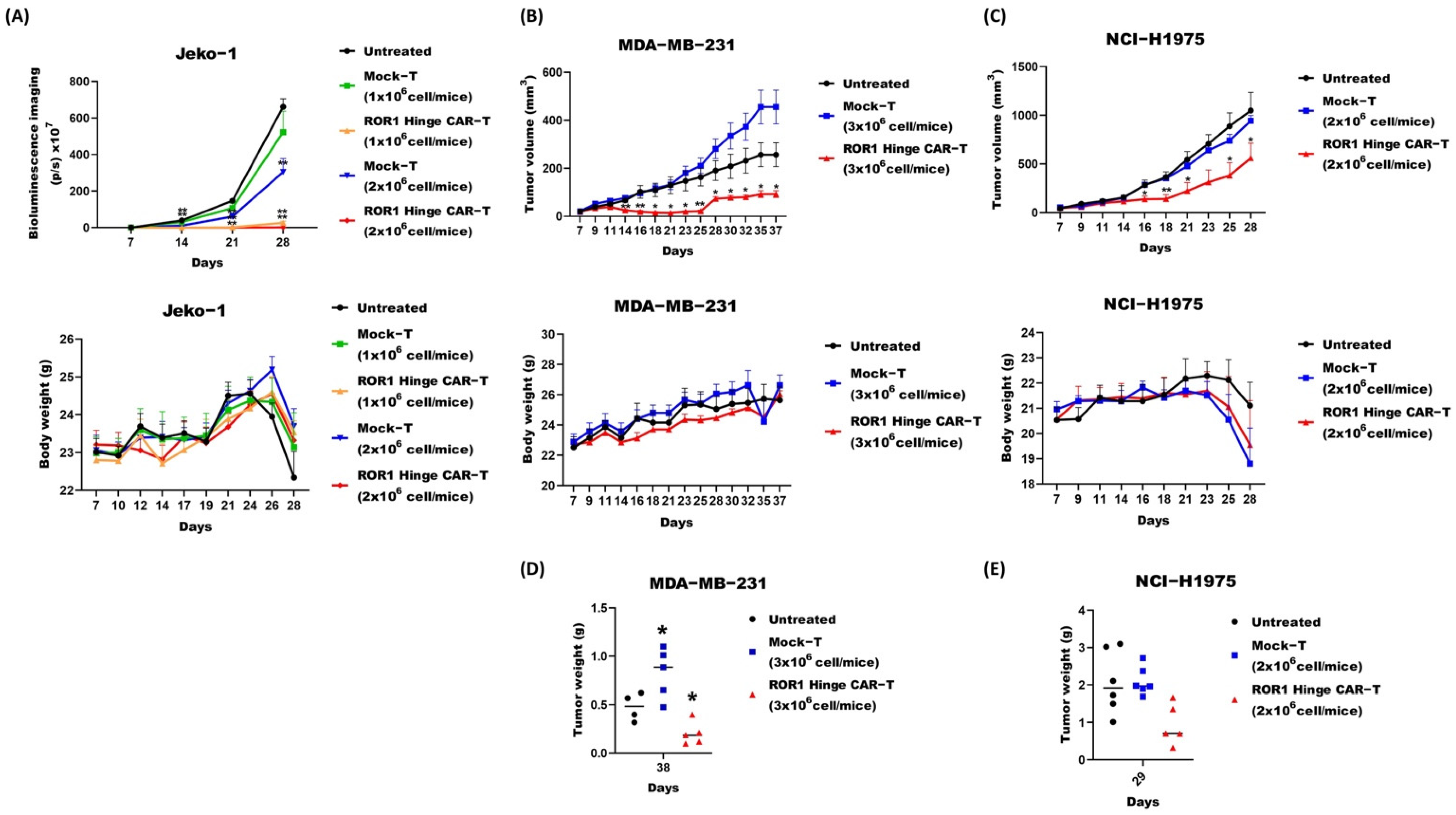
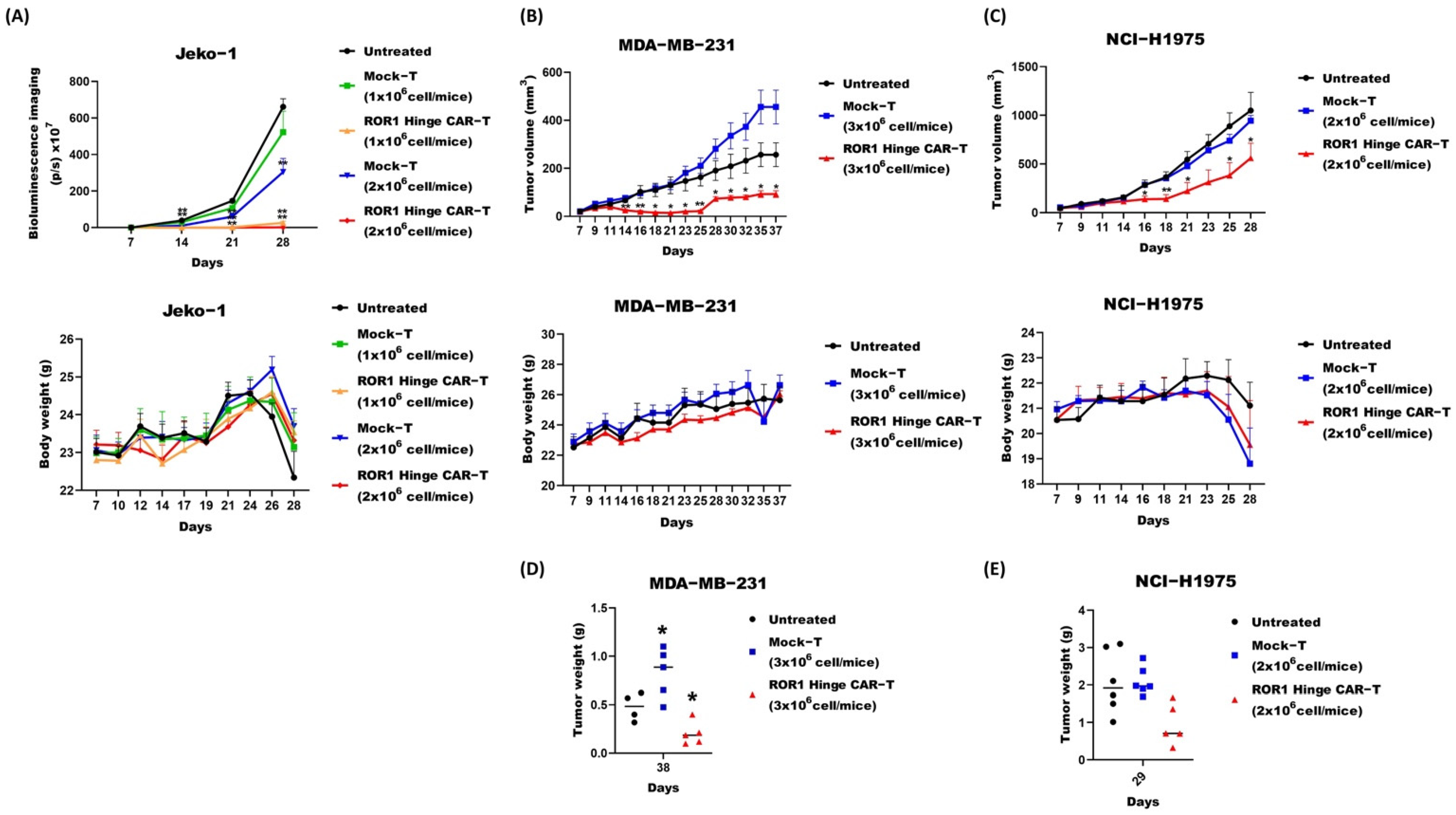
3.4. ROR1 Hinge CAR Cells also Demonstrated Promising Effect on Murine Solid Tumor Models
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. eBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokarew, N.; Ogonek, J.; Endres, S.; Von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Shabani, M.; Naseri, J.; Shokri, F. Receptor tyrosine kinase-like orphan receptor 1: A novel target for cancer immunotherapy. Expert Opin. Ther. Targets 2015, 19, 941–955. [Google Scholar] [CrossRef]
- Matsuda, T.; Nomi, M.; Ikeya, M.; Kani, S.; Oishi, I.; Terashima, T.; Takada, S.; Minami, Y. Expression of the receptor tyrosine kinase genes, Ror1 and Ror2, during mouse development. Mech. Dev. 2001, 105, 153–156. [Google Scholar] [CrossRef]
- Stricker, S.; Rauschenberger, V.; Schambony, A. ROR-Family Receptor Tyrosine Kinases. Curr. Top. Dev. Biol. 2017, 123, 105–142. [Google Scholar]
- Balakrishnan, A.; Goodpaster, T.; Randolph-Habecker, J.; Hoffstrom, B.G.; Jalikis, F.G.; Koch, L.K.; Berger, C.; Kosasih, P.L.; Rajan, A.; Sommermeyer, D.; et al. Analysis of ROR1 Protein Expression in Human Cancer and Normal Tissues. Clin. Cancer Res. 2017, 23, 3061–3071. [Google Scholar] [CrossRef] [Green Version]
- Hudecek, M.; Schmitt, T.M.; Baskar, S.; Lupo-Stanghellini, M.T.; Nishida, T.; Yamamoto, T.N.; Bleakley, M.; Turtle, C.J.; Chang, W.C.; Greisman, H.A.; et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood 2010, 116, 4532–4541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Zhou, S.; Wang, H.; Li, Y.; Xiao, X.; Yang, J. Stable silencing of ROR1 regulates cell cycle, apoptosis, and autophagy in a lung adenocarcinoma cell line. Int. J. Clin. Exp. Pathol. 2020, 13, 1108–1120. [Google Scholar] [PubMed]
- Zhang, S.; Cui, B.; Lai, H.; Liu, G.; Ghia, E.M.; Widhopf, G.F.; Zhang, Z.; Wu, C.C.; Chen, L.; Wu, R.; et al. Ovarian cancer stem cells express ROR1, which can be targeted for anti-cancer-stem-cell therapy. Proc. Natl. Acad. Sci. USA 2014, 111, 17266–17271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhang, H.; Ghia, E.M.; Huang, J.; Wu, L.; Zhang, J.; Lam, S.; Lei, Y.; He, J.; Cui, B.; et al. Inhibition of chemotherapy resistant breast cancer stem cells by a ROR1 specific antibody. Proc. Natl. Acad. Sci. USA 2019, 116, 1370–1377. [Google Scholar] [CrossRef] [Green Version]
- Mani, R.; Mao, Y.; Frissora, F.W.; Chiang, C.-L.; Wang, J.; Zhao, Y.; Wu, Y.; Yu, B.; Yan, R.; Mo, X.; et al. Tumor antigen ROR1 targeted drug delivery mediated selective leukemic but not normal B-cell cytotoxicity in chronic lymphocytic leukemia. Leukemia 2015, 29, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Daneshmanesh, A.H.; Hojjat-Farsangi, M.; Khan, A.S.; Jeddi-Tehrani, M.; Akhondi, M.M.; A Bayat, A.; Ghods, R.; Mahmoudi, A.-R.; Hadavi, R.; Österborg, A.; et al. Monoclonal antibodies against ROR1 induce apoptosis of chronic lymphocytic leukemia (CLL) cells. Leukemia 2012, 26, 1348–1355. [Google Scholar] [CrossRef] [Green Version]
- Vaisitti, T.; Arruga, F.; Vitale, N.; Lee, T.-T.; Ko, M.; Chadburn, A.; Braggio, E.; Di Napoli, A.; Iannello, A.; Allan, J.N.; et al. ROR1 targeting with the antibody-drug conjugate VLS-101 is effective in Richter syndrome patient-derived xenograft mouse models. Blood 2021, 137, 3365–3377. [Google Scholar] [CrossRef]
- Berger, C.; Sommermeyer, D.; Hudecek, M.; Berger, M.; Balakrishnan, A.; Paszkiewicz, P.J.; Kosasih, P.L.; Rader, C.; Riddell, S.R. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol. Res. 2015, 3, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Salter, A.I.; Liggitt, D.; Yechan-Gunja, S.; Sarvothama, M.; Cooper, K.; Smythe, K.S.; Dudakov, J.A.; Pierce, R.H.; Rader, C.; et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 2019, 35, 489–503.e8. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.Y.; Widhopf, G.F., II; Wu, C.C.; Cui, B.; Lao, F.; Sadarangani, A.; Cavagnaro, J.; Prussak, C.; Carson, D.A.; Jamieson, C.; et al. Pre-clinical Specificity and Safety of UC-961, a First-In-Class Monoclonal Antibody Targeting ROR1. Clin. Lymphoma Myeloma Leuk. 2015, 15, S167–S169. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Choi, M.Y.; Siddiqi, T.; Barrientos, J.C.; Wierda, W.G.; Isufi, I.; Tuscano, J.M.; Lamanna, N.; Subbiah, S.; Koff, J.L.; et al. Phase 1/2 study of cirmtuzumab and ibrutinib in mantle cell lymphoma (MCL) or chronic lymphocytic leukemia (CLL), in 2021 ASCO Annual Meeting 2021. J. Clin. Oncol. 2021, 39, 7556. [Google Scholar] [CrossRef]
- Guest, R.D.; Hawkins, R.E.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O’Neill, A.; Irlam, J.; Chester, K.A.; Kemshead, J.T.; Shaw, D.M.; et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: Evaluation of four different scFvs and antigens. J. Immunother. 2005, 28, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.Y.; Widhopf, G.F., 2nd; Ghia, E.M.; Kidwell, R.; Hasan, K.; Yu, J.; Rassenti, L.Z.; Chen, L.; Chen, Y.; Pittman, E.; et al. Phase I Trial: Cirmtuzumab Inhibits ROR1 Signaling and Stemness Signatures in Patients with Chronic Lymphocytic Leukemia. Cell Stem Cell 2018, 22, 951–959.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [Green Version]
- Wallstabe, L.; Göttlich, C.; Nelke, L.C.; Kühnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L.; et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight 2019, 4, e126345. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Furlan, S.N.; Jaeger-Ruckstuhl, C.A.; Sarvothama, M.; Berger, C.; Smythe, K.S.; Garrison, S.M.; Specht, J.M.; Lee, S.M.; Amezquita, R.A.; et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy when Combined with Checkpoint Blockade. Cancer Cell 2021, 39, 193–208.e10. [Google Scholar] [CrossRef]
- Wilkie, S.; Picco, G.; Foster, J.; Davies, D.M.; Julien, S.; Cooper, L.; Arif, S.; Mather, S.J.; Taylor-Papadimitriou, J.; Burchell, J.M.; et al. Retargeting of human T cells to tumor-associated MUC1: The evolution of a chimeric antigen receptor. J. Immunol. 2008, 180, 4901–4909. [Google Scholar] [CrossRef] [Green Version]
- James, S.E.; Greenberg, P.D.; Jensen, M.C.; Lin, Y.; Wang, J.; Till, B.G.; Raubitschek, A.A.; Forman, S.J.; Press, O.W. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J. Immunol. 2008, 180, 7028–7038. [Google Scholar] [CrossRef] [Green Version]
- Galva, L.D.C.; Jiang, X.; Hussein, M.S.; Zhang, H.; Mao, R.; Brody, P.; Peng, Y.; He, A.R.; Kehinde-Ige, M.; Sadek, R.; et al. Novel low-avidity glypican-3 specific CARTs resist exhaustion and mediate durable antitumor effects against HCC. Hepatology 2021, 76, 330–344. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, B.k.; Wan, Y.; Chin, Z.l.; Deng, L.; Deng, M.; Leung, T.m.; Hua, J.; Zhang, H. Developing ROR1 Targeting CAR-T Cells against Solid Tumors in Preclinical Studies. Cancers 2022, 14, 3618. https://doi.org/10.3390/cancers14153618
Lee Bk, Wan Y, Chin Zl, Deng L, Deng M, Leung Tm, Hua J, Zhang H. Developing ROR1 Targeting CAR-T Cells against Solid Tumors in Preclinical Studies. Cancers. 2022; 14(15):3618. https://doi.org/10.3390/cancers14153618
Chicago/Turabian StyleLee, Boon kiat, Yuhua Wan, Zan lynn Chin, Linyan Deng, Mo Deng, Tze ming Leung, Jian Hua, and Hua Zhang. 2022. "Developing ROR1 Targeting CAR-T Cells against Solid Tumors in Preclinical Studies" Cancers 14, no. 15: 3618. https://doi.org/10.3390/cancers14153618
APA StyleLee, B. k., Wan, Y., Chin, Z. l., Deng, L., Deng, M., Leung, T. m., Hua, J., & Zhang, H. (2022). Developing ROR1 Targeting CAR-T Cells against Solid Tumors in Preclinical Studies. Cancers, 14(15), 3618. https://doi.org/10.3390/cancers14153618